
Abstract
Spinal cord injury (SCI) remains a major unsolved problem that permanently impairs the lives of innumerable individuals worldwide. Although advances in the basic, pre-clinical and clinical sciences of SCI hold promise for patients, clinicians may lack a full insight into the relevant cellular and molecular events, and laboratory researchers may underappreciate how cellular and molecular phenomena translate into meaningful functional outcomes. To help bridge these perspectives, we first review the American Spinal Injury Association (ASIA) Impairment Scale (AIS) grade, which is the principal instrument used to gauge clinical outcomes in SCI, and the clinically important concept of AIS grade “conversion” (improvement), which occurs in some but not all patients. We then review underlying mechanisms that contribute to the AIS grade and its conversion, including mechanisms of transient neurological dysfunction (neuronal and axonal “stunning”), mechanisms of secondary cell loss (apoptosis, pyroptosis, and necroptosis), and mechanisms of axonal loss (primary axotomy and secondary axonal degeneration). Finally, we briefly review approaches to clinical management that may ameliorate identified mechanisms of secondary tissue loss and neurological dysfunction following SCI.
Introduction
Spinal cord injury
Spinal cord injury (SCI) is a devastating pathological event resulting from mechanical damage to the spinal cord. The most common etiologies are falls and motor vehicle accidents, although sports accidents and penetrating injuries also contribute. 1 SCI has a worldwide prevalence of approximately 20.6 million cases with ∼4/100,000 yearly incident cases in the United States. 2 While epidemiological profiles of SCI vary across countries, in general, the number of male patients exceeds females, 3 with the average age of injury at 59.2 years. 1 Notably, over the past few decades, the average patient age and the proportion of SCIs attributable to falls have increased, whereas the proportion of functionally complete to incomplete injury and the number of patients with fracture dislocation have decreased.4–6
SCI is a leading cause of long-term disability, responsible for more than 6.2 million years of life lived with disability, a dramatic increase in the last 3 decades. 7 Long-term sequelae of SCI may include locomotor dysfunction, loss of hand function, respiratory complications, chronic pain, bowel and bladder incontinence, impaired sexual function, impaired kidney function, and psychiatric abnormalities. 8
SCI also imposes a large economic burden. Direct costs of acute and long-term care vary with level and grade of injury, location of care, and methodology, but, in the United States, they range from $111,780 to $1,156,410. 9 The indirect cost of SCI is perhaps more alarming, with the average annual cost per patient estimated above $29,000. The lifetime economic impact of a single patient with SCI is between $0.5 and $2.3 million, with younger age of injury contributing to a greater total lifetime economic burden. Employment rate post-SCI decreases by an estimated 52%. 10 While the cost to society is substantial, much of the economic burden is borne by SCI patients and their families.
A bench-to-bedside disconnect in SCI research
The current treatment for SCI consists of timely and adequate surgical decompression and critical care with blood pressure augmentation. A large and fast-growing body of clinical research seeks to improve outcomes in SCI. Areas of current interest include the role of early surgical decompression, optimization of surgical timing for adequate decompression, the use of intraoperative imaging including ultrasound, and advanced neurophysiology monitoring including spinal cord perfusion pressure monitoring.11,12 Simultaneously, a growing body of basic science research aims to understand and prevent mechanisms of neuron and axon loss after SCI. This work has yielded a list of exciting (albeit many still experimental) therapies, including calpain inhibitors, 13 sodium-calcium exchanger (NCX)1 inhibitors, 14 voltage-gated sodium channel (VGSC) inhibitors,15–17 sulfonylurea receptor 1–transient receptor potential melastatin 4 (SUR1-TRPM4) inhibitors,18,19 cell-based therapies, 20 novel neurorehabilitation methods, 21 and innovative techniques involving brain–computer interface. 22
While these advances hold great promise for patients with SCI, there often exists a disconnect between clinical and basic science research. Clinicians and therapists may not fully appreciate the cellular and molecular events that mediate improvements or constitute impediments to functional recovery following SCI. Similarly, laboratory researchers may not fully appreciate how cellular or molecular phenomena translate into functional outcomes and quality of life. In this review, we seek to remedy this disconnect by bridging these two perspectives. We first discuss how function is commonly quantified in patients with SCI and then correlate these grading scales with the cellular and molecular events that occur after injury.
Basic Spinal Cord Anatomy
Gross anatomy
The spinal cord is composed of a grey matter core encased in white matter that forms an “H” or butterfly shape cross-sectionally. The spinal cord is predominantly (∼85%) comprised of white matter. 23 The spinal cord’s gray matter is comprised of motor and sensory neuron cell bodies, interneurons, unmyelinated axons, and neuroglia organized into the lateral, dorsal, and ventral horns. The lateral horns, present mainly in the thoracic region of the cord, contain preganglionic autonomic neurons, whereas the dorsal horns contain neurons that receive sensory input from the spinal nerves’ dorsal roots and Clarke’s columns that process proprioceptive information. The ventral horns contain cell bodies of motor neurons terminating on skeletal muscle. Grey matter is further defined by its laminar distribution into 10 distinct subgroups based on topography and density. 24 The white matter exterior of the spinal cord is comprised of mostly myelinated sensory and motor tracts. The ascending sensory tracts include the dorsal columns, spinothalamic, and spinocerebellar tracts, which carry pressure, vibration, pain, temperature, touch, and proprioception information from the body to higher levels of the central nervous system (CNS). Descending tracts relay information related to conscious skeletal muscle control, posture, balance, and muscle tone via the corticospinal, vestibulospinal, rubrospinal, and reticulospinal tracts. In general, sensory tracts are found dorsally, preganglionic visceral motor tracts laterally, and somatic motor tracts in the ventral region of the cord. 25
Grossly, the spinal cord can be divided into four distinct regions defined by the vertebral exit point of 31 spinal nerves: the cervical, thoracic, lumbar, and sacral regions. The first 7 cervical nerves exit the cord superior to the corresponding vertebrae, whereas the remaining 24 nerves exit inferiorly. 24 After exiting the cord, spinal nerves within a region merge to form a network called a spinal plexus. The severity, prognosis, and treatment options for SCI are in part determined by the affected region of spinal cord and plexuses. 26
Axon anatomy
White matter and axons comprise nearly 85% of the total spinal cord volume in an adult human male. 23 Therefore, axon physiology and pathology is intimately associated with spinal cord function. Axons are highly specialized subcellular compartments, described previously as a “cell within a cell.” Interestingly, in certain species of wasp, some axons exist independently of their nuclei, which lyze and die during development. 27 Axon dimensions vary greatly: axon diameters range from 0.1 to 30 microns in vertebrates and axon lengths can be up to 1 m in humans. Larger axonal diameters support higher firing rates at the expense of greater energy and space requirements.28,29
Ultrastructurally, axons contain a cytoskeleton comprised of a microtubule and neurofilament core surrounded by an actin cortex. 29 Microtubules are ∼25 nm thick, polar filaments of variable length comprised of polymerized α/β tubulin heterodimers arranged longitudinally in overlapping bundles. Microtubules serve as a roadway for axonal transport and contribute to structural stability and branching. 30 Neurofilaments are ∼10 nm thick, nonpolar filaments comprised of polymerized neurofilament hetero-oligomeric subunits and are arranged longitudinally alongside the microtubule bundles. Neurofilaments form an inert hydrogel “solvent” that influences axon diameter by expanding the cross-sectional area.29,31 The axon cortex is comprised of periodically spaced F-actin rings interlocked by spectrin cross-linkers that form a “corset” around the cytoskeletal core. The actin cortex serves as a tension buffer that organizes the axon plasmalemma.30,32–34
Axons contain an array of organelles. Mitochondria are present in large numbers to support the high axonal energy demand, 35 since even at rest a human neuron consumes ∼4.7 billion ATP molecules per second. 36 Axonal mitochondria are regulated by cytosolic calcium and contribute to calcium buffering.37–39 Axons also harbor an extensive reticulated network of thin (∼10 µm) smooth endoplasmic reticulum (SER) tubules. 40 Axonal SER supports the lipid metabolism and lipogenesis required for axon repair and growth. 41 Axonal SER is also an important calcium store and conducts intra-axonal signaling between the plasmalemma and organelles or between the axon and the neuronal somata.42–44 Axons also contain a robust endosomal and peroxisomal system. 30
The axolemma of myelinated central axons exhibits regular periodicity in its composition. Nodes of Ranvier, where axonal segments are “bare” and not covered with myelin, are interspersed with axonal internodes, where the investing myelin becomes compacted. 45 In the regions immediately adjacent to the nodes of Ranvier, the surrounding myelin forms paranodal loops, where cytoplasm fills the myelin lamellae. 45 VGSCs are abundant at nodes of Ranvier, 46 whereas potassium channels are primarily present at juxtaparanodal regions. 47
SCI AIS Grade Conversion
SCI grading
The most widely used and best validated clinical scoring system to assess SCI and establish prognosis is the International Standards for Neurological Classification of Spinal Cord Injury (ISNCSCI) and the American Spinal Injury Association (ASIA) Impairment Scale (AIS). 48 This grading scale was first published in 1982 and has undergone several revisions. 49 Strength is quantified by motor scores and sensation (light touch and pinprick) by sensory scores. The ISNCSCI helps to determine the neurological level of injury (NLI), and the AIS grade summarizes the extent or completeness of injury. AIS grade A injuries have no preservation of motor or sensory function in the S4-5 sacral segments, AIS B injuries have preservation of sensation below the NLI including S4-5, AIS C and D have increasingly preserved motor function below the NLI, and AIS E represents a patient with intact gross motor and sensory function. Of note, the NLI itself does not directly influence the AIS grade.
AIS grade conversion
Patients presenting AIS grade A–D may later recover some degree of motor and sensory function leading to improvement in the AIS grade, a phenomenon referred to as AIS grade conversion. 50 The precise definition of AIS grade conversion varies in the literature, but here, we use the term to refer to any upward change in AIS grade after the initial presenting assessment.
There are conflicting opinions and data regarding the reliability and stability of AIS grade obtained acutely after SCI. Some have reported early worsening in the AIS grade during the first 72 h after injury, with later (>72 h) AIS grade assessments correlating better with outcome than early (24 h) assessments.51,52 Similarly, others have shown that AIS grade assessments captured at very early time points (<4 h) are more likely to exhibit later grade conversion. 53 However, other investigators have demonstrated and argued that early (<72 h) AIS grade assessments can be reliably correlated with outcome.54,55 Notwithstanding the ongoing debate regarding the prognostic utility of early (<72 h) AIS grade assessments, a subset of SCI patients do appear to exhibit AIS grade fluctuations acutely after injury.
Upward AIS grade conversions can also occur at later timepoints. Approximately 25–30% of patients who are initially categorized as AIS grade A group later convert to AIS grade B or better, ∼65–75% of AIS grade B and C patients convert to improved grades, and ∼10% of AIS grade D patients convert to E.56–58 Importantly, conversion does not imply complete neurological recovery since generally, when patients exhibit AIS grade conversion, they convert to the next better AIS grade rather than leap multiple grades. 56 While most (∼50%) upward conversion occurs within the first month, some patients experience conversion throughout the first year after injury, with decreasing likelihood thereafter. 56
The practical impact of AIS grade conversion upon patient quality of life must be considered. Patients with SCI place particularly high importance on recovery of hand function, walking, and bowel and bladder function.59,60 Recovery of these functions depends heavily on the initial AIS grade. For example, patients who are initially AIS grade A are highly unlikely to recover walking, as only 14% of those who exhibit grade conversion from A being able to achieve functional walking. 61 Furthermore, if walking is achieved in AIS A patients, they are likely to have low thoracic or lumbar injuries, and the walking is likely to be slow and energy intensive.62,63 The odds of walking recovery are better in AIS grade B, C, and D patients, rising to 33%, 75%, and 100%, respectively. 61 Interestingly, while AIS grade conversion indicates improved function, grade conversion does not correlate well with recovery of walking function, indicating that more targeted neurological outcomes measures are necessary.61,64 Total motor score is predictive of bowel function, which is more severely impacted in complete versus incomplete injuries. 65 Completeness of injury and thus AIS grade is also highly correlated with the likelihood of recovery of hand function. 66
Clinical factors associated with AIS grade conversion
Various factors have been linked to AIS grade conversion. Broadly, these factors can be categorized as modifiable versus nonmodifiable. Nonmodifiable factors are related to patient characteristics including demographics, injury mechanism, injury morphology, and presenting neurological status. For example, the initial AIS grade is strongly predictive of grade conversion. As discussed above, patients who are AIS grades A and D are less likely to convert than grades B or C, although for different reasons. The low rate of conversion for AIS A is likely due to injury severity, whereas the low rates of AIS D conversion are probably reflective of a “ceiling effect” inherent to the AIS grading scale itself. 56 Level of injury is also predictive of grade conversion, with lumbar injuries being the most likely to undergo grade conversion, followed by cervical and finally thoracic injuries. 50 These segmental differences in vulnerability arise in part from differences in blood supply and grey matter composition. 67 Other nonmodifiable factors include patient age, mechanism of injury (penetrating versus blunt, motor vehicle collision versus ground-level fall), and genetic polymorphisms. 50 The extent of the sensory zone with partial preservation or the number of levels below the sensory NLI that retain partial sensation is also predictive of conversion, as preservation of more than three levels is linked to a greater likelihood of conversion. 68
Modifiable factors are of greater interest to the medical community. Blood pressure augmentation and careful avoidance of hypotension are critical components of the acute management of SCI and have been linked with improved outcomes.69,70 The AANS/CNS Joint Section on Spine and Peripheral Nerves recommends that the mean arterial pressure be maintained at 85–90 mm Hg for 7 days after injury.71,72 Early (<24 h after injury) and adequate surgical decompression has also been linked with improved outcome.58,73–75 Additional experimental interventions, including intrathecal pressure monitoring, have been proposed to help guide blood pressure management. Overall, these modifiable factors likely contributed to the improved rates of AIS grade conversion recently observed versus historical controls. 76
The precise mechanisms responsible for AIS grade conversion are often vaguely defined, with many authors alluding to improved tissue perfusion and reduction of secondary injury. However, this lack of mechanistic detail presents a barrier to the development of novel targeted therapies. Here, we discuss known mechanisms that underlie AIS grade conversion.
Mechanisms Underlying AIS Grade Conversion
AIS grade conversion is not due to healing
Many patients and even some practitioners may witness AIS grade conversion and assume that these patients are simply healing from their injury. However, since the CNS mostly lacks regenerative properties, this cannot be the case. Parenthetically, while endogenous repair mechanisms such as axonal sprouting have been proposed to contribute to functional recovery, their role in motor recovery in human SCI remains unclear.77,78 Rather, recovery of function after SCI primarily results from axons and neurons that become temporarily silenced following injury and that later recover function. During this period of neuronal silencing or stunning, secondary injury and the resultant permanent loss of additional axons and neurons may occur, lowering the total pool of potentially viable neural elements and limiting functional recovery. Therefore, AIS grade conversion reflects the integrated effects of two phenomena: (i) secondary injury with ongoing loss of neurons and axons and (ii) temporary “stunning” of the anatomically preserved axons and neurons due to conduction block and hyperpolarization, respectively, also referred to as transient neuropraxia and spinal cord concussion. These phenomena and their relevance to AIS grade conversion are discussed in this section.
Neuronal stunning creates a “fog of war” in SCI
Following SCI, a subpopulation of injured spinal cord axons and neurons becomes temporarily nonfunctional, a phenomenon that we refer to here as “neuronal stunning” but also referred to as transient neuropraxia and spinal cord concussion. Stunned axons and neurons remain viable and are potentially salvageable but cannot support normal signal transduction. Mechanisms of stunning differ between axons and neuronal somata. Temporary axon dysfunction secondary to conduction block has been observed in perilesional axons in animal models of SCI.79–81 Several factors contribute to axon conduction block, including demyelination of perilesional axons,80,82 secondary axonal degeneration (SAD), a form of delayed axonal loss that is reversible in early phases and results in the formation of axonal spheroid formation, 83 edema and hemorrhage with subsequent neuroinflammation, generation of nitrous oxide and free radicals, and localized mass effect resulting in impaired tissue perfusion. 84 Resolution of conduction block is associated with functional improvement. 80 Temporary neuronal somata dysfunction is primarily due to post-injury hyperpolarization and decreased excitability. 85 Of note, a related phenomenon is so-called “spinal shock,” which refers to the temporary loss of spinal cord reflexes caudal to the level of injury secondary to the loss of descending synaptic input.86,87 Stunned axons can potentially exist both in the T2-weighted MRI hyperintense lesion itself and in the perilesional tissues that appear radiographically normal.
Neuronal stunning is a temporary situation in which the extent of interrupted neurological function may or may not predict the underlying extent of permanent tissue loss. In essence, neuronal stunning creates a “fog of war” in SCI. The phrase “fog of war” was coined by Sir Lonsdale Augustus Hale in 1896 as “the state of ignorance in which commanders frequently find themselves as regards the real strength and position, not only of their foes, but also of their friends.” 88 Similarly, due to neuronal stunning, after SCI the actual number of viable axons and neurons is hidden and ongoing losses are obscured. Neuronal stunning therefore complicates baseline assessments in SCI clinical trials and may cloud early prognostication. Neuronal stunning is highly relevant to AIS grade conversion in that its resolution can contribute to AIS grade improvement.
Tissue bridges highlight functional versus anatomical completeness of injury
Importantly, due to neuronal stunning, what may be thought to be a “complete” SCI (with the designation of completeness based upon the initial neurological examination) may turn out to be an incomplete injury that later exhibits partial functional recovery. For this reason, a careful distinction should be made between “functional completeness,” where completeness of injury is based upon the neurological examination, and “anatomical completeness,” which corresponds to anatomical tissue loss that would preclude neurological recovery. Therefore, an initially functionally complete SCI patient may not harbor an anatomically complete injury such as a cord transection but rather have stunned white matter with later partial functional recovery as neuronal stunning resolves. Similarly, patients with incomplete neurological deficits may eventually have complete return of function with no underlying neurological tissue loss at all (e.g., transient neuropraxia in the setting of congenitally narrow spinal canal).
The presence of intact tissue bridges on T2-weighted MRI after traumatic SCI is a clear example of intact white matter tracts that are stunned and may potentially recover. As discussed above, stunned axons and axons undergoing secondary degeneration can exist in both T2-weighted MRI hyperintense lesional tissue and the surrounding perilesional tissue. Multiple studies have validated the prognostic value of tissue bridges, showing that for each mm of preserved tissue, patients gained 7.8–12.1% of maximal sensorimotor recovery at 3 months.89,90 Here, we describe an index case that illustrates tissue bridges and highlights the difference between anatomical and functional completeness of injury.
Index case: A 56-year-old man suffered a recreational sport traumatic SCI and was rendered quadriplegic. He was airlifted to the R Adams Cowley Shock Trauma Center and was seen within 45 min in the Trauma Resuscitation Unit. Post-resuscitation ISNCSI exam revealed an ASIA motor score of 15 and ASIA Impairment grade of A. CT scan of the cervical spine obtained 2 h after trauma revealed a C5 tear-drop fracture (AO Class C; Fig. 1A) and T2-weighted MRI 6 h after trauma showed hematomyelia and spinal cord swelling (Fig. 1B). One-stage C5 corpectomy and C4 + C5 laminectomy and fusion (Fig. 1C) was performed. Post-operative MRI indicated spinal cord decompression and spinal cord swelling (Fig. 1D). MRI obtained nearly 6 months after injury indicated a small syrinx at the C5 level (Fig. 1E). Closer examination of MRIs indicates clear tissue bridges traversing the injury pre-operatively (Fig. 2A) that were preserved immediately post-operatively (Fig. 2B) and at 6-month follow-up (Fig. 2C). At 6-month follow-up, the patient’s ASIA motor score improved to 56 and AIS grade C.

Index case. Midsagittal CT cuts indicating C5 tear-drop fracture (arrow) with translation of C5 body into spinal canal
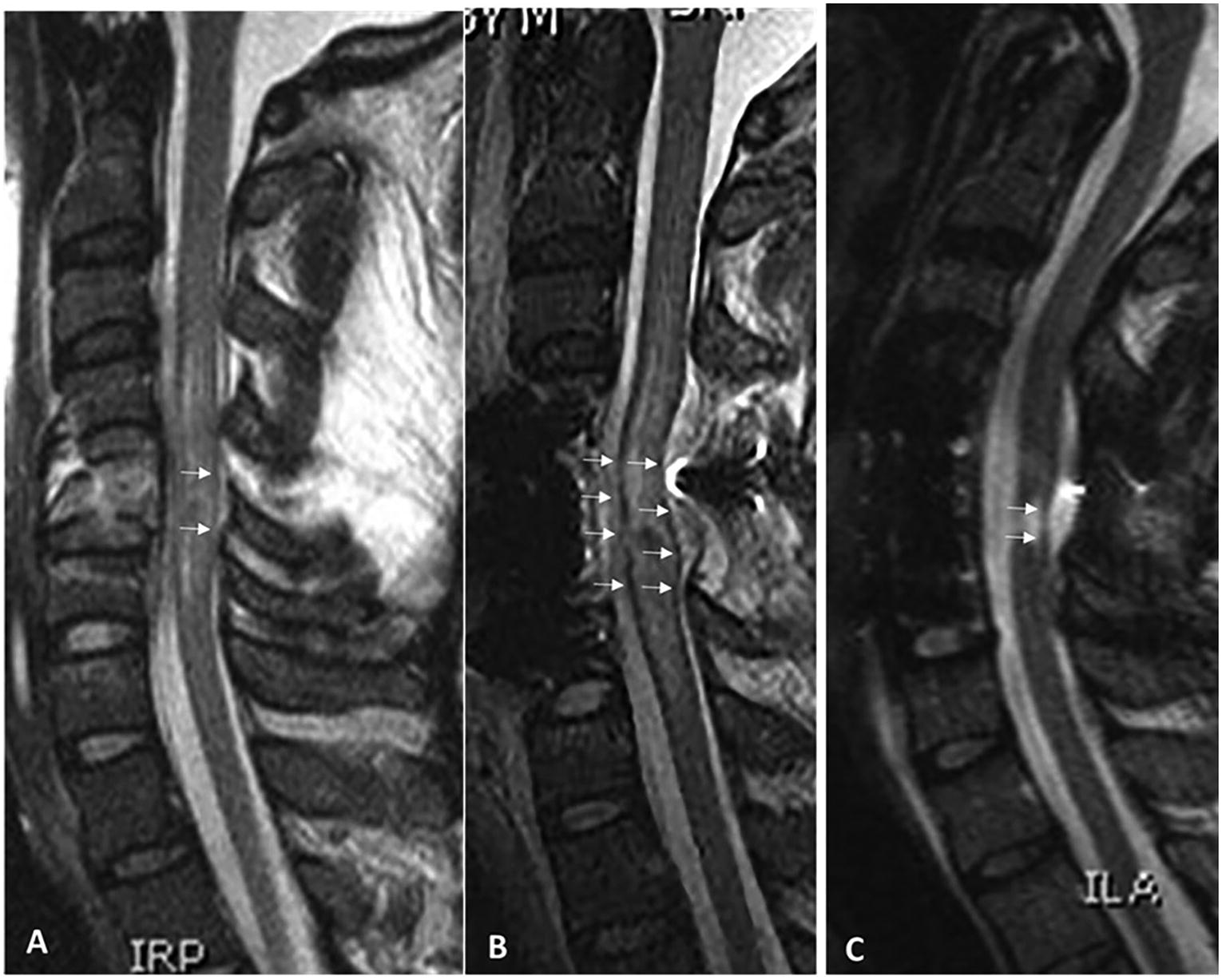
Index case tissue bridges. Midsagittal T2-weighted MRI cut indicating faint tissue bridges (arrows) in preoperative MRI
Careless use of nomenclature may have resulted in the index patient being erroneously labeled as harboring a “complete SCI,” resulting in an overly grim prognosis delivered to the patient and his family. Instead, due to the phenomenon of neuronal stunning of axons in the preserved tissue bridges, this patient harbored a functionally complete yet anatomically incomplete injury. Assisted by proper medical and surgical management, he later underwent AIS grade conversion. Careful use of nomenclature is essential to avoid unwarranted clinical nihilism.
Basic concepts of secondary injury
Secondary injury is widely recognized to be an important contributor to tissue loss after SCI. The concept of secondary injury originated in 1911 from experiments conducted by Major Alfred Reginald Allen, who developed the first experimental model of SCI. Dr. Allen noticed that evacuation of the contused hemorrhagic tissue led to functional improvement and postulated that some “noxious agent” was released by the injured tissue that led to autodestruction of surrounding neural tissue. 91 Tragically, he died at the age of 42 from a shrapnel injury during the Meuse-Argonne Offensive of World War I. Later ultrastructural studies showed that, following experimental spinal cord contusion, initially intact tissue surrounding the lesion undergoes progressive changes including loss of endothelial integrity, erythrocyte extravasation, axonal swelling and degeneration, edema formation, and cystic degeneration.18,19,92,93
Despite its long history, the term secondary injury remains nebulously defined. Phenomena including ischemia, progression of hemorrhage, oxidative stress, and neuroinflammation have all been referred to as secondary injury in the literature. However, these phenomena represent causes or mechanisms of secondary injury rather than secondary injury itself. We define secondary injury as the delayed loss of functional neural tissue, including axons or neuronal somata, and the delayed loss of supportive cells such as glia or vasculature, which can contribute to neuronal loss. The time course of secondary injury after SCI varies depending on the mechanism. For example, in animal models, axonal swelling and loss is a relatively early event, with spheroid formation occurring within 1 h of injury, 70% of axonal loss occurring within the first day after injury, and continued loss throughout 2 weeks. 94 In contrast, apoptotic cells are most frequently observed at ∼1 week after injury, although apoptosis is detectable as early as 6 h after injury. 95
AIS grade conversion is an integration of neuronal stunning and secondary injury
Secondary injury reduces probably the upward AIS grade conversion by progressively reducing the underlying population of potentially viable neural tissue. Since a mere 5–10% salvage of white matter volume can translate to substantial improvement in functional outcome,96,97 the aggressive prevention of tissue losses from secondary injury is of the highest priority. Secondary injury may co-associate with neuronal stunning, which may both mimic and potentially “hide” ongoing secondary injury, so that it is not always possible at the bedside to distinguish secondary injury from transient neuronal stunning. Only after the “fog of war” of neuronal stunning has subsided can the true extent of primary and secondary injury be fully appreciated, although it remains difficult to determine the amount of potentially salvageable neural tissue that is lost (see above). The integration of these phenomena therefore manifests as AIS grade conversion (Fig. 3). As a consequence of the entanglement of secondary injury and neuronal stunning, neurological deficits cannot be simply ascribed to stunning but rather should be aggressively treated as if they may become permanent without medical intervention. In the following sections, key mechanisms of secondary injury are individually described.

Neurological function is an integration of neuronal stunning, primary injury, and secondary injury. Illustration showing that neurological impairment at each time point and the neurological outcome after spinal cord injury reflects the integrated effects of primary injury, secondary injury, and neuronal stunning. Neuronal stunning, which arises from conduction block and hyperpolarization, is a reversible phenomenon. Secondary injury due to SAD and programmed cell death is potentially preventable yet irreversible once it occurs and primary injury is neither preventable nor reversible. Neuronal stunning may obscure the extent of primary injury and ongoing secondary injury, i.e., the “fog of war,” resulting in neurological examinations that do not fully predict potential functional recovery. SAD, secondary axonal degeneration.
Programmed cell death contributes to secondary injury
Multiple mechanisms of cellular death contribute to neuronal loss after SCI. While reviews exist that describe these mechanisms in detail,98,99 here, we briefly discuss their relevance to secondary injury after SCI. Of note, the multifactorial nature and variety of mechanisms of cell death after SCI may explain the lack of success of specific inhibitors.
Apoptosis, a form of programmed cell death that involves membrane blebbing, nuclear fragmentation, cell shrinkage, and plasmalemmal exposure of phosphatidylserine, may be triggered by intrinsic or extrinsic factors. 98 During intrinsic (mitochondrial) apoptosis, intracellular stress precipitates permeabilization of the outer mitochondrial membrane, cytochrome C release, and activation of the apoptosome and caspase-9, -7, and -3, with subsequent execution. 98 Extrinsic apoptosis is triggered by signaling death ligands including fas ligand or tumor necrosis factor (TNF), which activates caspase-8 and subsequently activates caspases-7 and -3, with execution. 98 Importantly, apoptosis does not include cell lysis and is therefore less inflammatory than other forms of programmed cell death. Apoptosis is widely detected in multiple different cell types after experimental and human SCI. 100 Neuronal apoptosis peaks at early time points within the first day after injury, whereas a second wave of apoptosis occurs in glial white matter cells several days after injury and is associated with white matter tract degeneration.101–103
Pyroptosis is a form of inflammatory cell death originally characterized in bacterial infections. In pyroptosis, extracellular markers of infection or disease, such as pathogen- or damage-associated molecular patterns (DAMPs), engage cell surface receptors, resulting in activation of large complexes called inflammasomes. These inflammasomes activate caspase-1, resulting in plasmalemmal pore formation, cell swelling, and membrane rupture. 98 After SCI, markers of pyroptosis are upregulated,104,105 and pyroptosis has been shown to contribute to post-SCI neuronal death. 106
Necroptosis is a caspase-independent form of programmed cell death that is triggered classically by TNFα signaling and inflammation. Necroptosis exhibits many features similar to necrosis, with early membrane permeabilization and cellular and organelle swelling, but unlike necrosis relies on an active signaling cascade mechanism.107,108 Necroptosis has been shown to contribute to neuronal and glial cell death in SCI and worsens functional outcome.109–111
Secondary axotomy contributes to secondary injury
Axon disconnection after injury can be caused by primary axotomy, which refers to the direct mechanical rending of axons that occurs at the time of injury, or secondary axotomy, a delayed form of axotomy that occurs as a consequence of SAD. Primary axotomy is not amenable to therapeutic intervention since the disconnection occurs at the time of injury (an ultrastructural analog of anatomical completeness). SAD is morphologically evident by the formation of periodic swellings or spheroids along the axons involved. Unlike primary axotomy, since SAD remains reversible in the initial stages and occurs in the hours after injury, it may be amenable to therapeutic intervention.112–114
SAD was originally described in traumatic brain injury (TBI). While axon retraction bulbs and axon swellings were observed in human specimens as early as 1900, they were presumed to be caused by direct mechanical rending of axons. 115 In 1956, Sabina Strich described five patients with diffuse degeneration of the white matter and poor neurological outcome following TBI, despite a lack of intracranial hematoma or raised ICP. 116 This led Thomas Gennarelli and others in the 1980s to describe and coin the term diffuse axonal injury (DAI). 117 However, even at this time, axon degeneration after DAI was still presumed to be caused by direct mechanical rending of axons. 118 It is only since the late 1980s and 1990s when John Povlishock and others showed that many axons remain initially in continuity after TBI but later undergo disconnection and that the role of SAD was appreciated. 119 Primary axotomy is now believed to only occur after the most severe TBIs, whereas secondary axotomy likely represents the dominant cause of axon disconnection.
Secondary axotomy and SAD also occur in SCI, 94 where axons undergoing SAD exhibit similar morphological changes to those seen after TBI, including formation of axon swellings and endbulbs.94,113 As in TBI, the axons undergoing SAD after SCI are potentially salvageable in the early stages, with one study showing spontaneous resolution of ∼40% of spheroids after injury. 94 Given the importance of white matter to functional outcomes after SCI,96,97 SAD is an important target for therapeutic intervention. Of note, SAD and secondary axotomy may be particularly relevant in tissue bridges in that these processes would shrink the pool of potentially recoverable neural elements. Since SAD and secondary axotomy are also triggered by hypoxia, timely surgical decompression and avoidance of systemic hypotension may improve outcomes in part through axonoprotection. 120 While the detailed molecular mechanisms currently remain unclear, SAD appears to occur in three major phases discussed below, which may be part of a general axonal response to mechanical injury (Fig. 4).

Phases of SAD. Illustration showing the phases of SAD. In phase 1, axoplasmic ionic dysregulation occurs with maladaptive overload of axoplasmic sodium and calcium. In phase 2, axoplasmic calcium overload triggers activation of calcium-sensitive proteases and lipases and mitochondrial permeability transition pore (mPTP) permeabilization, which further contributes to calcium overload and energy failure. In phase 3, protease-mediated degradation of the axon cytoskeleton results in spheroid formation and eventually in secondary axotomy. Prior to axotomy, this process is potentially reversible. SAD, secondary axonal degeneration.
Phase 1: Axoplasm ionic dysregulation
The first phase of SAD is the dysregulation of axoplasm ion homeostasis. Within minutes of injury, axoplasmic sodium quickly elevates. 121 There are multiple potential routes for sodium influx. The so-called mechanoporation of the axolemma, where temporary pores form in the normally impermeable plasma membrane, may allow for translocation of ions and larger molecules such as charged proteins such as horseradish peroxidase.122–124 Notably, mechanoporation remains controversial, with some authors arguing that these observations may simply arise from the severed and open ends of primarily axotomized axons.125,126 Sodium overload also arises from inhibition of the Na+/K+/ATPase secondary to energy failure and transmembrane influx through VGSC.127,128 VGSCs are mechanosensitive,129,130 and when axonal stretch is applied, these channels convert from slow to fast current kinetics. Pharmacological inhibition of VGSCs following axonal injury has been shown to reduce sodium and calcium influx.121,125,131 Interestingly, activation of VGSCs is augmented after injury due to calcium-mediated proteolysis of an intracellular loop, resulting in the loss of inactivation.132–134
Sodium influx precedes and causes calcium influx,131,132 which is a key event after axonal injury. Axoplasmic calcium overload occurs through influx across the axolemma and also through intracellular store release. 135 Axoplasmic calcium overload is synchronous with a decrease of extracellular calcium, highlighting the importance of extracellular sources.121,125,136,137 This occurs through multiple plasmalemmal channels and transporters. NCX exchangers typically function in the so-called forward mode where calcium is exported and sodium imported. Normally, NCX1 is localized to perinodal regions where it contributes to calcium buffering. 138 After injury, NCX1 contributes to axon calcium overload125,133,139,140 and SAD. 141 During intracellular sodium overload, NCX exchanges operate in reverse mode, where sodium efflux becomes linked to calcium influx. Multiple subtypes of voltage gated calcium channel (VGCC) and N-methyl-D-aspartate (NMDA) receptors are also implicated in neuron degeneration and calcium influx after axon injury.125,142–144 Intracellular release of calcium stores is also a major contributor to calcium overload. Calcium release from the axoplasmic reticulum contributes to calcium overload after injury, and inhibition of axoplasmic reticulum calcium release is protective in models of SCI.145–150 Axonal mitochondria also release calcium after injury, further contributing to overload. 145
Phase 2: Immediate sequelae of calcium overload
Axoplasmic calcium overload is a key pathological event in SAD. After SCI, dysregulation of intracellular calcium is independently predictive of axonal loss, and depletion of extracellular calcium is axonoprotective. 113 Calcium overload triggers an array of sequelae that contribute to axon degeneration, including activation of calcium-sensitive proteases and lipases, including calpains, caspases, phospholipase A2, lipoxygenase, and cyclooxygenase.151,152 Mitochondrial dysregulation and protease activation are two immediate sequelae of calcium overload that are particularly pertinent to axon injury and represent potential targets of therapeutic intervention.
Calcium overload leads to mitochondria dysregulation, which contributes to axonal degeneration. Axoplasmic calcium overload triggers permeabilization of the mPTP, which results in loss of the mitochondrial membrane potential, energetic failure, and additional release of intracellular calcium.153,154 Inhibition of mitochondria permeability transition pore (mPTP) protects axons from degeneration and contributes to axon degeneration through release of cytochrome-c and activation of caspases and calpains.155,156
Axoplasmic calcium overload also triggers calpain protease activation. Calpains are a family of calcium-activated intracellular proteases that are activated after axon injury and contribute to SAD. 157 The most common calpain isoforms are µ- and m-calpain, which are activated by micromolar and millimolar levels of calcium, respectively. 157 All neurofilament isoforms, microtubules, and spectrin are calpain substrates, and calpain-mediated cytoskeletal breakdown represents a key event in SAD. 157 After axonal injury, calcium influx is spatially correlated with calpain activation, which is detected in axons as early as 90 min after injury and is present up to 1 week after injury.124,158,159 Blockade of sodium and calcium influx protects against calpain activation. 144 Calpain activation is correlated with cytoskeletal degradation and neurofilament compaction, and calpain blockade protects against axonal degeneration.113,160–165 Importantly, pan-calpain inhibition may not be therapeutic, as some calpain isoforms yield neuroprotective effects. 166
Phase 3: Axon spheroid formation and secondary axotomy
As SAD progresses, a subset of injured axons demonstrate morphological changes, wherein focal regions of axon swelling form, a phenomenon also referred to as axon spheroid formation. Spheroid formation was first described in 1983 in a cat model of mild TBI 167 and later confirmed in human specimens. 168 In these tissues, direct mechanical disconnection of axons, microhemorrhage, and parenchymal disruption is often absent.119,167,169
Axon swellings primarily occur at internodal regions and reflect an abnormal local accumulation of organelles, ultimately caused by a local breakdown of anterograde transport due to cytoskeletal disruption.170,171 Calpain-mediated cytoskeletal breakdown plays a major role in this process. 124 Nodal regions also exhibit other early morphological changes, including formation of nodal blebs and loss of subaxolemmal density. 172
Starting at 3–6 h and peaking at 12–24 h after injury, a subset of axon swellings exhibit constrictions and multilobulations.167,169,172 These constrictions deepen, eventually dividing the axon and completing the process of SAD with secondary axotomy, with subsequent degradation of the distal part of the axon. Interestingly, while secondary axotomy is most frequent during the hours-to-days after injury, axonal swellings are also detected at more chronic time points after injury, indicating ongoing delayed degeneration.173,174 While the majority of studies have been conducted in human TBI and TBI animal models, similar changes occur after SCI and peripheral nerve injury,94,175 indicating that SAD is a general form of delayed axonal degeneration.
Immunohistochemical markers have been developed to detect secondary axotomy in postmortem tissues. β amyloid precursor protein (β-APP), a widely expressed anterograde transported axonal protein, is currently the most sensitive and best validated marker for SAD.176–178 β-APP immunolabeling accumulates at regions of axonal swellings and disconnection due to local breakdown of anterograde transport and is a useful marker for the aforementioned morphological changes.170,179 There are several important limitations for β-APP immunolabeling that should be considered. For unclear reasons, β-APP immunolabeling only identifies a subset of traumatically injured axons, 180 is lost after a few days in the proximal axon, 181 is not present in the distal axon, 169 and may therefore underrepresent total axonal damage in some cases. 182
SAD and spheroid formation is reversible during the early stages prior to axon disconnection.112–114,183 Axons that exhibit focal swellings are vulnerable but remain potentially viable. In one study of SAD following experimental SCI, more than 40% of axons exhibiting focal swellings exhibited spontaneous recovery during the course of the experiment. 94 While SAD contributes to secondary injury by driving secondary axotomy, it also contributes to neuronal stunning in that axons undergoing early stages of SAD suffer from conduction block. Since up to 40% of axons exhibiting spheroid formation can recover, this conduction block may be resolved, manifesting as AIS grade conversion.
Factors that worsen or ameliorate secondary injury
Programmed cell death and secondary axotomy represent the core underlying mechanisms of secondary injury. Associated pathophysiological processes including ischemia, inflammation, edema, and hemorrhage exacerbate these processes by creating a hostile extracellular milieu in which the chance of axon and cell survival is diminished. Many of these factors are also potentially modifiable with clinical interventions.
Tissue ischemia and hypoperfusion are major drivers of secondary injury after SCI. Following SCI, tissue perfusion becomes progressively impaired in both grey and white matter.184–188 In axons and neurons, ischemia results in increased intracellular sodium and loss of intracellular potassium consistent with dysregulation of the Na/K/ATPase.127,128 This culminates in increased intracellular calcium and degeneration, which further worsens programmed cell death and secondary axotomy as described above.127,128 Ischemia and hypoxia therefore greatly compound post-traumatic neuronal and axonal loss. 189 Early and complete surgical decompression and avoidance of hypotension after SCI are the current mainstays of treatment to mitigate the negative effects of ischemia and hypoperfusion and promote survival of neural tissue. There is ongoing debate regarding the precise timing and method of surgical decompression. The STASCIS trial showed that early (within 24 h from injury) surgical decompression improved the odds of AIS grade conversion in patients with cervical SCI, 74 a finding that has been reproduced in subsequent studies.190–192 It remains unclear whether additional benefits might be reaped from ultra-early surgery, in which surgery is pursued at earlier timepoints after injury. 191 With regard to surgical technique for decompression, posterior laminectomy yields increased decompression rates versus anterior fixation. 193
Under physiological conditions, the spinal cord is an immune privileged organ. However, injury precipitates a local release of DAMPs, which stimulate the innate immune system. This triggers a wave of neutrophils and later monocytes and macrophages to invade the damaged spinal cord. 194 At later timepoints, the adaptive immune system also contributes to neuroinflammation by the presence of NK-, B-, and T-cells. For reasons that are not entirely clear, SCI generates a relatively greater magnitude of immune response compared with TBI.195–197 Although neuroinflammation contributes to debris clearance and is not purely maladaptive, many immune cells also have cytotoxic effects. Neutrophil degranulation releases myeloperoxidase, which generate free radicals within the contusion, contributing to glial and neuronal apoptosis beyond the injury epicenter. 198 Neutrophil extracellular traps composed of granule proteins and chromatin are expelled from the neutrophils and cause additional apoptosis and fibrosis.199,200 Classically activated monocytes and macrophages also contribute to secondary injury by secreting neurotoxic compounds including interleukin (IL)-1, TNF-α, and IL-6, which further propagate inflammation, contribute to blood–spinal cord barrier breakdown, and stimulate astrogliosis and glial scarring.201–204 These monocytes and macrophages also secrete nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and nitric oxide synthase (NOS2), which lead to oxidative stress.203,205,206 Axon dieback, the phenomenon in which axonal growth cones undergo rapid retraction, has been connected to the presence of pro-inflammatory macrophages in the injured spinal cord.207,208 While the national acute spinal cord injury study (NASCIS) trials have studied methylprednisolone to reduce neuroinflammation in the NASCIS trials,209,210 controversy over potential lack of efficacy and side effects have prevented widespread adoption.
Contusion expansion and edema formation are key aspects that promote secondary injury in SCI. Contusive lesions appear within minutes after SCI and progressively expand due to progressive hemorrhagic necrosis.18,19,211 In the hours after injury, small petechial hemorrhages form in the normal parenchyma surrounding an existing contusion. These petechial hemorrhages may grow in size and merge, thereby expanding the front of the original contusion. Mechanistically, maladaptive de novo upregulation of the SUR1-TRPM4 channel by peri-lesional capillaries mediates oncotic endothelial death and capillary rupture due to influx of water.19,212 Edema accumulates in the spinal cord within minutes of injury and persists for 20 or more days.213,214 The extent of edema is proportional to injury severity. 215 Mechanisms driving edema formation are diverse and reviewed elsewhere.216,217 The presence and expansion of progressive hemorrhagic necrosis and edema can be observed radiographically in human SCI as a region of T2 hyperintensity centered at the lesion epicenter. This area of T2 signal is usually measured and summarized by the intramedullary lesion length (IMLL) or BASIC score. In human SCI, IMLL expands at a rate of approximately 1000 µm/h in patients with AIS grades A and B injuries and 21 µm/h in AIS grades C and D injuries.218,219 Edema and hemorrhage together worsen the odds of a positive AIS grade conversion. IMLL expansion is strongly associated with AIS grade conversion, with a 10-mm increase in IMLL being linked with a 40% decreased odds of AIS grade conversion. 58 Blood itself contains multiple directly neurotoxic components that are active at physiological concentrations. 220 As a consequence, spinal cord hemorrhage directly results in axon and neuronal death. 221 Both edema and hemorrhage contribute to increased intrathecal pressure and can worsen tissue perfusion, indirectly contributing to secondary injury. 214
Conclusions
For patients with SCI, the possibility of undergoing AIS grade conversion represents a flame of hope during what is likely the worst period of their lives. AIS grade conversion is an integration of the effects of primary injury, secondary injury, and neurological stunning and does not represent “healing” of neurological tissues. The key modifiable factor contributing to AIS grade conversion is secondary injury, which is comprised of programmed cell death of neuronal somata and SAD of axons. Currently, the only interventions to decrease secondary injury are timely and complete surgical decompression and avoidance of hypotension. These interventions are mechanistically nonspecific and have remained unchanged for decades. While we have developed some insight into the mechanisms of programmed cell death and SAD after SCI, more work is needed before targeted therapies can be developed and translated into widespread clinical use.
Transparency, Rigor, and Reproducibility Summary
Since this article is a review article, it was not preregistered. No animals were used for this study. No statistical testing, blinding, or power analysis was required for this study. Further information is available on request of the corresponding author.
Footnotes
Acknowledgments
The authors would like to thank Tina Wang for her generation of original illustrations for this review article.
Authors’ Contributions
J.A.S. and J.M.S.: Conceptualization; V.G. and J.M.S.: Resources; B.A.: Data curation; J.A.S.: Writing—original draft; J.A.S., R.S., N.G., B.W., T.J.C., G.S., B.A., V.G., and J.M.S.: Writing—review and editing; V.G., J.M.S., T.J.C., G.S., and B.A.: Supervision; V.G. and J.M.S.: Funding acquisition.
Author Disclosure Statement
The authors deny any relevant financial conflicts of interest. There are no personal interests to disclose.
Funding Information
V.G. supported by a grant from the National Institute of Neurological Disorders and Stroke (NINDS; R01NS107262). J.M.S. supported by a grant from the National Institute of Neurological Disorders and Stroke (NINDS; R01NS127986).