
Abstract
This study examined the role of oxygen radicals in pial arteriolar changes during cortical spreading depression (CSD). CSD was induced by microinjection of 5% KCl in anesthetized adult rabbits. Pial diameter was measured with a closed cranial window and intravital microscopy. During control CSD (n = 12), the dilation amplitude and area were 55 ± 14% and 693 ± 69 mm2 (baseline = 76 ± 14 μm), respectively. Oxygen radical scavengers, superoxide dismutase (SOD; 105 U/ml, topical application; n = 5) or oxypurinol (50 mg/kg i.v.; n = 7), did not alter the dilation amplitude and area or change onset latency during CSD. Further, SOD and oxypurinol did not prevent NG-nitro-L-arginine from attenuating arteriolar dilation during CSD (n = 12). We conclude that oxygen radicals do not play a role in the transient dilation of cerebral arterioles during CSD.
Mechanisms underlying cerebrovascular dilation during cortical spreading depression (CSD) are complex and involve competing dilator and constrictor influences. On the one hand, arteriolar dilation is promoted by actions of nitric oxide (NO) and calcitonin gene-related peptide (CGRP) in rabbits (Colonna et al., 1994a, b ) and cats (Goadsby et al., 1992; Wahl et al., 1994), although conflicting evidence concerning NO is available for rats (Duckrow, 1993; Fabricius and Lauritzen, 1994; Zhang et al., 1994). On the other, cyclooxygenase-dependent mechanisms appear to restrain arteriolar dilation during CSD in all species studied (Lauritzen, 1987; Shibata et al., 1990, 1991a). Thus, blockade of cyclooxygenase by indomethacin potentiates CSD-induced arteriolar dilation. In addition to blocking prostanoid production, indomethacin also prevents the enhanced production of superoxide anion under several conditions that involve metabolic or anoxic depolarization of neurons (Armstead et al., 1989; Pourcyrous et al., 1990; Bauknight et al., 1992). Superoxide anion could restrain cerebrovascular dilation during CSD by inhibiting NO synthase (NOS) (Rengasamy and Johns, 1993), inactivating NO before it can reach vascular smooth muscle (Rubanyi and Vanhoutte, 1986; Nelson et al., 1992), or by directly promoting arteriolar constriction (Katusic and Vanhoutte, 1989). However, the participation of superoxide anion in cerebrovascular responses during spreading depression has not been investigated.
The purpose of this study was to examine the role of superoxide anion in mediating arteriolar changes during CSD. We tested the hypothesis that superoxide anion exerts a restrictive influence on cerebrovascular dilation during CSD. In addition, we investigated whether superoxide anion scavengers would block attenuation by NG-nitro-L-arginine (L-NNA) of cerebral arteriolar dilation during CSD. Rosenblum et al. (1992) have reported that superoxide dismutase (SOD) prevents at least one NOS inhibitor from eliminating apparent NO-induced cerebrovascular dilation.
MATERIALS AND METHODS
Female New Zealand rabbits (2.1–2.5 kg) were anesthetized with sodium thiopental (30–50 mg/kg i.v.) and then urethane (1.6–1.8 mg/kg i.p.). Supplemental doses were added to maintain the required level of anesthesia. Rabbits were intubated and a femoral artery and vein were cannulated. Arterial blood pH and gases were within normal values during the experiments. The electroencephalogram was recorded to document elicitation of CSD. A stainless-steel and glass cranial window was inserted into the skull as described previously (Shibata et al., 1990) and was filled with artificial CSF that was warmed to 37°C and equilibrated with 6% O2/6.5% CO2 in N2. Arteriolar diameter was measured using intravital microscopy. CSD was induced by injecting 10 μl of 5% KCl onto the anterior cortex.
Arteriolar diameter was measured continuously. In addition to presenting peak dilation values, we quantified the degree of dilation over time by presenting dilation area. Dilation area, presented in square millimeters, is the area under the dilation curve and above the baseline diameter. Typically, as we have shown before (Shibata et al., 1990), arteriolar diameter increases over baseline for 1–2 min before returning to baseline. Dilation area was measured with the HP DeskScan II and NIH Image 1.43 software.
Experimental protocols
First, a control CSD was induced by microinjection of 5% KCl. In the SOD group (n = 5), SOD (105 U/ml CSF; Sigma) was topically applied five to six times for a 60-min period and allowed to remain under the window during the second CSD. Pial arteriolar responses are virtually identical during at least four repetitive CSDs (Colonna et al., 1994a, b ). We have shown previously that approximately one-half of this dose of SOD is sufficient to scavenge superoxide anions following ischemia or asphyxia (Armstead et al., 1989; Pourcyrous et al., 1990, 1993). In the oxypurinol group (n = 7), oxypurinol (Sigma) was injected intravenously 30 min prior to the second CSD. We have shown this dose of oxypurinol to be an effective scavenger of superoxide anions (Pourcyrous et al., 1993).
Following the second CSD in both groups, we administered 15 mg/kg L-NNA (Sigma) intravenously (10 mg/kg initially plus 5 mg/kg given 30 min later). Another CSD was induced after waiting an additional 30 min (60 min from the first L-NNA administration). We have shown previously that this dose of L-NNA results in ∼90% inhibition of NOS activity in rabbit cortex (Meng et al., 1995).
Statisical analysis
All values are reported as means ± SD. Paired t tests were used for comparing data before and during/after treatments. A value of p < 0.05 was considered as statistically significant.
RESULTS
Effects of SOD on CSD-induced dilation
Topically applied SOD neither altered baseline diameter (79 ± 7 vs. 78 ± 9 μm for pretreatment and SOD treatment, respectively) nor altered the amplitude of cerebral arteriolar dilation during CSD (Fig. 1). In addition, the dilation area was not altered by topical SOD (Fig. 1). Speed of CSD was not altered by SOD. Mean arterial blood pressure was not changed by the SOD treatment (81 ± 9 mm Hg before vs. 78 ± 9 mm Hg after SOD application).
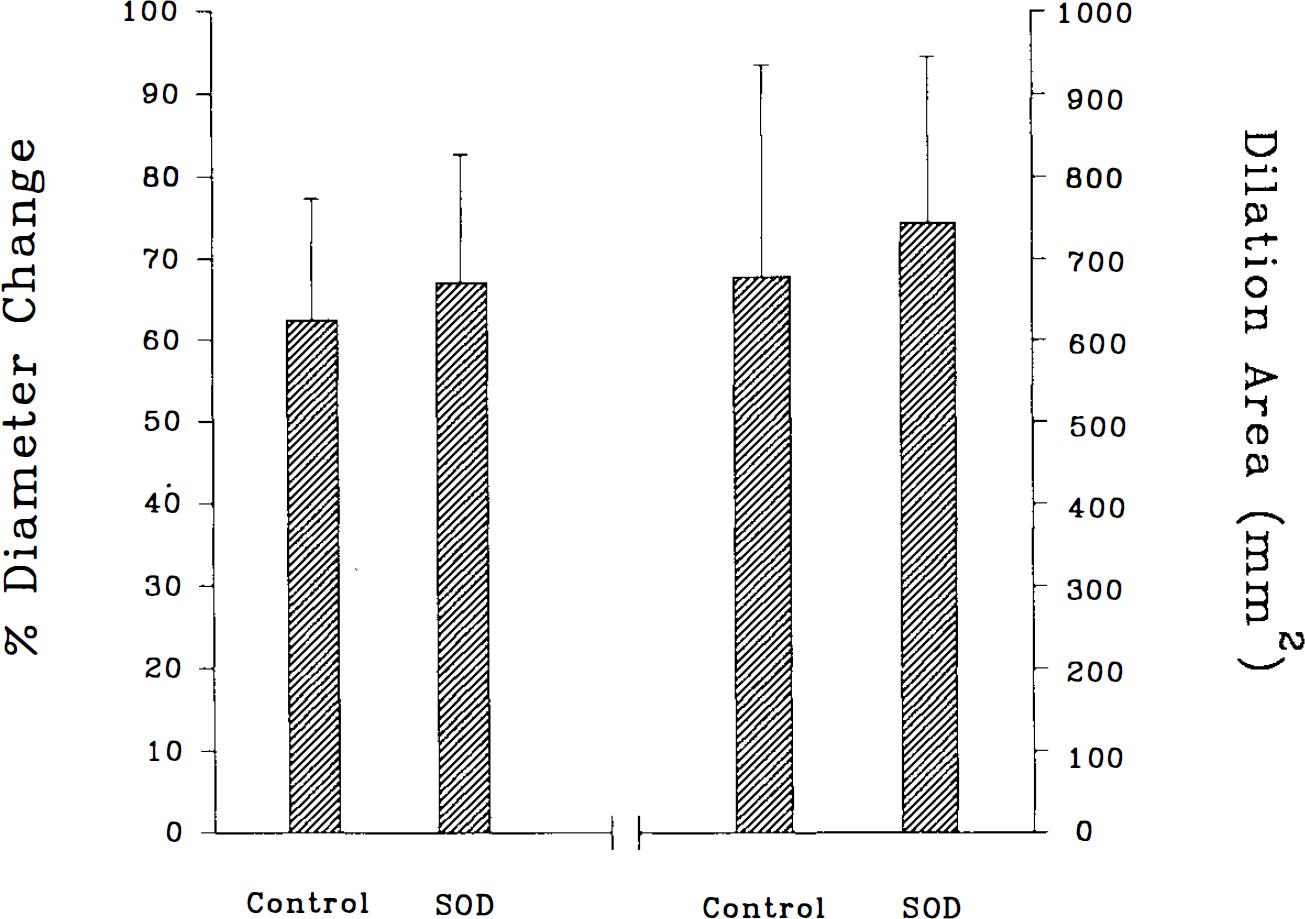
Changes of maximal amplitude and dilation area of the pial arteriolar dilation during cortical spreading depression (CSD) before and after topical application of superoxide dismutase (SOD; 105 U/ml). Topical SOD did not alter arteriolar responses to CSD. Values are means ± SD, n = 5.
Effects of oxypurinol on CSD-induced dilation
Intravenously administered oxypurinol neither altered baseline diameter (74 ± 16 vs. 72 ± 24 μm for pretreatment and oxypurinol treatment, respectively) nor altered the amplitude of cerebral arteriolar dilation during CSD (Fig. 2). In addition, the dilation area was not altered by intravenous oxypurinol (Fig. 2). The speed of CSD was not altered by oxypurinol. Mean arterial blood pressure was not changed by oxypurinol treatment (75 ± 11 mm Hg before vs. 80 ± 11 mm Hg after oxypurinol administration).
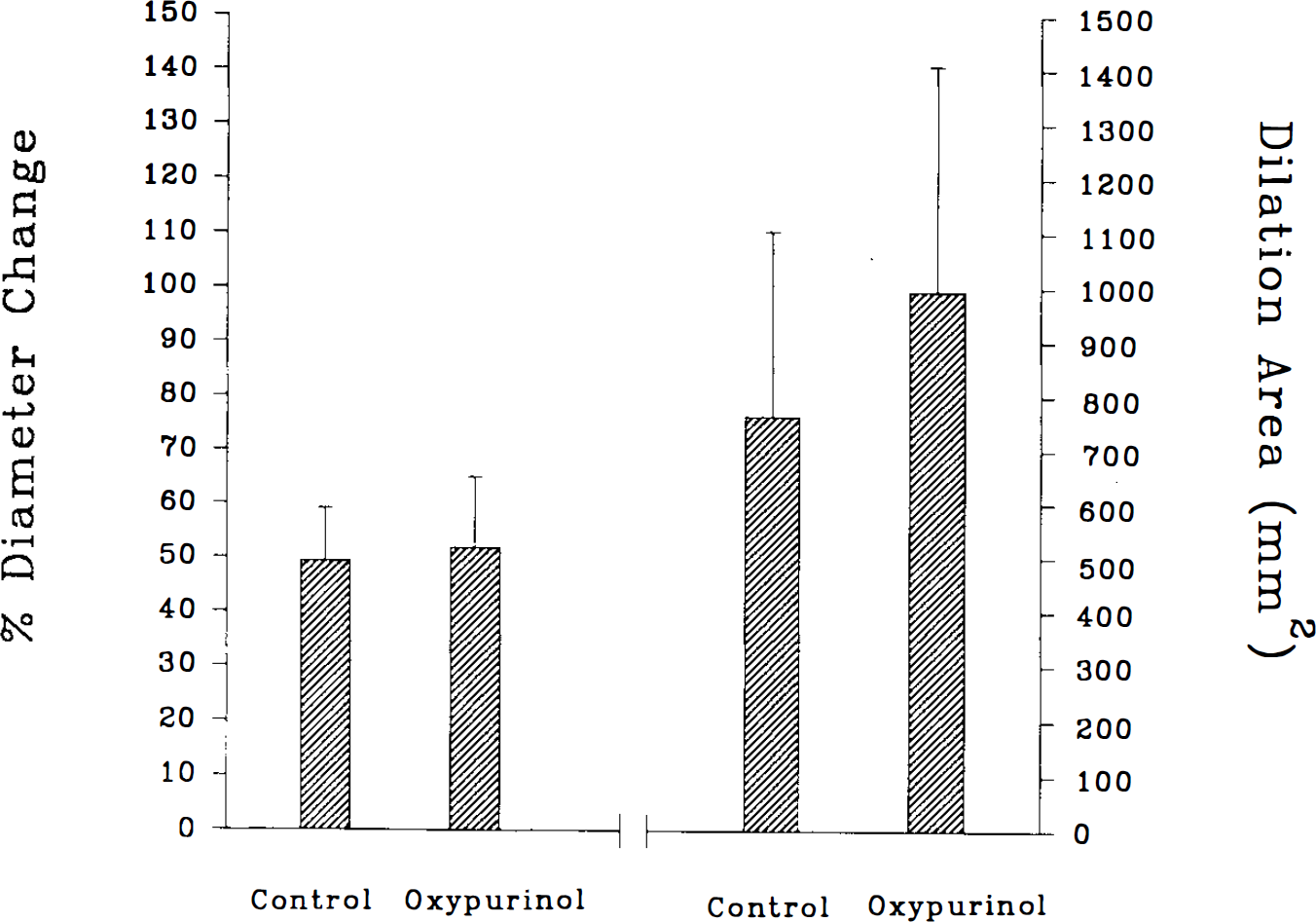
Changes of maximal amplitude and dilation area of the pial arteriolar dilation during cortical spreading depression (CSD) before and after intravenous administration of oxypurinol (50 mg/kg). Oxypurinol did not alter arteriolar responses to CSD. Values are means ± SD, n = 7.
Effects of SOD and oxypurinol on attenuation by L-NNA of CSD-induced arteriolar dilation
In the presence of SOD or oxypurinol (n = 12), CSD dilated cerebral arterioles (peak dilation = 58 ± 17%; dilation area = 890 ± 353 mm2). Following additional administration of L-NNA, CSD-induced dilation was significantly attenuated (peak dilation = 37 ± 17%; dilation area = 647 ± 312 mm2; p < 0.05 for both compared with values in the absence of L-NNA). Arteriolar diameter was 75 ± 24 μm before L-NNA administration and 77 ± 24 μm afterward.
DISCUSSION
The major finding of the current study is that topical application of SOD or intravenous oxypurinol does not alter CSD-induced cerebral arteriolar dilation. Thus, it appears that oxygen radicals such as superoxide anion do not play a major role in this response.
Arteriolar dilation and increased CBF are features of CSD in all species examined (Lauritzen, 1987; Shibata et al., 1990–1992; Goadsby et al., 1992; Duckrow, 1993; Colonna et al., 1994a, b ; Wahl et al., 1994). We have shown previously that cerebral vasodilation is closely associated spatially with spreading depression (Shibata et al., 1990) and that superfusion of the cortical surface with artificial CSF does not alter arteriolar dilation (Shibata et al., 1991b). Further, we and others have shown that in rabbits and cats with an intact prostanoid synthetic capability, NO and CGRP mediate almost all of the pial arteriolar dilation (Colonna et al., 1994a, b Wahl et al., 1994). The source of CGRP is probably from perivascular nerves (Mayberg et al., 1984), and NO could arise from endothelium, neurons, or astroglial cells (Pelligrino, 1993). It should be mentioned, however, that NO may not mediate cerebrovascular dilation during CSD in rats (Duckrow, 1993; Fabricius and Lauritzen, 1994; Zhang et al., 1994). Reasons for different findings appear to involve species differences or differences in experimental conditions (Meng et al., 1995).
Arteriolar dilation during CSD is opposed by one or several constrictor influences. We and others have previously shown that indomethacin is able to potentiate arteriolar dilation and increases in CBF during CSD (Lauritzen, 1987; Shibata et al., 1991a). However, the exact mechanism is unclear. One possibility, not completely tested, is that arteriolar dilation is restrained by constrictor prostanoids (Shibata et al., 1991a, 1992). Another hypothesis, tested in the current study, is that increased superoxide anion production is able to promote constriction, via either enhanced destruction of NO (Rubanyi and Vanhoutte, 1986; Nelson et al., 1992), impairment of NOS (Rengasamy and Johns, 1993), or direct constrictor effects on vascular smooth muscle (Katusic and Vanhoutte, 1989). Superoxide anion is produced on a one-to-one basis with prostaglandin H2 by cyclooxygenase (Kukreja et al., 1986), and increased release and metabolism of arachidonic acid are characteristics of CSD (Lauritzen et al., 1990; Shibata et al., 1991a, 1992). However, in contrast to findings with indomethacin, with administration of scavengers we were unable to detect an important influence of superoxide anion on cerebrovascular responses to CSD. Although there was a nonsignificant tendency for arteriolar dilator effects during CSD to be modestly greater following SOD or oxypurinol administration, the responses were not consistent among animals. Further, any trend for increased dilator responses following SOD or oxypurinol was relatively small compared with the effects of indomethacin on CSD-induced cerebral vasodilation (Lauritzen, 1987; Shibata et al., 1991a; Meng et al., 1995).
An additional finding was that scavengers were unable to prevent L-NNA from decreasing arteriolar dilator responses during CSD. Previously, Rosenblum et al. (1992) found that oxygen radical scavengers were able to prevent an NOS inhibitor from attenuating acetylcholine-induced dilation of mouse cerebral arterioles. Thus, in the rabbit, L-NNA apparently does not attenuate NO-induced cerebral arteriolar dilation via generation of superoxide anion.
We used two different agents as superoxide anion scavengers in this study. First, we topically applied SOD to the cortical surface. The dose of SOD that we used was twice the dose shown by Rosenblum et al. (1992) to be effective in mouse and similar to that shown to be effective in cats by Nelson et al. (1992) and Kontos et al. (1992). In addition, we have shown a lower dose of SOD to be an effective scavenger of superoxide anion following ischemia or asphyxia (Armstead et al., 1989; Pourcyrous et al., 1990, 1993). Second, we administered oxypurinol intravenously. Although recognized as a xanthine oxidase inhibitor, oxypurinol has been shown to be a potent scavenger of oxygen radicals (Moorhouse et al., 1987). Further, we have shown that oxypurinol at the dose given was able to prevent detection of superoxide anion in piglet cortex that occurs during the reventilation period following asphyxia (Pourcyrous et al., 1993) and to preserve arteriolar responses normally reduced following asphyxia (Busija and Meng, 1993). Similar scavenging effects of oxypurinol have been reported for other organs. An important role for xanthine oxidase in mediating oxygen radical production in brain is uncertain (Vettenrarta and Raivio, 1990; Betz et al., 1991).
We are unaware of other studies specifically examining the effects of oxygen radical scavengers on cerebral hemodynamics during CSD. Several studies have examined the effects of tirilazad mesylate on cerebral hemodynamics during CSD (Hall and Smith, 1991; Duckrow and Beard, 1992; Goadsby, 1992). However, this compound is a poor oxygen radical scavenger and exerts its influence via stabilization of membranes (Helfaer et al., 1994). Tirilazad does not alter increases in CBF during CSD.
In summary, administration of SOD and oxypurinol does not affect cerebral arteriolar dilation during CSD. Thus, superoxide anion does not appear to play an important role in cerebral arteriolar responses under these conditions.
Footnotes
Acknowledgment:
This study was supported by grants HL-30260 and HL-46558 from the Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD, U.S.A.