
Abstract
Significance:
The binding of cytokines and growth factors to heparan sulfate (HS) chains on proteoglycans generates gradients that control development and regulate wound healing. Syndecan-1 (sdc1) is an integral membrane HS proteoglycan. Its structure allows it to bind with cytosolic, transmembrane, and extracellular matrix (ECM) proteins. It plays important roles in mediating key events during wound healing because it regulates a number of important processes, including cell adhesion, cell migration, endocytosis, exosome formation, and fibrosis.
Recent Advances:
Recent studies reveal that sdc1 regulates wound healing by altering integrin activation. Differences in integrin activation lead to cell-type-specific changes in the rate of cell migration and ECM assembly. Sdc1 also regulates endocytosis and the formation and release of exosomes.
Critical Issues:
Understanding how sdc1 facilitates wound healing and resolution will improve treatment options for elderly and diabetic patients with delayed wound healing. Studies showing that sdc1 function is altered in cancer are relevant to those interested in controlling fibrosis and scarring.
Future Directions:
The key to understanding the various functions ascribed to sdc1 is resolving how it interacts with its numerous binding partners. The role played by chondroitin sulfate glycosaminoglycan (GAG) chains on the ability of sdc1 to associate with its ligands needs further investigation. At wound sites heparanase can cleave the HS GAG chains of sdc1, alter its ability to bind cytokines, and induce shedding of the ectodomain. This review will discuss how the unique structure of sdc1 allows it to play key roles in cell signaling, ECM assembly, and wound healing.
Scope and Significance
S
Translational Relevance
Most of the published studies on sdc1 focus on its involvement within a variety of cancer types where it is either overexpressed or downregulated. Since all four sdc family members are upregulated during fibrosis, cancer studies are relevant to those interested in suppressing fibrosis and scarring during wound healing.
Clinical Relevance
Many of the important events that take place during wound healing, such as cell migration, cell proliferation, and inflammation, are regulated by sdc1. Further, sdc1 is upregulated during fibrosis. Delayed wound healing occurs in the elderly and patients with diabetes; therefore, understanding the role sdc1 plays in the wound healing process is of direct clinical relevance. Studies that focus on the biology of the sdc1 proteoglycan are relevant to those interested in suppressing fibrosis and scarring during wound healing.
Background
Sdc1 was cloned and named by Merton Bernfield's group in 1989. 1 The involvement of sdc1 in wound healing was reported shortly after discovery of the proteoglycan. 2 Sdc1-null mice were generously provided to our group by Dr. Bernfield. We used our corneal model to determine whether the wound-healing delay seen in the skin was caused by reductions in migration and/or proliferation of sdc1-null keratinocytes or by other factors, such as changes in angiogenesis or wound contraction. In 2002, we showed that sdc1-null mice are viable, but displayed delays in both skin and corneal wound healing. These delays were due to defects in keratinocyte migration, not reduced proliferation. 3 Following our report, sdc1-null mice on two different genetic backgrounds (BALB/cJ and C57BL6J) have been widely used by numerous groups. 4
The purpose of this review is to describe the relevance of sdc1 in epithelial homeostasis and wound repair. Sdc1 shares structural domains with 3 additional family members called sdc2–4 and as a result, data obtained studying one sdc family member often apply to all four. Excellent reviews that focus on different aspects of sdc biology and function have appeared. 4 –8 Those more interested in sdc1 in various diseases should read the review by Teng et al. 4 whereas those interested in wound healing will want to see the review by Alexopoulou et al. 5 Transmembrane signaling, 6 glycosaminoglycan (GAG) chain functions, 7 and core-protein-mediated functions 8 are the focus of additional reviews. Here we will place emphasis on the newly described roles that sdc1 play in mediating endocytosis and exosome formation that impact epithelial tissue homeostasis, cell migration, and epidermal and corneal wound healing. We will also point out topics where additional study is needed in hopes of expanding interest in the study of sdc1 in wound healing.
Discussion of Findings and Relevant Literature
Sdc1 core protein structure
Sdc1 is an integral membrane proteoglycan
sdc1 is the founding member of a family of four integral membrane proteoglycans, sdc1–4, that are expressed in mammals. 2 The name syndecan was derived from the Latin word syndein, which means to bind together, and was chosen because the first function ascribed to syndecan involved the binding of cells to extracellular matrix (ECM) proteins. Sdc1 contains extracellular, transmembrane, and cytoplasmic domains, all of which have been shown to contribute to its various different functions (Fig. 1). In frogs, sdc1 is expressed at the 4-cell stage, on the basolateral cell surfaces of the embryonic ectoderm and endoderm during gastrulation, 9 and throughout organogenesis on ectodermal tissues. 10 In mammals, sdc1 is the major heparan sulfate proteoglycan (HSPG) on epithelial cells achieving expression levels estimated at 106 copies per cell. 11
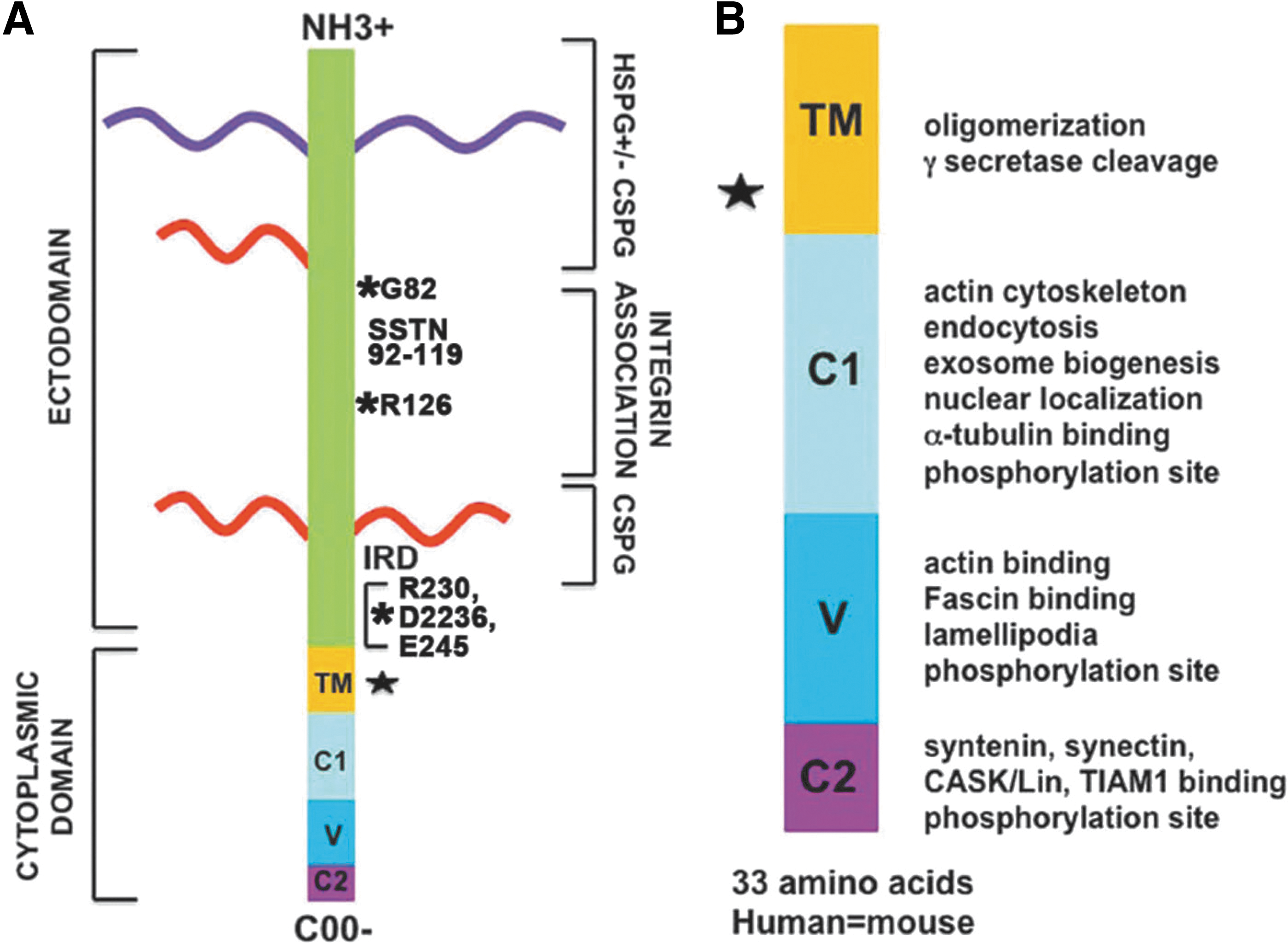
Syndecan-1 is an integral membrane proteoglycan. Panel
The HSPGs of sdc1 allow the proteoglycan to bind to the heparin-binding sites present on numerous ECM proteins, growth factors, cytokines, and other proteins. In all, there are more than 100 proteins capable of binding to the extracellular domain of sdc1. Table 1 lists the direct and indirect binding partners reported for the extracellular domain of sdc1.
Syndecan-1's extracellular contact list
All four sdcs contain short cytoplasmic tails. The cytoplasmic tail of sdc1 is 33-amino-acid long and is completely conserved between mice and humans. There are only five other reported integral membrane proteoglycans: NG2/CSPG4, CD44, neuropilin-1, betaglycan/TGFβRIII, and thrombomodulin. 6 Of these, NG2 and thrombomodulin are chondroitin sulfate proteoglycans (CSPGs). CD44, neuropilin-1, and betaglycan/TGFβRIII can be expressed either as glycoproteins or as CSPGs. It appears that the four sdc family members are the only known transmembrane HSPGs.
HS and CS GAG chains can be added to the sdc1 extracellular domain
The sdc1 extracellular domain has five sites where GAG chains can be attached. Three are toward the N-terminus and two are located close to the plasma membrane. GAG chains are repeating units of disaccharides added to the core protein in the ER and Golgi prior to expression at the cell surface. The GAG chains added to sdc1 are of two types: heparan sulfate (HS) and/or chondroitin sulfate (CS). While both HS and CS chains can be located at the N-terminus, only CS chains are present near the plasma membrane. Sdc1 always has one or more HS chains at its N-terminus. Cells can vary the number of the HS and CS GAG chains added. When Langford et al. varied the number of GAGs attached to the N-terminal sites using mutagenesis, they found that reducing the GAG chains reduced the ability of sdc1 to mediate invasion and cell–cell interaction. 12 Thus, differences in the numbers of GAG chains lead to changes in sdc1 function.
The length and sulfation states of the attached GAG side chains vary in tissue-specific ways due to the action of a suite of several functionally related enzymes that regulate glycan elongation and modification. 13 The extracellular regulation of HSPG length by heparanase is of particular importance. Heparanase is an enzyme that cleaves HSPGs leaving HS stubs on the extracellular domain. Heparanase is overexpressed in myeloma cells 14 and in tissues after wounding. 15 Sdc1 with HS stubs rather than longer HSPGs is more susceptible to proteases that mediate shedding. 16
The functions of the CS chains on sdc1 require additional study
While most studies focus on the role HSPGs play in the function of sdc1, the contributions of CS to the function of sdc1 have been largely unexplored. The presence of CS chains on the ectodomain near the plasma membrane could impact the ability of sdc1 to associate with other transmembrane receptors, alter its susceptibility to cleavage and shedding by proteases, and/or disrupt its clustering. Interestingly, the binding of a small bioactive sdc1 ligand called lacritin to sdc1 was recently shown to require the presence of HS stubs, indicating heparanase cleavage, and a CS chain. 17 Lacritin is a protein found in tears. It is reduced in expression in patients with dry-eye disease. 18 Reducing the length of the sdc1 HSPGs by heparanase reduces the binding of HSPG ligands and exposes binding sites on the core protein. Detailed studies of the impact of heparanase on the functions of both cell-surface-associated and shed sdc1 are needed.
Although there are currently no reports for the endogenous extracellular trimming of CS side chains in mammalian cells, CS chains can be shortened by an enzyme isolated from gut bacteria called chondroitinase ABC. Currently, there is intense interest in using chondroitinase ABC as a potential treatment for debilitating spinal cord injuries. 19 CS proteoglycans accumulate within glial scars after damage to the central nervous system (CNS) and prevent reinnervation. Injections of chondroitinase ABC enhance functional recovery after spinal cord injury in rodent models by reducing the presence of CS in glial scars. 19
Since HS, not CS, GAG chains are modified by heparanase in vivo, the function of the CS glycans on the sdc1 extracellular domain is of great importance since heparanase is upregulated in the wound environment and chondroitinase ABC is only made by bacterial cells. Since CSPGs inhibit the repair of nerves, 19 shifting GAG chains on sdc1 from primarily HSPGs to CSPGs is expected to significantly alter sdc1 function. The impact of differences in the length, identity (HS vs. CS), and sulfation state of sdc1 GAG chains and their effect on cell proliferation, cell migration, and ECM assembly in vivo and in vitro are not clear.
The shedding of the ectodomain
The extracellular portion of sdc1 can be shed by numerous extracellular and membrane-bound proteases of the metzincin family, which includes matrix metalloproteinases (MMPs), membrane-tethered MMPs (MT-MMPs), and A-disintegrin and-matrix-metalloproteinases (ADAMs). Additionally, sdc1 can also be cleaved by trypsin in vitro. During wound healing, the cleaved ectodomain of sdc1 accumulates in the wound fluid and granulation tissue in skin 5 and in the tears after corneal injury. 17 Following cleavage, the carboxy-terminal fragment (CTF) is left in the plasma membrane.
The exact protease cleavage sites used to induce shedding of the sdc1 ectodomain were recently determined. 20 Proteolysis takes place not at a single location but within a cluster of amino acids located 6–15 residues from the plasma membrane (Fig. 1). Because the exact protease cleavage site can vary, the C-terminal amino acids of the cleaved ectodomain and the N-terminal amino acids of the CTF differ depending on which site the protease cuts.
Sdc2, 3, and 4 share many but not all functions with sdc1
Sdc2 is involved in early development and patterning
Different cells express different sdc family members. Epithelial cells and plasma cells express primarily sdc1 and neuronal cells express primarily sdc3. Mesenchymal cells have sdc2 and sdc4 but can also express sdc1 as well. Null mice for sdc1, 3, and 4 exist and are currently available. 4 The function of sdc2 during early development was tested using frog and zebrafish model systems. In frogs, sdc2 is expressed on migrating ectoderm cells and functions in communicating information regarding the establishment of left-right asymmetry. 21 In zebrafish, knockdown of sdc2 alters angiogenesis during development 22 and the expression of the left-right asymmetry marker southpaw by a mechanism that involves binding of fibroblast growth factor-2 (FGF2) and the expression of Tbx16. 23 In addition, sdc2 plays a role in breast cancer cell survival and is being considered as a possible target for chemotherapy given that inhibiting sdc2 induced apoptosis and suppressed tumor cell growth. 24 Sdc2, like other sdcs, is upregulated during fibrosis. It is expressed by fibroblasts and upregulated by transforming growth factor β1 (TGFβ1) and insulin-like growth factor binding protein-3. 25
Sdc3 functions in the CNS and in skeletal muscle
Sdc3 is sometimes referred to as N-syndecan since it was originally described as an HSPG expressed in the developing brain and was implicated in neurite guidance and synaptic plasticity. Sdc3-null mice exhibit feeding behavior defects, 26 increased addiction to cocaine, 27 and impaired hippocampus-dependent memory. 28 Sdc3 is also expressed on skeletal-muscle-specific stem cells called satellite cells 29 and its deletion in satellite cells alters their ability to complete the cell cycle and these effects are mediated through cleavage of Notch by tumor necrosis factor α converting enzyme/ADAM17 (TACE/ADAM17). 30 In addition, overexpression of sdc3 is seen in the muscles of patients with muscular dystrophy. 29
Sdc4 mediates focal adhesion dynamics
The report that sdc4 was localized to focal adhesions by Woods and Couchman lead to new insight into the functions of the transmembrane and cytoplasmic domains of all four sdcs. 31 Sdc4-null mice exist and are viable. 32 Like sdc1-null mice, the sdc4-null mouse exhibits evidence of delayed wound healing. However, unlike the sdc1-null mice, the sdc4-null dermal fibroblasts exhibit reductions in cell migration rates. Subsequent studies using sdc4-null mice and/or cells showed that sdc4 is important in the regulation of transient receptor potential canonical 6 channels and fibrosis. 33 The extracellular domain of sdc4 mediates ECM adhesion via interaction with αv-containing and α5β1 integrins. 6 The cytoplasmic domain of sdc4 regulates phosphatidylinositol 4,5-bisphosphate and protein kinase C activity. 34 cSrc-mediated tyrosine phosphorylation of the sdc4 cytoplasmic tail is a control point for the endosomal recycling of integrins. Sdc4 has also been reported to control focal adhesion assembly. 35 Sdc4 expression is known to modulate canonical 36 and noncanonical 37 Wnt signaling. Modulating Wnt signaling is mediated by the sdc4 extracellular and transmembrane domains through association with fibronectin (FN) to form a Wnt signaling complex.
Proteomic analyses of focal adhesions from a kidney cell line overexpressing α4 integrin and from fibroblasts isolated from mouse kidneys revealed more than 400 different proteins but no syndecans. 38,39 Roper et al. make a compelling argument that sdc ectodomains are excluded from mature focal adhesions due to spatial constraints. 40 Integrins extend 10–17 nm from the plasma membrane; sdc HSPGs reach between 40 and 500 nm from the cell membrane. The distance between the plasma membrane and ECM within a focal adhesion is 10 nm. Intact sdc cannot fit within mature focal adhesion plaques; whether the sdc CTF could be present in focal adhesions is not clear. Data also show that sdc1 is important in the formation and the disassembly of focal adhesions in migrating epithelial cells. 41 The composition of focal adhesions will vary between mesenchymal cells that have been used for proteomic studies and epithelial cells where sdc1 expression is high. Therefore, additional proteomic studies using epithelial cells are needed to determine whether sdc1 or sdc4 CTFs accumulate in focal adhesions.
The functions of the sdc1 extracellular domain
Sdc1 mediates ECM adhesion and organization
Sdc1 has been reported to mediate adhesion to laminin (LN), collagen (CN), FN, and thrombospondin (TSP) via their heparin-binding domains. 2 Since many additional ECM proteins possess TSP and FN motifs, the number of ECM proteins that can interact with sdc1 is quite large (Table 1). The HepII domain of FN contains a heparin binding site that binds to the sdc1 GAGs and regulates CN matrix assembly by inhibiting αv-integrin-mediated signaling. 42 Keratinocytes and corneal stromal cells from sdc1-null mice show reduced CN deposition and FN fibril assembly. 43,44 By contrast, dermal fibroblasts lacking sdc1 show increased FN fibrillogenesis. 44 In control animals and people, chronic expression of sdc1 and sdc4 by stromal cells correlates with fibrosis, 6 while injury-induced fibrosis in sdc1- and sdc4-null mice is reduced. 4 A better understanding of the precise role the four sdcs play in fibrosis is needed.
Sdc1, integrins, and integral membrane proteins form complexes
Recently, Roper et al. compared the literature-curated interactomes between sdc1 and sdc4. 40 Although both sdcs bind to many of the same receptor proteins, including integrins (α2β1, αvβ3, and α6β4), ADAM proteins, MT-MMPs, MMP9, FGFR, and erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ErbB2), differences in integrin association do exist between sdc1 and sdc4. Sdc4 associates with α5β1 and α6β1 whereas sdc1 associates with αvβ5. The interaction of sdcs with receptors is initiated via cluster-induced binding of ECM proteins or growth factors. Definitive studies showed that sdc1, sdc2, and sdc4 interact with integrins via amino acids in their ectodomains. Sdc1 interacts with αv-containing integrins via a sequence of 27 amino acids located in the center of the sdc1 ectodomain. 45 A peptide derived from this region, termed synstatin, interferes with sdc1:αv integrin association. Neither sdc1 nor integrins possess kinase activity, yet both proteins induce kinase-mediated signaling. While integrins have been shown to associate with numerous kinases, including FAK and epidermal growth factor receptor (EGFR), thus far, sdc1 has only been shown to associate directly with the insulin-like growth factor 1 receptor (IGF1R); along with αv-containing integrin heterodimers, sdc1 and IGF1R form a ternary complex that induces cell signaling. 46
Association between sdc2 and integrin is more complex. A juxtamembrane sequence on the C1 domain of sdc2 associates with the integral membrane tyrosine phosphatase, CD148, which in turn associates with β1-family integrins. The sdc2:CD148:β1-family integrin complex then mediates cell adhesion via activation of Src kinases. 47 Given the sequence homology shared between all four sdcs in the C1 domain, it is likely that other sdc:integrin:phosphatase ternary complexes exist. Currently, there are no studies that demonstrate an association between sdc3 and integrins. However, sdc3 does associate with Notch and mediates its activation via ADAM-17/TACE, 30 which suggests the untested possibility that sdcs associate with the disintegrin domains of ADAM proteins.
Characterizing the shed ectodomain
Bacterial infections and sterile injuries induce the upregulation of sdc1, 4 heparanase, 15 and of proteases capable of cleaving sdc1 and releasing an ectodomain fragment. 48 The sdc1 ectodomain added to cells in culture suppressed the growth of tumor cells but had no impact on contact-inhibited cell lines. 49 This effect was mediated by the HS chains on the sdc1 proteoglycan. To further study the impact of shed sdc1, constitutively shed or uncleavable forms of sdc1 were expressed in MCF-7 breast cancer cells. 50 Proliferation and invasion assays showed that tumor cells expressing uncleavable membrane-bound sdc1 were more proliferative but less invasive whereas tumor cells expressing shed sdc1 ectodomain were less proliferative but more invasive. These events were regulated via sdc1 HS GAG chains. These data implicate overexpression and shedding of sdc1 in the switching of cancer cells from a proliferative to an invasive phenotype.
While sdc1 is elevated in some cancers, it is no longer found on advanced skin 51 and other cancers. 52 Insight into sdc1 and cancer was obtained by Langford et al. who showed that, in a short sequence of five amino acids (AVAAV) in the sdc1 ectodomain, only 26 amino acids from the transmembrane domain were responsible for the ability of intact sdc1 to inhibit tumor cell invasion. 53 Mutation in this site, called the invasion regulatory domain (IRD), permitted tumor cell invasion but did not impact the cell adhesion and spreading functions of sdc1. As discussed in “The shedding of the ectodomain” heading, and shown in Fig. 1A, the shed sdc1 contains the IRD domain. Studies that mutate the IRD in cells expressing constitutively shed sdc1 would show whether it plays a role in the function of the shed ectodomain.
While the aforementioned studies showed that HSPGs on sdc1 regulate tumor cell invasion, other experiments conducted using core proteins made without glycans showed that the sdc1 ectodomain inhibited sdc1-mediated cell spreading on vitronectin in the MDA-MB-231 breast cancer cell line. 54 These observations lead to additional experiments that showed that the sdc1 core protein interacts directly with αvβ3 integrin and that this interaction is independent of the GAG side chains. This lead to the characterization of synstatin, a sdc1-derived peptide that binds to integrins and reduces cancer cell invasion and tumor angiogenesis. 45,46,55
Sdc1-mediated activation of integrins impacts wound healing
Corneal wound healing studies performed using sdc1-null mice show delayed reepithelialization not caused by altered cell proliferation. 3 Cutaneous wound healing studies (full-thickness and dermabrasion) in sdc1-null mice confirmed wound-healing defects. Mouse corneas lacking sdc1 also showed increased recruitment of neutrophils to wound sites. 3,56 sdc1-null keratinocytes exhibited elevated surface expression of several integrins, were more adhesive, and less migratory compared with control keratinocytes. Data obtained by adding antibodies directed against the extracellular domains of β1, α6, and αv integrins revealed reduced activity of both α6β4- and αv-containing integrins in sdc1-null keratinocytes. The ECM deposited by sdc1-null keratinocytes contained less CN and the migration differences observed with these cells could be reversed by replating them onto FN/CN, laminin 332, or ECM produced by control keratinocytes. 43 Enzyme-linked immunosorbant assay (ELISA) and luciferase reporter assays showed that sdc1-null cells produced elevated levels of TGFβ1. Differences in integrin activity and matrix assembly were responsible for the differences in migratory rates of sdc1-null keratinocytes.
The changes in epithelial migration in sdc1-null cells confirm the importance of sdc1 in epithelial biology. The in vitro differences in cell migration were due, in part, to the lack of feedback from the cleaved ectodomain since adding GST-sdc1 but not GST-sdc4 or a scrambled peptide to fibroblast cell cultures reversed the cell migration phenotype seen in sdc1-null dermal fibroblasts (data not shown). However, the increased integrin surface expression on sdc1-null keratinocytes also suggests the possibility that reduced trafficking of integrins and other receptors from the cell surface may also play important roles. 43
The mechanism leading altered integrin activation in mice lacking sdc1 was explained through critical experiments performed by Beauvais et al. 55 These experiments showed that sdc1 and αvβ3 integrin are functionally coupled, but that this interaction does not require the sdc1 transmembrane or cytoplasmic domain since GPI-linked sdc1 also could couple with αvβ3. The sdc1–αvβ3 interaction could be disrupted with a recombinant sdc1 ectodomain lacking GAG side chains. Later studies showed that sdc1, αvβ3, and IGFR form a tripartite complex that mediated cell signaling and a small sdc1-derived peptide called synstatin could block this interaction. 45,46
Sdc1-null mice and sdc1-depleted cells are widely used to study the mechanism underlying many different types of injuries. The description of phenotypes seen in over 30 studies is presented in a recent review by Teng et al. 4 Collectively, these studies show that sdc1 aids in wound healing by enhancing cell migration, controls viral attachment and entry, regulates microbial defense, generates chemokine gradients, and modifies cell signaling.
The impact of sdc1 on cell migration rates is cell type specific
α6β4 is the hemidesmosomal integrin and its expression was upregulated in sdc1-null papillomas and on surfaces of normal and ras-transfected keratinocytes. 3,43 Blocking its function restored sdc1-null keratinocyte migration rates to those of control cells. 43 Wang et al. recently showed that the cytoplasmic domain of sdc1 interacts directly with α6β4 and forms a tripartite complex with ErbB2. 57 Binding of α6β4 to LN332 in A431 human squamous carcinoma cells leads to phosphorylation of cMet, Fyn, and the β4 subunit; Akt and PI3K signaling is also activated leading to cell spreading and protection from apoptosis. However, these signaling events were disrupted when sdc1 expression was blocked. Also, when the C2 domain of sdc1 was mutated, cMet and Fyn were phosphorylated although the β4 cytoplasmic tail was not. These results suggest that sdc1 engagement permits α6β4 to be phosphorylated. Numerous studies have shown that phosphorylation of β4 integrin in response to growth factors promotes hemidesmosome destabilization and thus cell migration. 58,59 Based on these observations, keratinocytes lacking sdc1 would be expected to show reduced phosphorylation of the β4 integrin cytoplasmic tail, which favors stable hemidesmosomes, and thus reduced migration rates as observed in sdc1-null keratinocytes. 43 Due, in part, to low levels of β4 integrin phosphorylation in primary keratinocytes, using immunoprecipitation and immunoblotting, we were unable to confirm differences in β4 integrin phosphorylation between control and sdc1-null keratinocytes (data not shown); a more sensitive approach such as mass spectroscopy would likely allow us to sort this out.
Keratinocyte migration rates are impacted by both low and high sdc1 expression in tissue-specific ways. Overexpressing sdc1 in vivo impaired wound healing in mice by reducing cell proliferation rates. 60 This defect was, at least in part, due to excess release of the sdc1 ectodomain and was partially rescued by depleting sdc1 at wound sites using an antibody or treating tissues with heparanase. 60 When sdc1 is inhibited in epithelial cells in vitro, cell migration rates can increase rather than decrease depending on the cell type and the ECM cells interact with. Bachy et al. performed elegant experiments using normal human keratinocytes and showed that reducing expression of sdc1 increased the ability of keratinocytes to migrate on processed LN332 but reduced migration on intact LN332. 61 In these studies, α3β1 integrin, not α6β4, was the key player in sdc1-mediated keratinocyte migration. LN332 is a trimer; two of its three chains (LNα3 and LNγ2) are processed after the protein is secreted and as cell migrates over it. 61 The processing of LN332 alters sdc1-mediated cell migration. This study shows, at a molecular level, that keratinocyte migration rates depend on sdc1 expression and ECM composition.
Using a lung epithelial-derived cell line called BEAS-2b, Chen et al. (2009) showed that reducing sdc1 expression increased cell migration rates. 62 On the BEAS-2b cells, sdc1 forms a complex with α2β1 integrin, a CN receptor, which results in slower cell migration. 63 Additional studies that use BEAS-2b cells showed that the association of the extracellular domain of sdc1 with α2β1 activates this CN receptor. 41 Using truncation mutants, these studies differentiated between the ability of sdc1 to induce integrin activation from its ability to accelerate cell migration. The sdc1 extracellular domain mediated activation of integrins, while the transmembrane and cytoplasmic domains regulated cell migration.
Several studies that show increased epithelial cell migration rates when sdc1 expression is reduced share a common feature: lack of expression of α6β4. Primary human bronchial epithelial cells and the human airway epithelial cell line express α6β4 whereas BEAS-2b cells do not. 64 We have assessed cell migration rates in control and sdc1-null dermal fibroblasts and corneal stromal cells, two cell types that do not endogenously express α6β4 44,65 and their migration rates both increased relative to control cells. Similar to sdc1-null keratinocytes, surface expression of α2β1 and α3β1 integrins was increased in sdc1-null dermal fibroblasts, and activation of β1- and αv-integrins was decreased. Together these studies confirm that sdc1 expression on cells that do not express α6β4 slows their migration rate via integrin activation. Recent studies showed that sdc1 knockdown in breast cancer cells increased their migration rate on FN-coated surfaces in a manner that was dependent on α5β1- or αv-containing integrins. 66 Whether similar results would be seen if cells were grown on LN332-coated surfaces is unknown.
The importance of the C-terminal transmembrane and cytoplasmic domains
The transmembrane domain of sdc1
The transmembrane domain of sdc1, as well as sdcs2–4, contains a sequence (GxxxG) that is present in many other transmembrane proteins and mediates transmembrane protein α-helical oligimerization. 61 Structure function studies show that sdc1 is less likely to form homodimers than sdcs2–4. 67,68 While sdc1 can form heterodimers with sdc2 and sdc3, it cannot form them with sdc4. The biological implications of the varied affinities between the sdc family members are unclear. 68 Altemeier et al. found that the transmembrane domain alone was sufficient in reducing cell migration rates via alterations in the rate of focal adhesion disassembly and the recycling of paxillin and FAK to focal adhesions. 63
The cytoplasmic tail of sdc1
The cytoplasmic tail of sdc1 contains three distinct domains called C1, V, and C2. C1 and C2 are conserved, while V is the variable domain that differs significantly in its sequences between all four family members. The numbers of cytoplasmic proteins associating directly and indirectly with the transmembrane and cytoplasmic domains of sdc1 are large and growing (Table 2). There is considerable overlap among sdc family members due to similarities in the primary amino acid sequences contained in the C1 and C2 regions of their cytoplasmic domains. Each of the three domains contains a tyrosine residue that is conserved among all four family members and can be phosphorylated to regulate signaling. All four sdcs also contain a single conserved serine residue in their C1 domains that can be phosphorylated. The V domain mediates specific functions unique to each sdc; however, it is also the least characterized of the three cytoplasmic domains. No data currently exist for V-region-specific functions for sdc2 and 3. However, a site in the V region of sdc4 interacts with PIP2, PKCα, and α-actinin and regulates focal adhesion dynamics. 6,69 The V region of sdc1 supports bundling of fascin with actin and lamellipodial-mediated cell spreading, 65 which are critical events regulating cell migration. The fact that the V region of sdc1 and sdc4 mediates the association of actin with the plasma membrane supports important and overlapping roles for both sdcs in the regulation of cell migration.
Syndecan-1's transmembrane and cytoplasmic domain contact list
The C1 domain is closest to the plasma membrane and mediates association with ezrin, Src, cortactin, and γ-secretase. It contains the juxtamembrane sequence RMKKK that mediates tubulin association, nuclear translocation, and exosome formation, and is a consensus sequence for γ-secretase-mediated cleavage. Presenilin forms a complex with γ-secretase and is required for γ-secretase cleavage of target proteins. Studies using cells lacking expression of presenilin showed decreased accumulation of sdc3 fragments compared with control cells. 70 Further analysis showed that these fragments, called syndecan intracellular domains (SICDs), were generated by cleavage of the sdc3 CTF by the presenilin–γ-secretase complex. The presenilin–γ-secretase complex did not cleave intact sdc3; it only cleaved the CTF generated in these experiments by trypsin cleavage. The sdc3 SICD regulates the subcellular localization of the adaptor protein CASK, which can bind to the PDZ site on all four sdcs. Based on homology of the cytoplasmic domains of sdc family members, it is likely that all four could serve as targets for γ-secretase-mediated cleavage; however, it is unclear whether the sdc1 CTF is targeted for additional cleavage by γ-secretase.
The C2 domain contains a PDZ binding motif that associates with syntenin, CASK, and TIAM1. Syntenin binding mediates exosome formation and is discussed in the following heading.
Endocytosis and exosome formation and the sdc1 cytoplasmic domain
Insight into the function of the sdc1 CTF was obtained from research initiated to study lipoprotein uptake by cells. Oswald et al. demonstrated that HSPGs could bind to and mediate internalization of triglyceride-phospholipid emulsions enriched in the lipoprotein apo E via a noncanonical mechanism. 71 Fuki et al. generated a chimeric sdc1 receptor containing an IgG extracellular domain fused to the sdc1 transmembrane and cytoplasmic domain. 11 They demonstrated that clustering of the sdc1 transmembrane and cytoplasmic domain mediated the binding, internalization, and delivery of lipoproteins to lysosomes. The sdc1-null mouse exhibits elevated levels of plasma triglycerides 72 and studies by Foley and Esko confirmed that sdc1 mediated hepatic triglyceride clearance. 73 Subsequent studies by Fuki et al. showed that the chimeric sdc1 C-terminal domain mediated the internalization of the EGF receptor. 74
Migrating cells move integrins from the rear or trailing edge to the front of the cell via organelles called endosomes. 75 Sdc1 plays important roles in the formation and trafficking of endosomes within cells. 69 Since sdc1 regulates endosome formation and interacts with integrins, it may play a role in regulating integrin trafficking. Chen and Williams characterized the sequence in the C1 domain on the sdc1 CTF that mediates endocytosis. 76 Two distinct phases were described and are illustrated schematically in Fig. 2. When sdc1 is not clustered into rafts, tubulin associates with the RMKKK sequence in its C1 domain. In the first phase of endocytosis, an sdc multivalent ligand clusters sdc1 and triggers the activation of ERK and movement of sdc1 into rafts. Proteins listed in Table 1 can associate with the sdc1 extracellular domain and can serve as multivalent sdc ligands to initiate clustering. These include growth factors bound to the sdc1 HSPGs and its cognate growth factor receptor, integrins, or inactive metzincin proteases. Multiple different sdc1–ligand interactions can take place within rafts as sdc1 clusters. Movement of sdc1 into rafts leads to the dissociation of tubulin from the sdc1 RMKKK sequence.
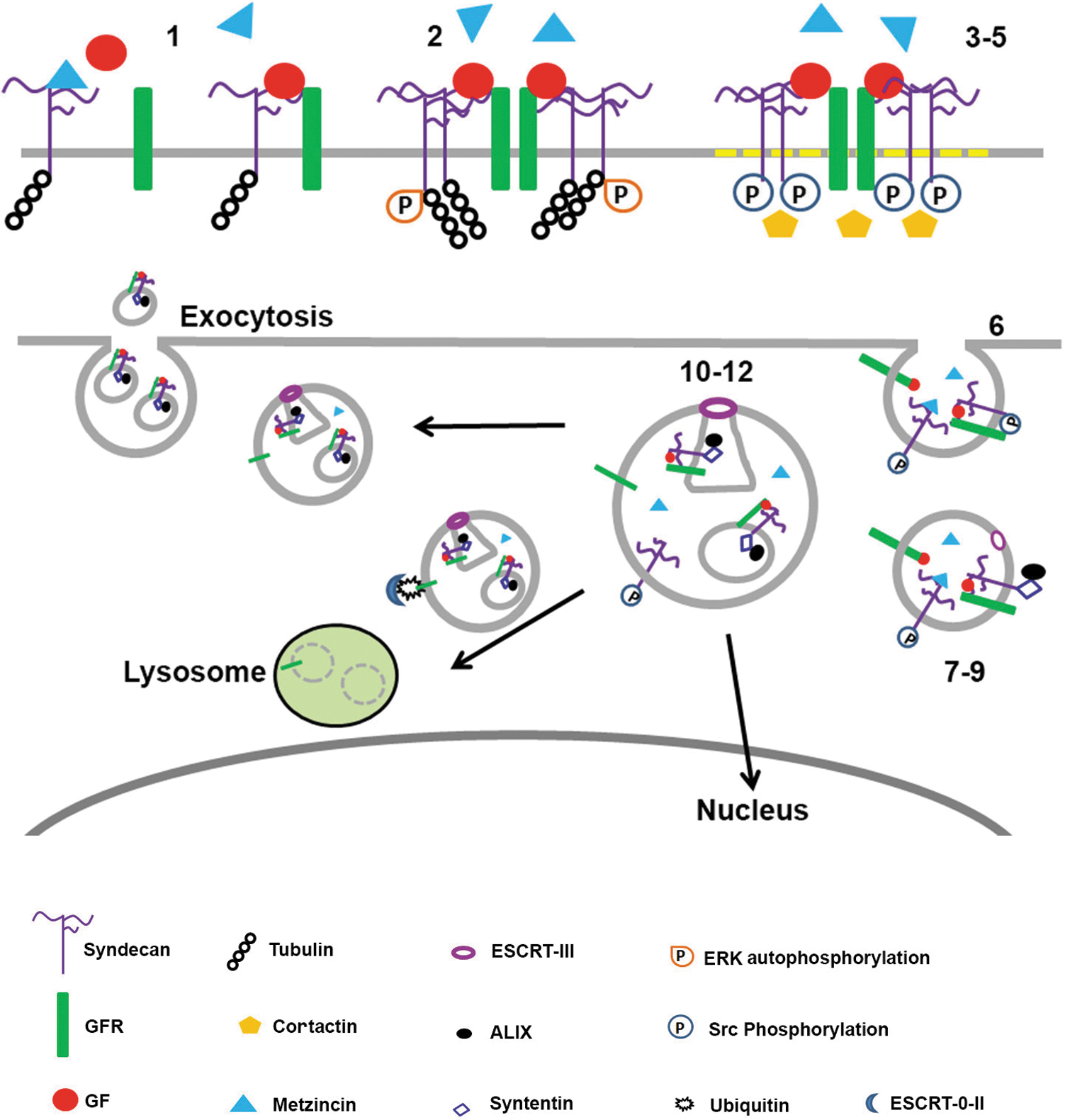
Schema describing how distinct domains on the sdc1 core protein regulate endocytosis, exosome formation and release, and endosomal translocation to the nucleus. 1. GF binding mediates sdc1–GFR clustering. 2. GFR and sdc1 clustering induces ERK autophosphorylation. 3. Sdc1:GFR:GF clusters move into cholesterol-rich membrane domains (rafts). 4. Src phosphorylates tyrosine residues on the sdc1 CTF. 5. Sdc1 phosphorylation recruits cortactin. 6. Actin-dependent endocytosis occurs. Trapped within endosomes are inactive proforms of Metzincin family proteases present in the extracellular space bound to sdc1 GAGs and the enzyme heparanase. Both are endocytosed along with sdc1, GF, and GFR and other integral membrane proteins, including integrins. 7. Rab5 mediates syntenin binding to sdc1 C2 domain on sdc1 CTF on endosomes. 8. Syntenin recruits ALIX. 9. ALIX recruits the ESCRT-III complex. 10. The endosomal limiting membrane invaginates forming ILV's/MVB/s; luminal proteins include inactive Metzincin family proteases. Intact sdc1 is required. 11. Heparanase trims HSPG GAGs on sdc1 and Metzincin proteases become activated (potentially by acidification of the MVBs) and cleave some of the sdc1 on their limiting membrane releasing the sdc1 ectodomain into the lumen of the MVB and within exosomes and sdc1 CTFs in the MBV and exosome limiting membranes. 12. Cargo sorting occurs. Some endosomes/MVBs go to the nucleus taking GF and intact sdc1. Some MVBs fuse with lysosomes and their contents get degraded. Others fuse with plasma membrane and release exosomes along with both intact and shed sdc1 into the environment. The mechanisms that control cargo sorting and MVB trafficking to specific compartments within cells are an active topic for research. The schema above summarizes work described in the following studies: the role of sdc1 in endocytosis was described by Chen and Williams, 76 and the involvement of sdc1 in exosome formation was demonstrated by Baietti et al. 79 and summarized by Hurley and Odorizzi. 80 Not shown, but also important, is that the HSPGs on sdc1 on cell surfaces, but not on the exosomes, mediate exosome adhesion and internalization by cancer cells. 80 Data implicate ERK signaling making it likely that the same events that regulate endocytosis of GFRs are involved in endocytosis of exosomes.
A second phase of sdc1-mediated endocytosis involves activation of Src, phosphorylation of the sdc1 cytoplasmic domain, and recruitment of cortactin. These events are followed by actin-mediated endocytosis. Immunoblots demonstrating phosphorylated sdc1 in isolated endosomes showed discrete bands rather than the broad smears obtained with the intact sdc1 HSPG. 76 This observation suggests that some sdc1 is cleaved during endocytosis. Studies by Zong et al. also showed that sdc1 and tubulin associate at the RMKKK site and that this association is required for endocytosis. 77 They demonstrated, using mesenchymal tumor cells, sdc1-mediated endosomal transport of FGF2 and sdc1 to the nucleus. Interestingly, FGFR became excluded from endosomes containing sdc1 and was trafficked to lysosomes. A role for sdc1 in sorting endosomal cargo to various cellular compartments cannot be ruled out.
The PDZ domain on the sdc1 cytoplasmic tail binds syntenin, which is involved in exosome formation and release. 78 Exosome formation starts within late endosomes. The endosomal membrane invaginates to generate multivesicular bodies (MVBs) containing intraluminal vesicles (ILVs). When MVBs fuse with the plasma membrane, they release ILVs as exosomes. Studies by Baietti et al. elegantly showed that endocytosis, which generates ILVs, and the release of exosomes from MVBs are regulated by the binding of sdc1 with syntenin. 79 Truncations of sdc1 lacking the ectodomain were unable to substitute for the intact molecule in generating exosomes. Both the sdc1 ectodomain and CTF are necessary for exosome formation. When they reduced sdc1 CTF generation using protease inhibitors, they reduced exosome formation and accumulation of sdc1 in exosomes. Intact sdc1 is needed to form exosomes, but metzincin proteases capable of cleaving sdc1 upon activation are present within exosomes and most of the sdc1 present in MVBs and exosomes in these studies was cleaved. Yet, endosomes and/or MVBs with the sdc1 ectodomain can be transported to the nucleus along with heparanase and growth factors like FGF2. 77 While data presented in the endocytosis, exosome, and nuclear transport studies discussed previously were generated using sdc1, the critical sequences needed for vesicle transportation to the nucleus (C1), binding to tubulin (C1), and for binding to syntenin (C2) are shared by all four sdcs. Importantly, sdc-mediated endocytosis and MVB fusion and exosome release are exploited by parasites and viruses to infect host cells. Additionally, retroviruses, such as HIV, use sdc-mediated endocytosis, MVB fusion, and exosome release to escape infected cells. 76,80 Figure 2 shows schematically the events that take place during endocytosis and exosome formation and release.
Summary
There is clearly more to learn about sdc1 and its role in wound healing. In the years since the Bernfield group first described sdc1, over 2,600 articles have reported on different aspects of sdc1 biology and function. Of those, approximately 1,000 are related to the role of sdc1 in cancer and fewer than 100 relate to wound healing. Wound repair studies will continue to gain insight from data being generated by our colleagues in the cancer community.
The integrin heterodimers expressed on cells, the ECM the cells are interacting with, and the amount of shed sdc1 in the environment together determine whether the expression of sdc1 by cells will increase or decrease cell migration. The composition of the ECM is determined both by the cells and by the environment. Cells produce their own ECM and migrate over granulation tissue during the early stages of wound repair. Sdc1 expression enhances keratinocyte migration rates but impedes stromal cell migration during this stage of wound healing. In migrating keratinocytes, sdc1 expression and LN332 secretion are upregulated, active proteases are present, and growth-factor-mediated signaling favors α6β4 phosphorylation and hemidesmosome destabilization. In stromal cells, sdc1 expression increases integrin activation and stabilizes focal adhesions. Enhanced heparanase and protease activity at wound sites causes sdc1 to be shed from cell surfaces and accumulate in the environment. Once keratinocyte migration ceases, the bulk of the LN332 cells encounter will be processed, growth-factor-mediated signaling will be reduced, and intact sdc1 will increase in expression at the cell surface.
Epithelial-derived cancers show significant differences in the expression of sdc1 as the cancer progresses. Most histopathological studies of sdc1 expression in cancer use antibodies that identify the ectodomain. The lack of the sdc1 ectodomain on tumor cells cannot be assumed to correlate with loss of sdc1 expression since protease-mediated shedding could take place. How much of the variability reported in sdc1 expression in cancer is due to differences in shedding and not expression is unclear. In the future, studies should be performed using both sdc1 ectodomain and CTF-directed antibodies.
In nonmelanoma skin cancer, reduced sdc1 is associated with more invasive tumors 51 but expression is increased, often in the tumor stroma, at advanced stages in skin and other cancers. 52 These observations prompted a number of ongoing studies that target sdc1 expression in attempts to treat different types of cancer, including myeloma, breast, colon, and pancreatic cancers. 52
During wound healing, MMP expression, sdc1, and heparanase are increased; the bulk of sdc1 present at wound sites is likely to be cleaved to generate a bioactive ectodomain and a CTF that can be further cleaved by γ-secretase. While we have learned a lot since sdc1 was first described, there are still many important questions we do not know the answers to. Does the shed ectodomain facilitate or impede optimal wound healing? Does it remain behind bound to provisional matrix or to cell surfaces? Do the HSPGs get cleaved to stubs by heparanase? Do the CSPGs on sdc1 play roles in wound repair and impact reinnervation? Would treatments aimed at helping cancer patients also help patients with poor wound healing?
Over the years, more and more proteins have been shown to associate with sdc1 and its functions in cells have expanded to include the formation and release of exosomes and control of endosomal-mediated trafficking. By mastering our understanding of how sdc1 manages to control all of its “contacts” under homeostatic and stressful conditions caused by tissue damage and cancer, we will uncover new ways to help patients manage these life-threatening conditions.
• The ectodomain of sdc1 and 4 regulates integrin activation.
• The intracellular domains of sdc1 and 4 regulate focal adhesion dynamics.
• Sdc ectodomains can be shed and the sdc1 ectodomain functions in regulating cell signaling.
• Heparanase-mediated GAG-chain trimming can alter the functions of both cell-surface-associated and shed sdc1.
• The functions of the CS chains on sdc1 require additional study.
• The impact of sdc1 on cell migration rates is cell type specific.
• All four sdcs potentially can serve as targets for γ-secretase-mediated cleavage.
• Endocytosis and exosome formation are mediated by sequences in the sdc1 cytoplasmic domain.
Footnotes
Acknowledgments and Funding Sources
The authors would like to thank all of our colleagues who have patiently answered questions and taken time to respond to our questions. The research done by our lab on syndecan-1 was funded by NIH grants EY008512, EY021784, and EY023106.
Author Disclosure and Ghostwriting
Stephen Gee, PhD, a postdoctoral fellow at the Wilmer Eye Institute of The Johns Hopkins University School of Medicine, was involved in the editing of this article. There was no ghost writing involved in the preparation of this article.
About the Authors