
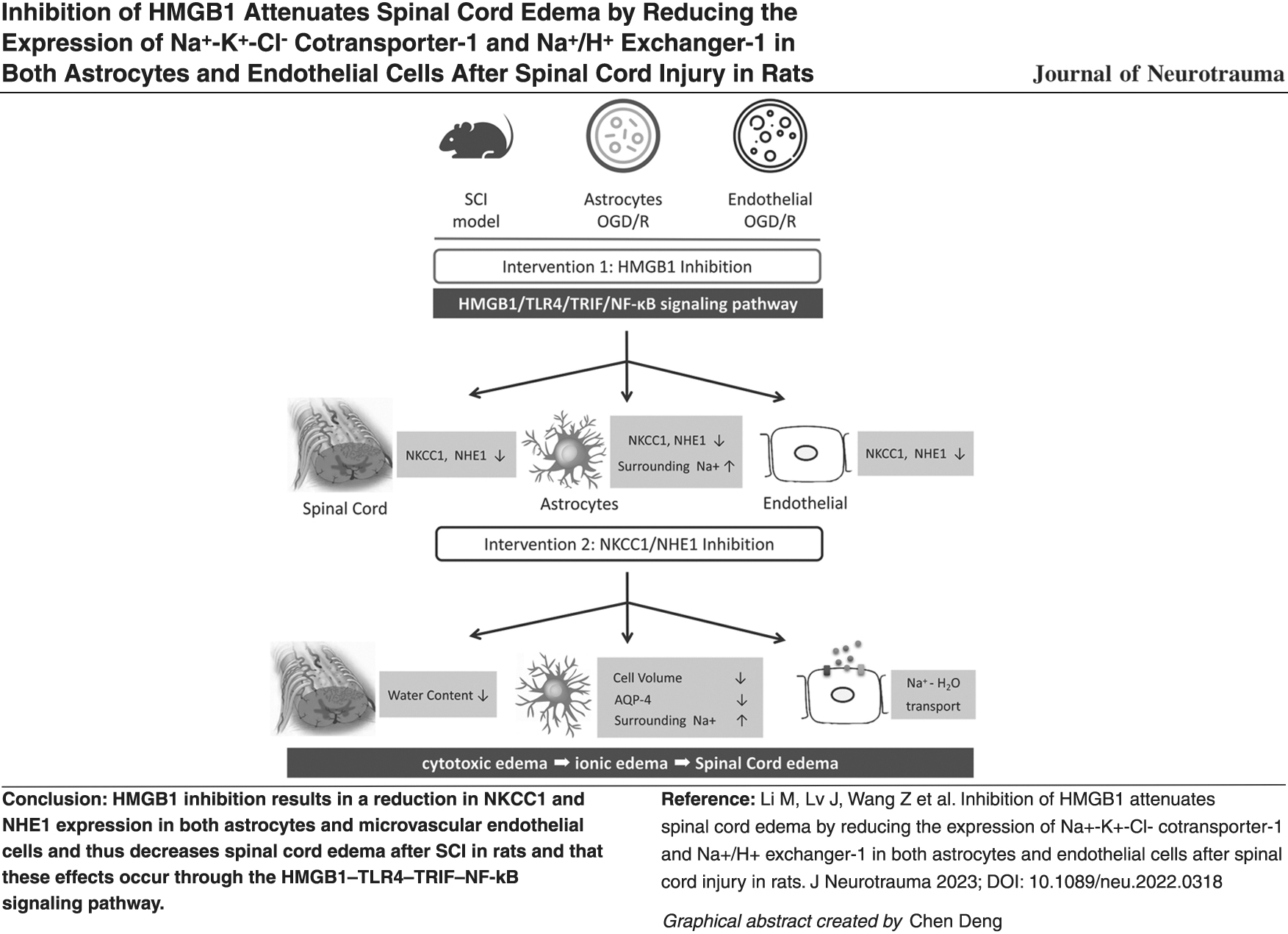
Abstract
Sodium/water transport through Na+-K+-Cl- cotransporter-1 (NKCC1) and sodium/hydrogen exchanger-1 (NHE1) in both astrocytes and endothelial cells is critical to cytotoxic and ionic edema following spinal cord injury (SCI). High-mobility group box-1 (HMGB1) promotes spinal cord edema after SCI. Accordingly, we sought to identify both the role of HMGB1 and the mechanism of its effect on NKCC1 and NHE1 expression in astrocytes and endothelial cells as well as the role of the regulation of spinal cord edema after SCI. An SCI model was generated in adult female rats using a heavy falling object, and an in vitro oxygen-glucose deprivation/reoxygenation (OGD/R) model was generated in rat spinal cord astrocytes and microvascular endothelial cells. The inhibition of HMGB1 reduced NKCC1 and NHE1 expression in the spinal cord of SCI rats, in cultured spinal cord astrocytes, and in cultured microvascular endothelial cells. The effects of HMGB1 on NKCC1 and NHE1 expression were mediated—at least in part—by activation of the Toll-like receptor 4 (TLR4)–Toll/interleukin-1 receptor domain-containing adapter inducing interferon-β (TRIF)–nuclear factor-kappa B (NF-κB) signaling pathway. The inhibition of NKCC1 or NHE1 decreased the spinal cord water content in rats following SCI, increased the Na+ concentration in the medium of cultured astrocytes after OGD/R, and reduced the astrocytic cell volume and AQP4 expression. These results imply that HMGB1 inhibition results in a reduction in NKCC1 and NHE1 expression in both astrocytes and microvascular endothelial cells and thus decreases spinal cord edema after SCI in rats and that these effects occur through the HMGB1–TLR4–TRIF–NF-κB signaling pathway.
Introduction
The development of edema is a main secondary injury event in spinal cord injury (SCI) and may lead to elevated intrathecal pressure, reduced blood flow, increased tissue damage, and aggravated functional deficits. 1 –3 Pathological edema in the central nervous system (CNS) has recently been reclassified into three categories: cytotoxic edema (oncotic cell swelling), ionic edema (the earliest stage of endothelial dysfunction and transcapillary sodium/water influx), and vasogenic edema (plasma protein leakage and edema through transvascular fluid movement). 4 –6
Cytotoxic edema is the primary component of edema and generates the driving force for ionic edema; it is through ionic edema that water from the vasculature begins to perfuse the interstitial tissue of the CNS. 4,7 Cytotoxic edema is particularly prominent in astrocytes following CNS injury; its mechanisms involve Na+ overload with the secondary participation of Cl– and water, acidosis-induced astrocyte swelling, and so on. 7 –10 Potential energy in the transendothelial Na+ gradient generated by cytotoxic edema drives osmotic agents and water extravasation during the development of ionized edema. 4,11 Na+-K+-Cl- cotransporter isoform 1 (NKCC1), a secondary active transporter, and NHE1, a member of the sodium/hydrogen exchanger (NHE) family, are constitutively expressed by both astrocytes and microvascular endothelial cells in the CNS and promote sodium and water transport during edema development. 12 –14
High-mobility group box-1 (HMGB1) is a conserved non-histone DNA-binding protein that is released from necrotic cells and secreted from reactive astrocytes and microglia into the extracellular environment of the CNS as a main agent of inflammation, immunity, and cell function. 15 –18 Extracellular HMGB1 activates numerous receptors in immunocompetent cells, astrocytes, and neurons in the CNS; through these receptors, HMGB1 signaling results in the activation of Toll/interleukin-1 receptor domain-containing adapter inducing interferon-β (TRIF) followed by nuclear factor-kappa B (NF-κB) transcription factors. 15,19 –21 Increases in HMGB1 and components of its signaling pathway are observed after SCI in both humans and an SCI rodent model. 22,23
Our previous studies indicated that the inhibition of HMGB1 improves motor function and decreases early spinal cord edema in SCI rats and reduces aquaporin-4 (AQP4) expression and cell swelling in cultured spinal cord astrocytes in oxygen-glucose deprivation/reoxygenation (OGD/R) injury in vitro, and these effects are mediated by the HMGB1–TLR4–NF-κB signaling pathway. 24,25 However, because HMGB1 inhibition attenuates spinal cord edema in SCI rats, little is known about: (1) HMGB1 in astrocytic Na+ overload and acidosis-induced cell swelling, which generate a transmembrane osmotic gradient in astrocytes driving water influx; (2) the relationship between HMGB1 and transendothelial extravasation of sodium and water; and (3) their potential regulatory mechanisms.
Here, we sought to ascertain whether HMGB1 is an important potential regulator of NKCC1 and NHE1 expression in the spinal cord of SCI rats, in cultured spinal cord astrocytes, and in cultured microvascular endothelial cells as well as the underlying mechanisms. We also studied the role of NKCC1 and NHE1 in the regulation of both spinal cord edema after SCI and sodium/water transport in spinal cord astrocytes in vitro after OGD/R.
Methods
Experimental protocol
Experiment 1:
Effects of HMGB1 on NKCC1 and NHE1 expression in the spinal cord of SCI rats, in cultured spinal cord astrocytes after OGD/R, and in cultured microvascular endothelial cells of the CNS after OGD/R.
First, NKCC1 and NHE1 expression in the spinal cord was analyzed in normal rats and in rats at 12 h, 1 day, and 3 days after SCI. Additionally, beginning 1 day after SCI, the following groups were established for a rat HMGB1 inhibition experiment: sham, SCI, SCI + EP (ethyl pyruvate, an HMGB1 inhibitor, 50 mg/kg 24,26 ), and SCI + GL (glycyrrhizin, another HMGB1 inhibitor, 100 mg/kg 24,27 ). The rats in the sham group underwent laminectomy alone. The rats in the SCI group underwent laminectomy followed by SCI and received 0.9% saline via an intraperitoneal injection. The rats in the SCI + EP group and the SCI + GL group received EP and GL, respectively, by intraperitoneal injection immediately following SCI. The measurements included the expression of NKCC1 and NHE1 in the spinal cord.
Second, the expression of NKCC1 and NHE1 in cultured spinal cord astrocytes was analyzed after 6, 12, 24, and 48 h of reoxygenation following OGD. In cultured spinal astrocytes used for HMGB1 inhibition experiments, the following groups were established: normal, OGD/R, OGD/R + HMGB1 short hairpin RNA (shRNA), 25,27 OGD/R + non-targeted shRNA, and OGD/R + EP (12 μM 25,27 ). Measurements of all the groups except the normal group were performed after 12 h of reoxygenation after OGD. These measurements included NKCC1 and NHE1 expression.
Third, astrocyte-conditioned media (ACMs) were harvested after 6, 12, 24, and 48 h of reoxygenation following OGD, and ACMs were also harvested from the OGD/R, OGD/R + HMGB1 shRNA, and OGD/R + non-targeted shRNA groups after 12 h of reoxygenation following OGD. The expression of NKCC1 and NHE1 in microvascular endothelial cells (Fig. 1) of the CNS was analyzed after 6, 12, 24, and 48 h of reoxygenation following OGD. The different ACMs from these time-points were applied to the microvascular endothelial cells in different groups at the time of reoxygenation after OGD. The following groups of microvascular endothelial cells were then established for the HMGB1 inhibition experiment: normal, OGD/R + OGD/R ACM, OGD/R + HMGB1 shRNA ACM, and OGD/R + non-targeted shRNA ACM. The different ACMs were added to the microvascular endothelial cells at the time of reoxygenation after OGD. Measurements of all the groups except the normal group were performed after 12 h of reoxygenation. The measurements included the expression of NKCC1, NHE1, and TLR4. 4,28
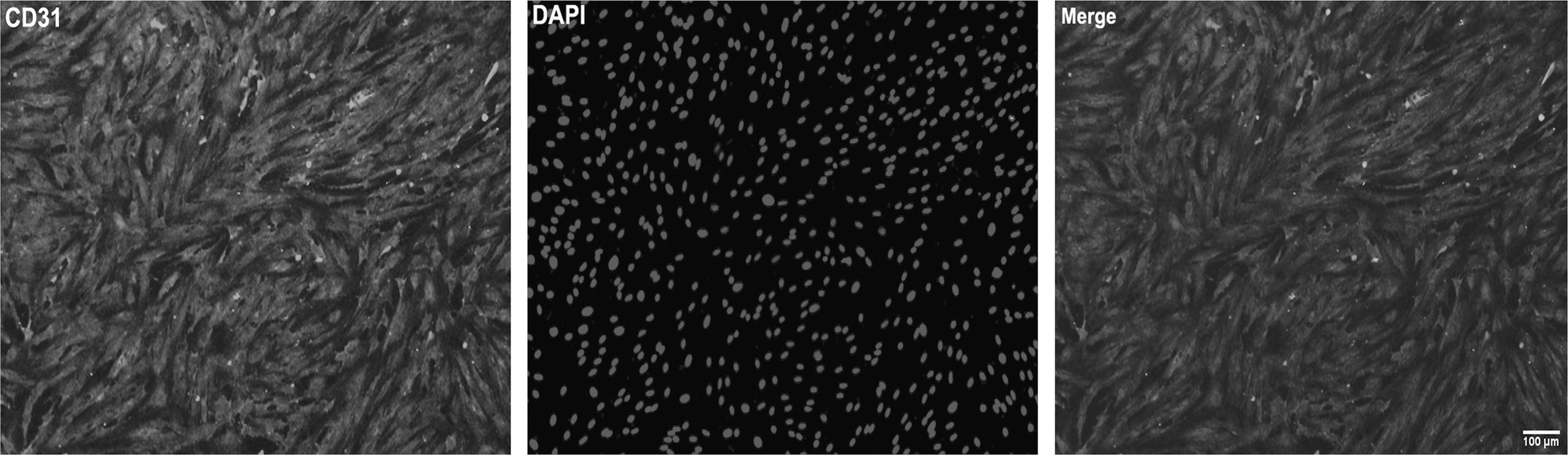
Immunofluorescence of spinal cord microvascular endothelial cells. The microvascular endothelial cells were stained with marker CD31. The microvascular endothelial cells were more than 95% of the total cells; n = 3.
We also evaluated the effects of recombinant HMGB1 (rHMGB1) on NKCC1 and NHE1 expression in spinal cord astrocytes and in microvascular endothelial cells of the CNS in vitro. Cultured astrocytes were exposed to rHMGB1 at various concentrations (0, 0.1, 1, 10, and 20 ng/mL), and NKCC1 and NHE1 expression was measured exactly 24 h after rHMGB1 exposure. Cultured microvascular endothelial cells were then treated with rHMGB1 at various concentrations (0, 0.1, 1, 10, and 20 ng/mL) in the culture medium starting from the time of reoxygenation after OGD. After 12 h of reoxygenation, the NKCC1 and NHE1 expression levels were measured.
Experiment 2:
Role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression upon HMGB1 inhibition in the spinal cord of rats after SCI and in spinal cord astrocytes and microvascular endothelial cells after OGD/R in vitro.
We studied the role of the TLR4–TRIF–NF-κB pathway in reducing NKCC1 and NHE1 expression as a result of HMGB1 inhibition. First, the rats were divided into the following groups: sham, SCI, SCI + lipopolysaccharide (LPS, a TLR4 agonist, 5 mg/kg), SCI + resveratrol (RES, a TRIF inhibitor, 200 mg/kg), and SCI + pyrrolidinedithiocarbamate (PDTC, an NF-κB inhibitor, 50 mg/kg). The rats received LPS, RES, or PDTC by intraperitoneal injection immediately following SCI. NKCC1, NHE1, TLR4, and TRIF expression and NF-κB activation in the spinal cord were measured 1 day after SCI.
We divided the following groups of cultured astrocytes: normal, OGD/R, OGD/R + CLI-095 (TLR4 inhibitor, 5 μM), OGD/R + C34 (another TLR4 inhibitor, 15 μM), OGD/R + RES (TRIF inhibitor, 40 μM), and OGD/R + Bay 11-7082 (NF-κB inhibitor, 5 μM). NKCC1, NHE1, TLR4, and TRIF expression and NF-κB activation in astrocytes were assessed after 12 h of reoxygenation following OGD. Different inhibitors were added to the medium prior to OGD injury and the time of reoxygenation.
Cultured microvascular endothelial cells were then divided into the following groups: normal, OGD/R + OGD/R ACM, OGD/R + OGD/R ACM + CLI-095 (5 μM), OGD/R + OGD/R ACM + C34 (15 μM), OGD/R + OGD/R ACM + RES (40 μM), and OGD/R + OGD/R ACM + Bay 11-7082 (5 μM). NKCC1, NHE1, TLR4, and TRIF expression and NF-κB activation in microvascular endothelial cells were measured after 12 h of reoxygenation following OGD. The OGD/R ACM was added to the microvascular endothelial cells at the time of reoxygenation after OGD. We added different inhibitors to the medium before OGD damage and during reoxygenation.
Experiment 3:
Effects of NKCC1 or NHE1 on the spinal cord water content in SCI rats and on cellular swelling and AQP4 expression in spinal cord astrocytes as well as the Na+ concentration in the surrounding medium after OGD/R in vitro.
Starting 1 day after SCI in rats, the following groups were established for an NKCC1 and NHE1 inhibition experiment: sham, SCI, SCI + azosemide (NKCC1 inhibitor, 10 mg/kg), and SCI + zoniporide hydrochloride hydrate (NHE1 inhibitor, 1 mg/kg). The rats in the SCI + azosemide group and the SCI + zoniporide hydrochloride hydrate group received azosemide and zoniporide hydrochloride hydrate, respectively, by intraperitoneal injection after SCI. NKCC1 and NHE1 expression in the spinal cord and the spinal cord water content were measured.
The following groups of cultured spinal cord astrocytes were established for the NKCC1 and NHE1 inhibition experiment: normal, OGD/R, OGD/R + bumetanide (NKCC1 inhibitor, 250 μM), and OGD/R + dimethylamiloride hydrochloride (DMA) (NHE1 inhibitor, 150 μM). We measured the volume of the astrocytes, the expression of NKCC1, NHE1, and AQP4 (a water channel protein expressed by astrocytes), and the concentration of Na+ in the surrounding medium. We measured the data after 12 h of reoxygenation following OGD in all groups except the normal group. We added different inhibitors to the medium before OGD damage and at the time of reoxygenation.
The rat experiments performed to assess the expression of NKCC1 and NHE1 in the spinal cord at 12 h, 1 day, and 3 days following SCI were repeated four times. The rat experiments aiming to explore the role of HMGB1–TLR4–TRIF–NF-κB on the expression of NKCC1 and NHE1 in the spinal cord were repeated five times. The rat experiments determining the effects of NKCC1 and NHE1 in the spinal cord at 1 day after SCI were repeated four times. All cell experiments were conducted at least three times using the same method in vitro. We applied a blinded method to establish the rat SCI model or cell OGD/R model, performed the experiments, and analyzed the images and values. Two rats died after SCI, and other rats were added as replacements. All chemical inhibitors and the LPS in the rat experiment were diluted in 0.9% saline, and all chemical inhibitors in the cell experiment were included in the medium with no other organic solvents. The chemical concentrations and treatment durations were based on pilot experiments. 24,25
Special chemicals, antibodies, and other materials/tools
Animals
We used the same animals and conditions as before, as detailed in Supplementary Appendix S1.
SCI model of animals
We used a revised form of Allen's weight-drop injury model to establish the SCI. 24,29,30 Detailed methods are provided in Supplementary Appendix S1.
Astrocyte culture
Primary cultured spinal cord astrocytes were prepared from post-natal day 1–2 SD rats (Shanxi Medical University, RRID: RGD_5508397, Taiyuan, China). 25 Detailed methods are provided in Supplementary Appendix S1.
Culture of microvascular endothelial cells from the CNS
Primary cultured microvascular endothelial cells from the CNS were prepared from post-natal day 10 SD rats (Shanxi Medical University, RRID: RGD_5508397). 31 Detailed methods are provided in Supplementary Appendix S1.
OGD/R procedure
We washed spinal cord astrocytes and microvascular endothelial cells with phosphate-buffered saline (PBS) three times. Cultured cells were placed in serum-free Dulbecco's modified eagle medium (DMEM) without glucose and then incubated for 6 h at 37°C in an anaerobic chamber bubbled with 1% O2, 5% CO2, and 94% N2. We next rinsed the cells with PBS and then placed the cells in normal conditions.
shRNA-mediated HMGB1 knockdown
Detailed methods and target sequences are provided in Supplementary Appendix S1.
Measurement of the spinal cord water content
We employed the wet weight/dry weight method to determine the water content in the spinal cord. A 10-mm spinal cord segment, including the epicenter of the injury, was collected. We first measured its wet weight and dried the segment at 80°C for 48 h to obtain its dry weight. The following equation was employed to obtain the spinal cord content: (wet weight – dry weight)/wet weight × 100%.
Western blots
We used previously reported methods and procedures described in Supplementary Appendix S1.
Immunofluorescence
We used previously reported methods and procedures described in Supplementary Appendix S1.
Immunohistochemistry
We used previously reported methods and procedures described in Supplementary Appendix S1.
PCR
Real-time–quantitative polymerase chain reaction (RT–qPCR) was used to determine the expression levels of NKCC1, NHE1, and HMGB1 messenger RNA (mRNA) relative to those of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Detailed description of the methods and the PCR primers are provided in Supplementary Appendix S1.
Na+ concentration in the medium
The Na+ concentration in the medium of spinal cord astrocytes at 12 h of reoxygenation following OGD in Experiment 2 was evaluated. Astrocyte culture medium was collected and centrifuged at 2500 rpm for 10 min. The supernatant was absorbed, and the concentration of sodium ions was measured using an automatic biochemical analyzer.
Astrocytic volume analysis
Detailed procedures are described in Supplementary Appendix S1.
Statistical analysis
All values are shown as the means ± standard deviations (StDevs), and statistical analyses were performed using the general-purpose statistical package SPSS 24.0 (IBM,
Results
Inhibiting HMGB1 reduces NKCC1 and NHE1 expression in the spinal cord in SCI rats
We studied the effect of SCI on NKCC1 and NHE1 expression in the spinal cord of rats by western blotting. Our western blot analysis demonstrated that NKCC1 and NHE1 expression in the spinal cord was significantly increased from 6 h to 3 days following SCI, with the peak increase observed at the 1-day time-point (p < 0.05, Fig. 2A,B).
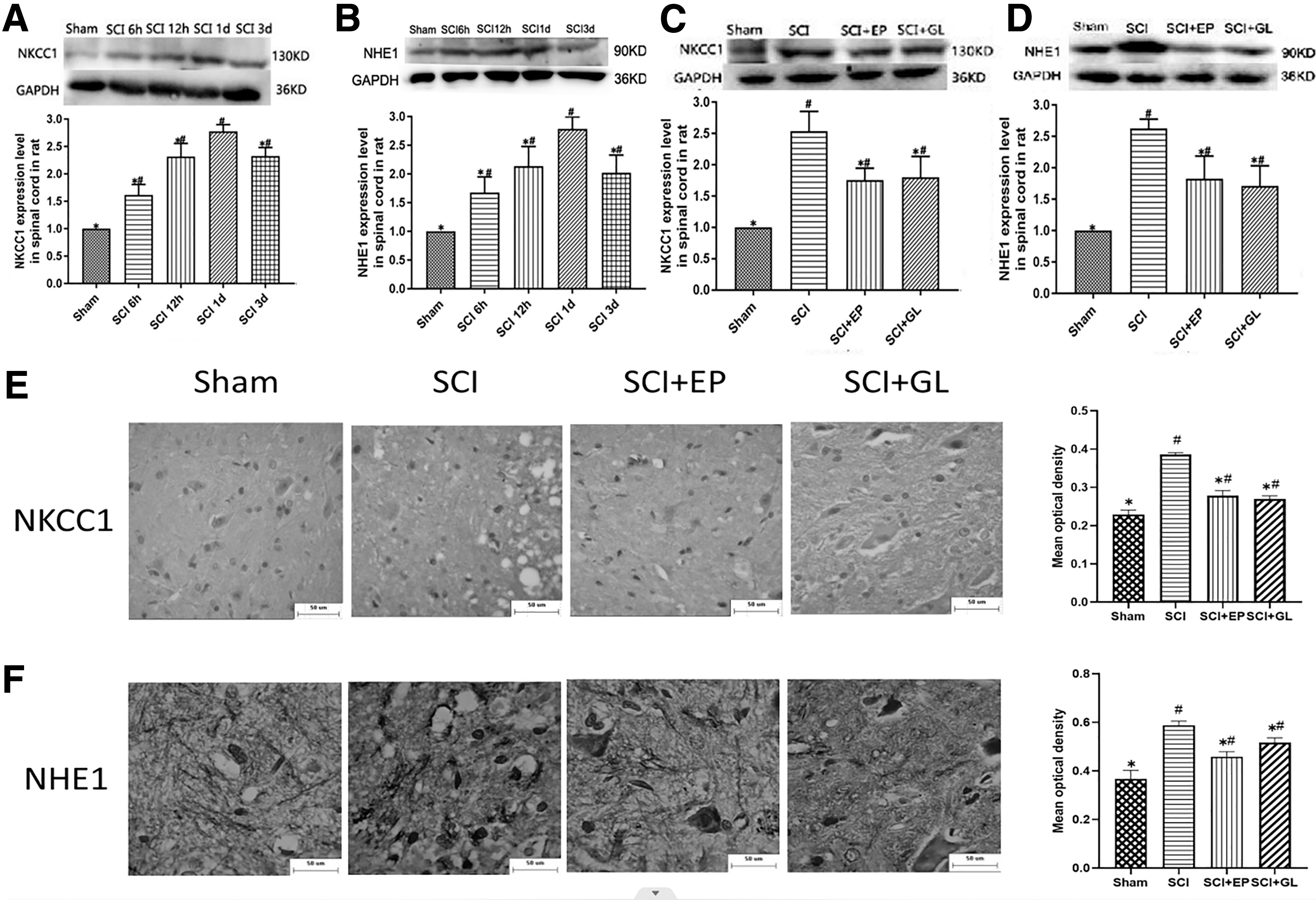
Inhibiting HMGB1 reduces NKCC1 and NHE1 expression in the spinal cord in rats with SCI.
We examined the effect of HMGB1 inhibition using EP and GL on spinal cord NKCC1 and NHE1 expression in rats at 1 day following SCI. Western blot analysis showed that the NKCC1 and NHE1 protein levels in the spinal cord were significantly increased in the SCI group. (p < 0.05), and this increase was significantly attenuated by HMGB1 inhibition (p < 0.05, Fig. 2C,D). The NKCC1 and NHE1 immunohistochemistry results are shown in Figure 2E,F. The mean optical density values for both NKCC1 and NHE1 were found to be significantly higher in the SCI group compared with the sham group (p < 0.05). In contrast, these values were significantly lower after EP or GL treatment. (p < 0.05).
The inhibition of HMGB1 suppresses NKCC1 and NHE1 expression in spinal cord astrocytes after OGD/R in vitro
Western blotting was performed to analyze NKCC1 and NHE1 expression in spinal cord astrocytes at various time-points after OGD/R. A western blot analysis demonstrated that significant increases in NKCC1 and NHE1 expression were observed at the time of the reoxygenation period and peaked at 12 h of reoxygenation (p < 0.05, Fig. 3A,B).
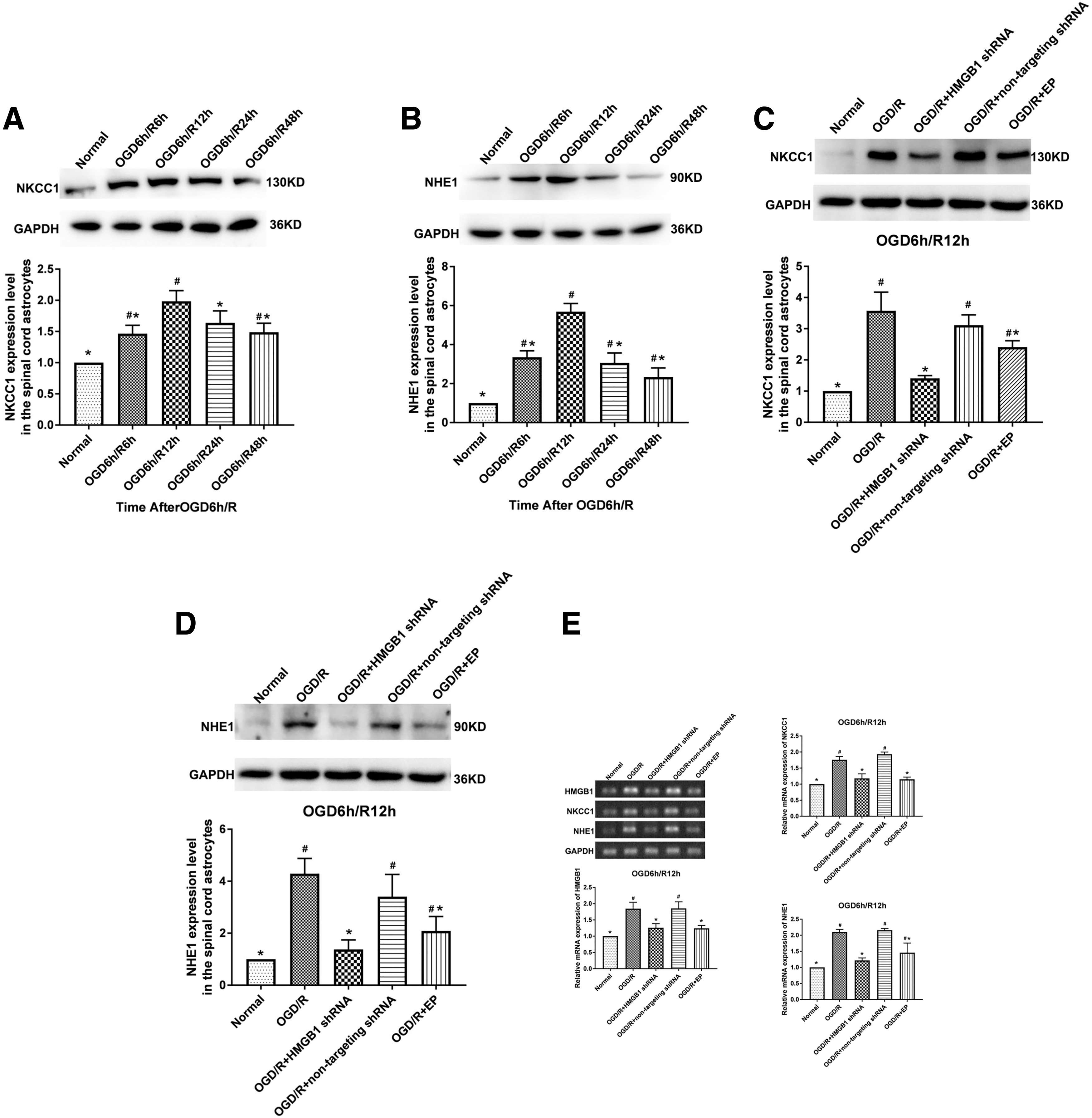
Inhibiting HMGB1 suppresses NKCC1 and NHE1 expression in spinal cord astrocytes after OGD/R in vitro.
We investigated the effect of HMGB1 inhibition on NKCC1 and NHE1 expression in spinal cord astrocytes at 12 h post-OGD. A western blot analysis demonstrated that the NKCC1 and NHE1 protein levels were significantly increased after OGD/R (p < 0.05) and that this increase could be significantly attenuated by HMGB1 inhibition (p < 0.05, Fig. 3C,D). PCR results demonstrated that the inhibition of HMGB1 reduced the increased RNA levels of HMGB1, NKCC1, and NHE1 in spinal cord astrocytes after OGD/R (p < 0.05, Fig. 3E).
Effects of HMGB1 on NKCC1, NHE1, and TLR4 expression in microvascular endothelial cells of the CNS after OGD/R in vitro
Different ACMs were harvested after 6, 12, 24, and 48 h of reoxygenation following OGD and were then added to CNS microvascular endothelial cells, and western blotting was performed to analyze NKCC1 and NHE1 expression at various time-points after OGD/R. Western blot analysis demonstrated that significant increases in NKCC1 and NHE1 expression were observed at various times during the reoxygenation period and peaked at 12 h of reoxygenation (p < 0.05, Fig. 4A,B).
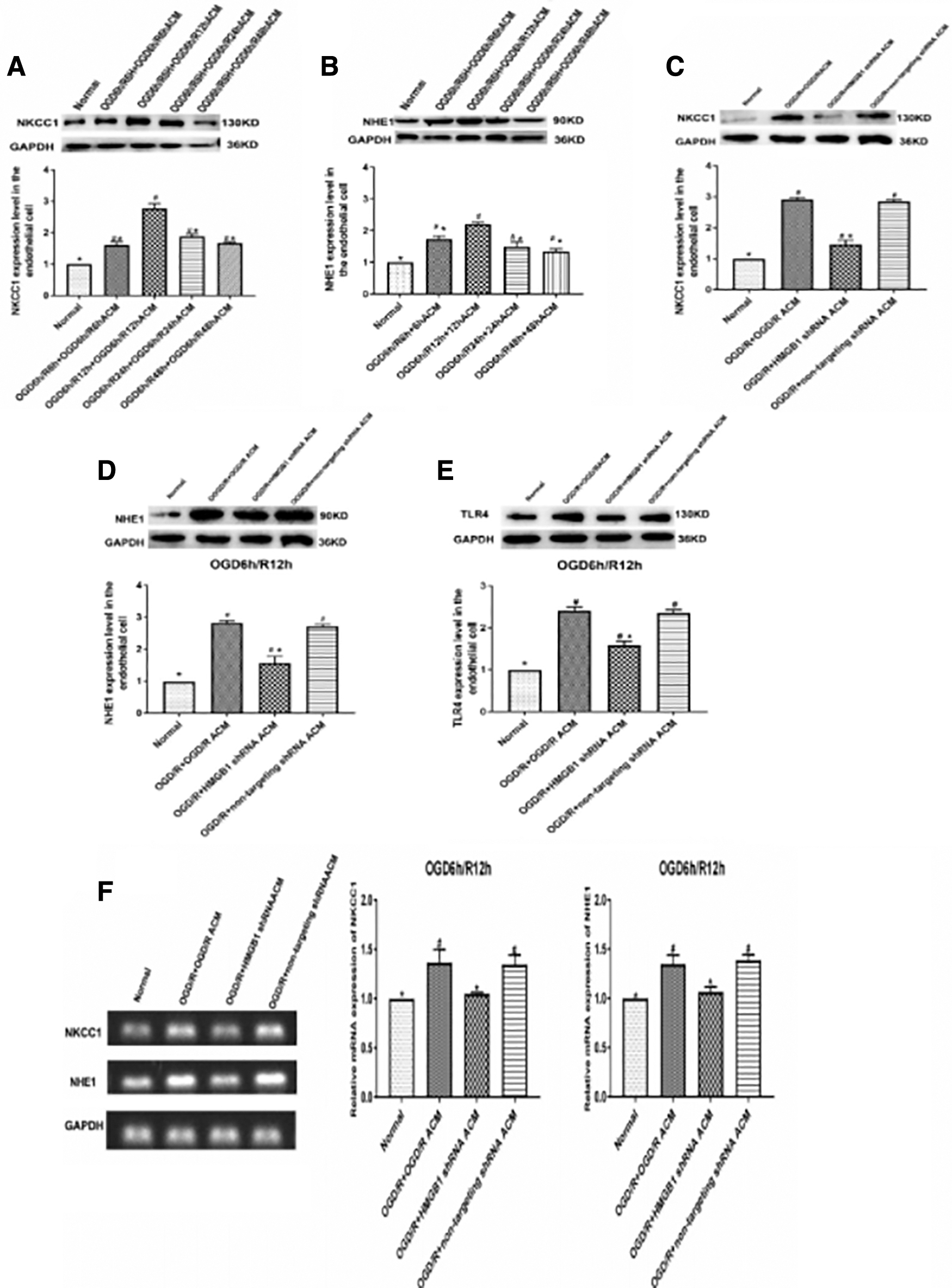
Effects of HMGB1 on NKCC1, NHE1, and TLR4 expression in microvascular endothelial cells of the CNS after OGD/R in vitro.
To illustrate the effect of HMGB1 inhibition on NKCC1, NHE1, and TLR4 expression in microvascular endothelial cells, ACMs were collected from the OGD/R, OGD/R + HMGB1 shRNA, and OGD/R + non-targeted shRNA spinal cord astrocytes after 12 h of reoxygenation post-OGD, and these media were applied to microvascular endothelial cells. After 12 h of reoxygenation following OGD, a western blot analysis demonstrated that the NKCC1, NHE1, and TLR4 expression levels were decreased in the OGD/R + HMGB1 shRNA ACM group compared with the OGD/R + OGD/R ACM group (p < 0.05, Fig. 4C–E). The PCR results demonstrated that the NKCC1 and NHE1 RNA levels were also decreased in the OGD/R + HMGB1 shRNA ACM group compared with the OGD/R + OGD/R ACM group (p < 0.05, Fig. 4F).
Effects of rHMGB1 on NKCC1 and NHE1 expression in spinal cord astrocytes and in microvascular endothelial cells of the CNS in vitro
A western blot analysis showed that a dose-dependent increase in NKCC1 and NHE1 expression was induced in spinal cord astrocytes cultured with rHMGB1 (0, 0.1, 1, 10, and 20 ng/mL) for 24 h (p < 0.05, Fig. 5A,B). Cultured microvascular endothelial cells were incubated with rHMGB1 at a range of concentrations (0, 0.1, 1, 10, and 20 ng/mL), and at 12 h of reoxygenation after OGD, a western blot analysis revealed increases in the NKCC1 and NHE1 expression levels (p < 0.05, Fig. 5C,D).
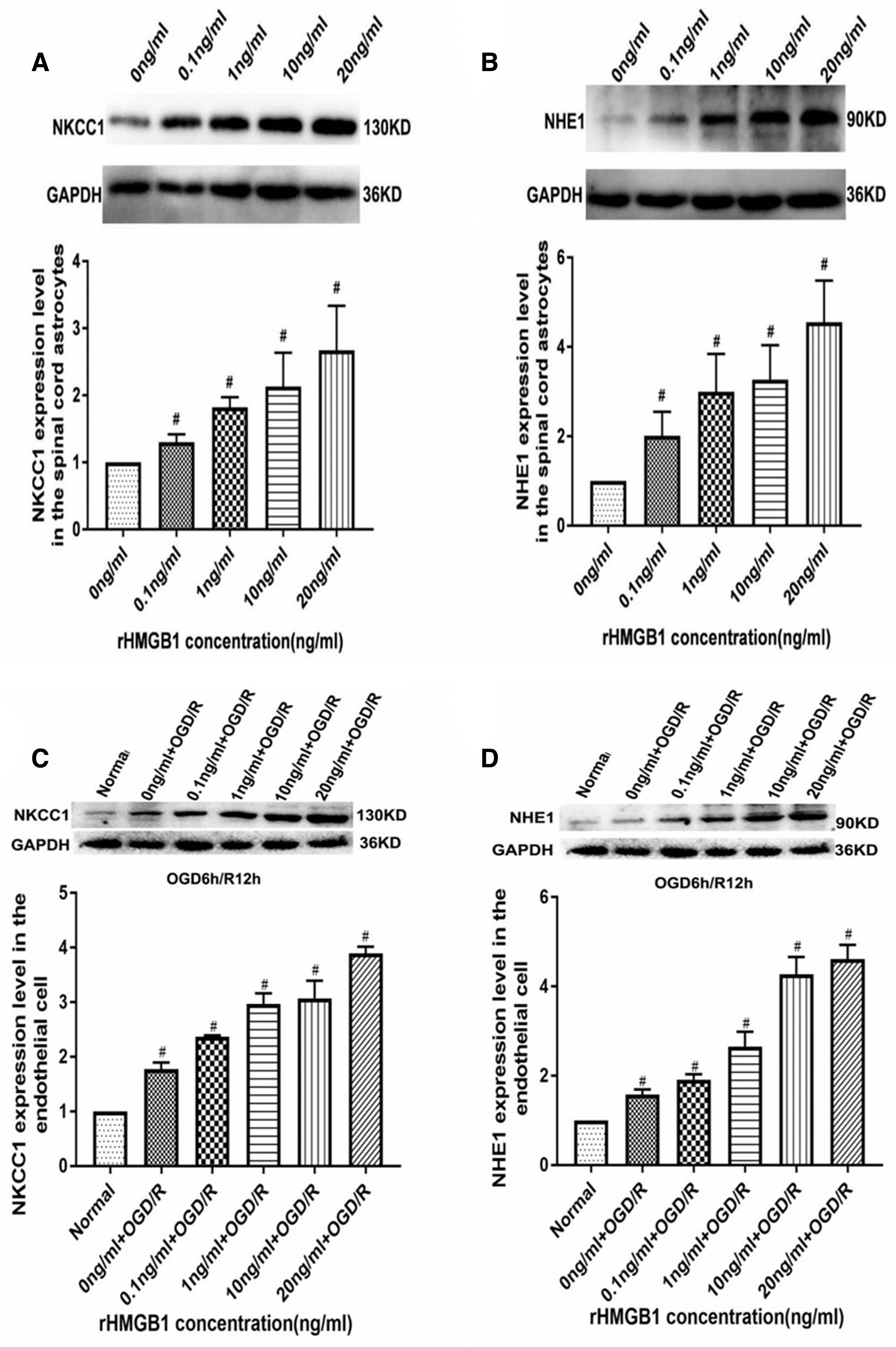
Effects of rHMGB1 on NKCC1 and NHE1 expression in spinal cord astrocytes and in microvascular endothelial cells of the CNS in vitro.
Role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression and the spinal cord water content induced by HMGB1 inhibition in SCI rats
Western blotting was performed to investigate the role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression induced by HMGB1 inhibition in SCI rats. The western blot analysis showed that the TLR4 agonist LPS increased NKCC1 and NHE1 expression and that the inhibition of TRIF or NF-κB reduced NKCC1 and NHE1 expression compared with the expression levels in the SCI group (p < 0.05, Fig. 6A,B). LPS increased TLR4, TRIF, and NF-κB expression, RES decreased TRIF and NF-κB expression, and PDTC decreased NF-κB expression in the spinal cords of SCI rats (p < 0.05, Fig. 6C–E). LPS also increased the spinal cord water content, and the inhibition of TRIF or NF-κB in rats decreased the spinal cord water content compared with that in the SCI group (p < 0.05, Fig. 6F).
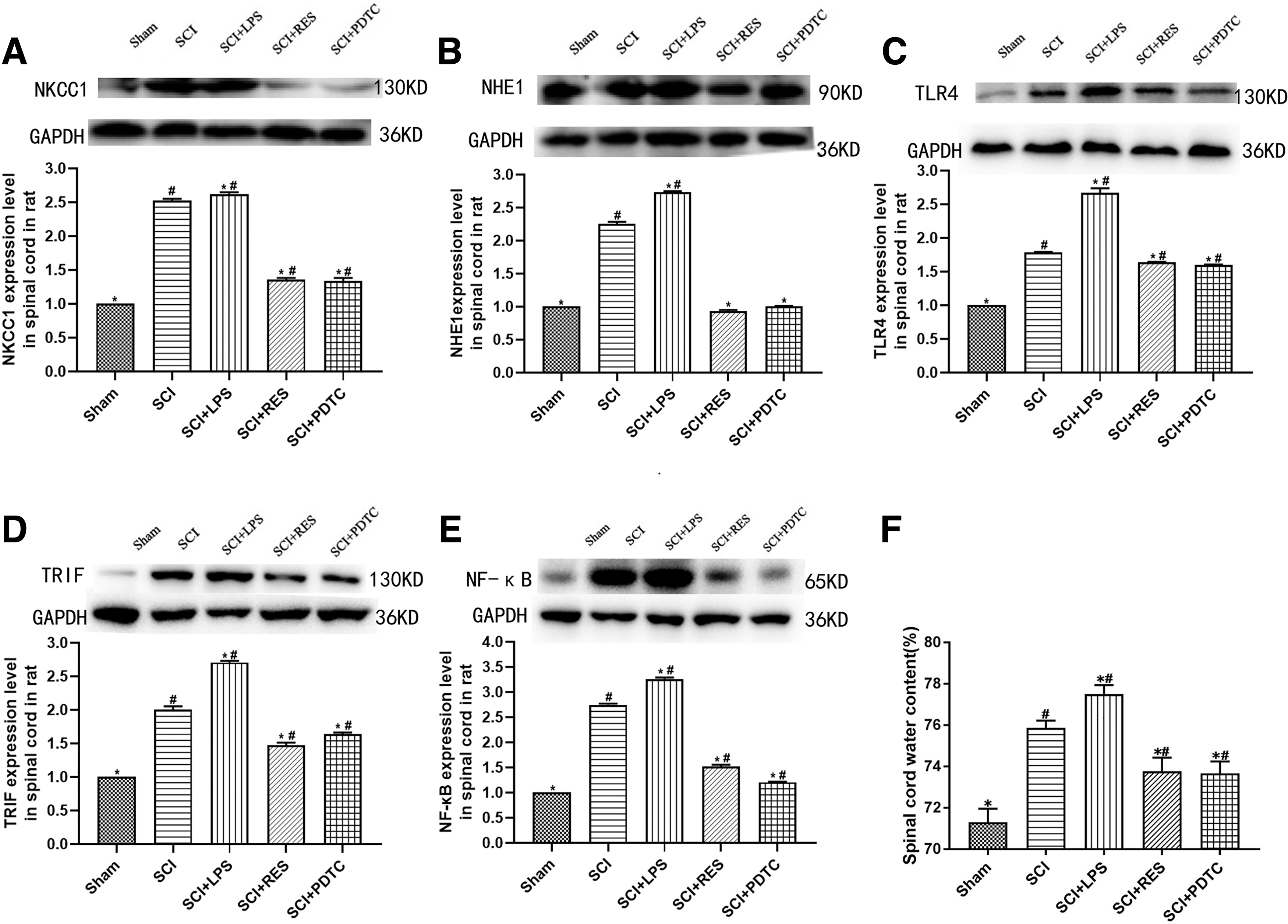
Role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression and spinal cord water content resulting from HMGB1 inhibition in rats after SCI.
Role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression following HMGB1 inhibition in spinal cord astrocytes subjected to OGD/R in vitro
The role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression in spinal cord astrocytes as a result of HMGB1 inhibition was investigated using western blotting after 12 h of reoxygenation following OGD. NKCC1 and NHE1 expression decreased after inhibition of TLR4, TRIF, and NF-κB (p < 0.05, Fig. 7A,B). CLI-095 and C34 reduced TLR4 expression, and the inhibition of TLR4 or RES reduced TRIF expression in astrocytes after 12 h of reoxygenation following OGD (p < 0.05, Fig. 7C,D). The activation of NF-κB in spinal cord astrocytes was revealed by the expression of nuclear NF-κB and p-IκBα. The results showed that the inhibition of TLR4/TRIF/NF-κB significantly attenuated the increases in NF-κB and phosphorylated-NF-kappa-B inhibitor alpha (p-IκBα) 12 h after reoxygenation compared with the OGD/R group (p < 0.05, Fig. 7E,F).
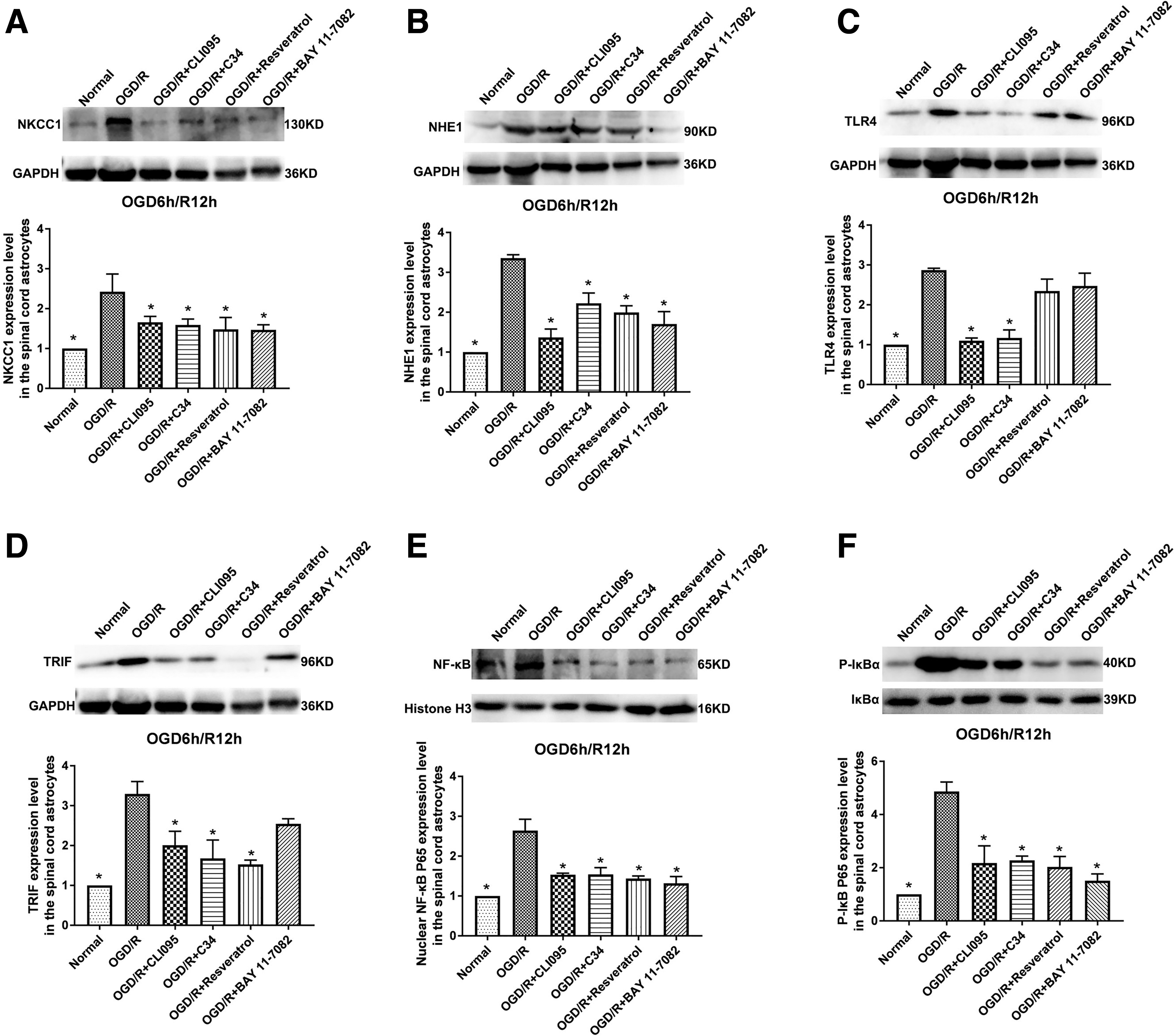
Role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression following HMGB1 inhibition in spinal cord astrocytes subjected to OGD/R in vitro.
Role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression following HMGB1 inhibition in CNS microvascular endothelial cells subjected to OGD/R in vitro
We also analyzed the role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression in microvascular endothelial cells as a result of HMGB1 inhibition; we performed western blotting after OGD-treated cells were exposed to OGD/R ACM and reoxygenated for 12 h. NKCC1 and NHE1 expression decreased after inhibition of TLR4, TRIF, and NF-κB compared with the levels in the OGD/R + OGD/R ACM group (p < 0.05, Fig. 8A,B). CLI-095 and C34 reduced TLR4 expression, and inhibiting TLR4 or RES reduced TRIF expression in endothelial cells incubated with OGD/R ACM after 12 h of reoxygenation following OGD (p < 0.05, Fig. 8C,D). The activation of NF-κB in microvascular endothelial cells was revealed by nuclear NF-κB and p-IκBα expression. The results showed that the increases in nuclear NF-κB and p-IκBα were significantly attenuated 12 h after reoxygenation compared with the OGD/R + OGD/R ACM group through the inhibition of TLR4/TRIF/NF-κB (p < 0.05, Fig. 8E,F).
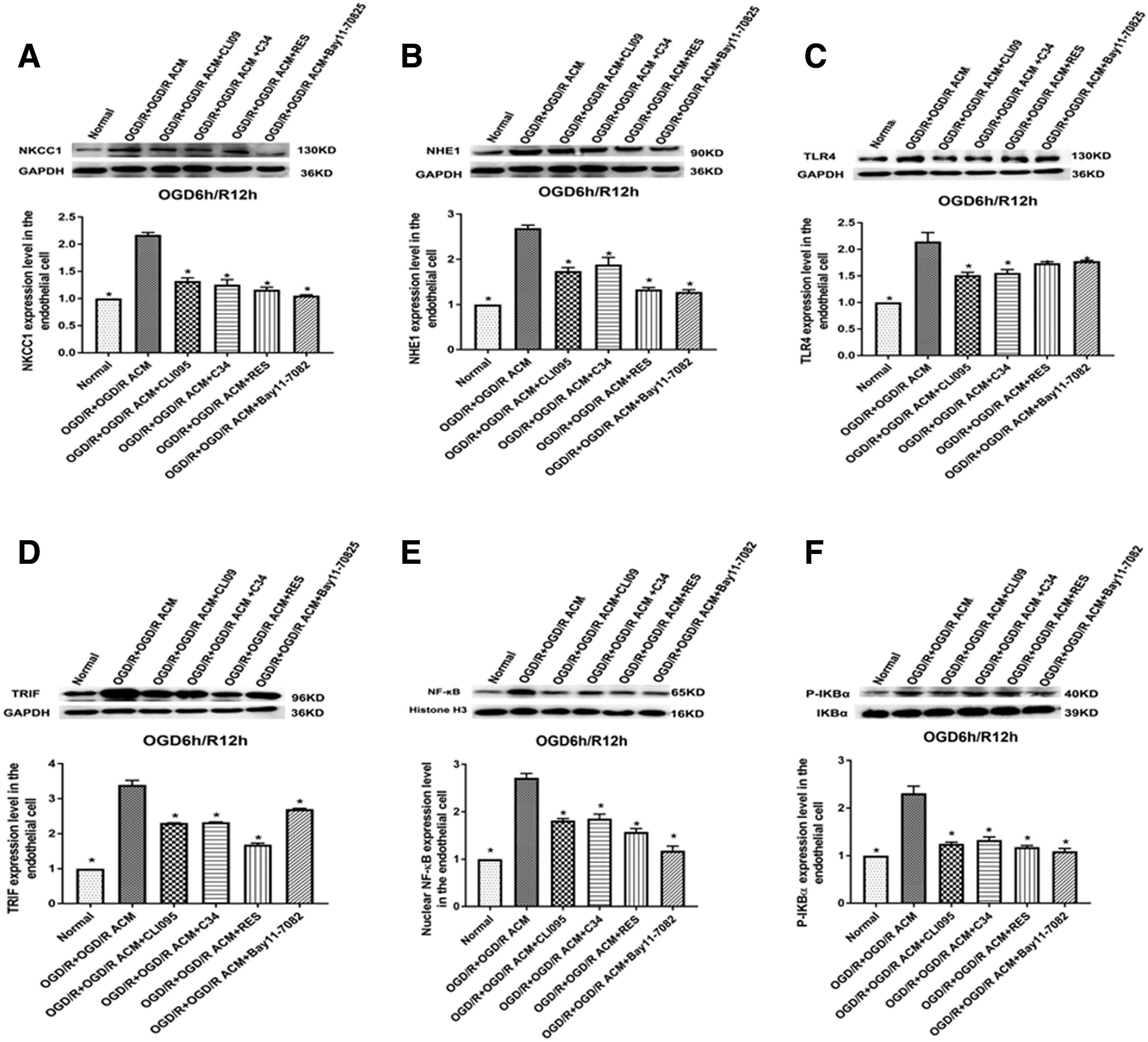
Role of the TLR4–TRIF–NF-κB signaling pathway in reducing NKCC1 and NHE1 expression following HMGB1 inhibition in CNS microvascular endothelial cells subjected to OGD/R in vitro. Western blots showed that after OGD-treated cells were exposed to OGD/R ACM and reoxygenated for 12 h,
Role of NKCC1 and NHE1 on the regulation of the spinal cord water content in SCI rats
NKCC1 inhibition by azosemide and NHE1 inhibition by zoniporide hydrochloride hydrate were performed in SCI rats. A western blot analysis suggested that the administration of azosemide markedly suppressed the increase in NKCC1 expression in the spinal cord of rats 1 day after SCI (p < 0.05, Fig. 9A). The increased NHE1 expression in the spinal cord was substantially suppressed following the administration of zoniporide hydrochloride hydrate in rats at 1 day following SCI (p < 0.05, Fig. 9B). The inhibition of either NKCC1 or NHE1 in rats reduced the water content of the spinal cord compared with that of the SCI group (p < 0.05, Fig. 9C).
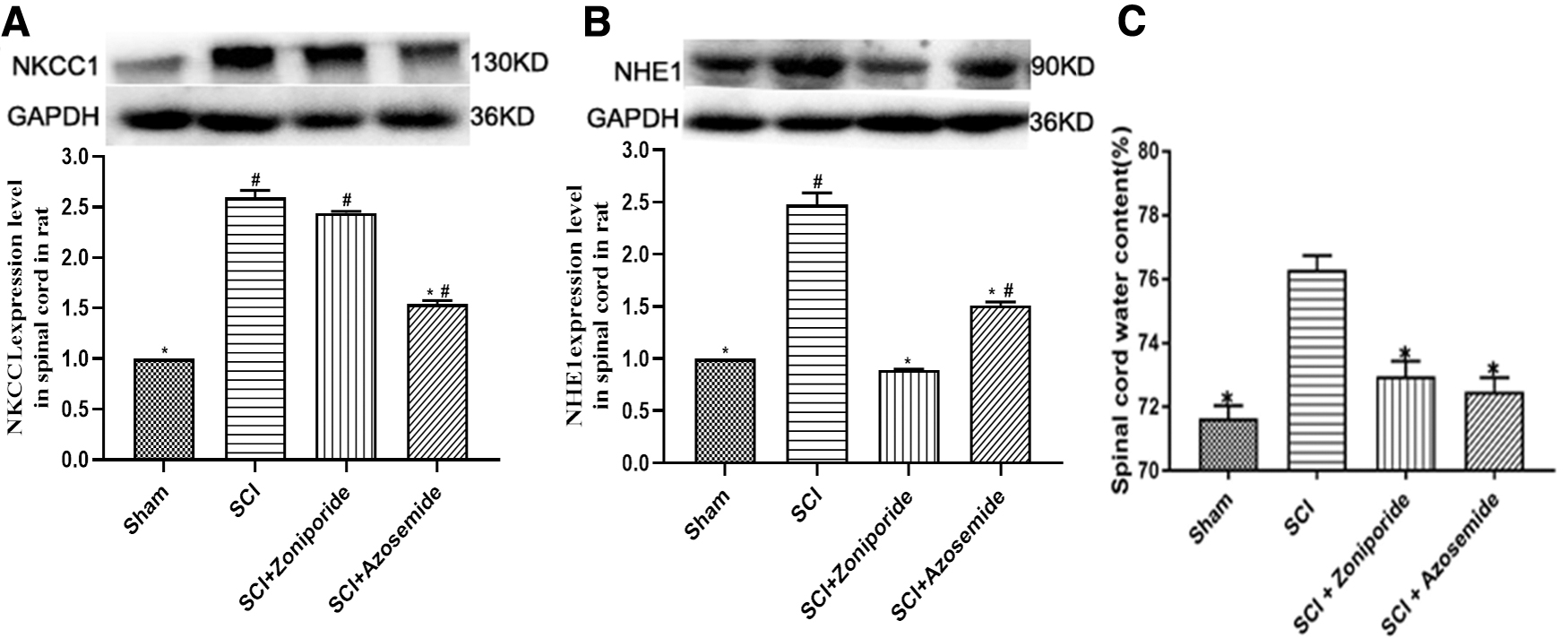
The role of NKCC1 and NHE1 regulation on spinal cord water content in rats after SCI.
NKCC1 and NHE1 regulation of cellular swelling and AQP4 expression in spinal cord astrocytes and the Na+ concentration in the culture medium after OGD/R in vitro
The effects of NKCC1 or NHE1 inhibition on NKCC1, NHE1, and AQP4 expression in spinal cord astrocytes after 12 h of reoxygenation following OGD were analyzed by western blotting, and the Na+ concentration in the surrounding medium was examined. Western blot analysis demonstrated that NKCC1 inhibition reduced the levels of NKCC1 and AQP4 expression compared with those in the OGD/R group (p < 0.05, Fig. 10A,C). NHE1 inhibition reduced the levels of NHE1 and AQP4 expression compared with those in the OGD/R group (p < 0.05, Fig. 10B,C). The Na+ concentration in the surrounding medium increased after NKCC1 and NHE1 inhibition compared with that in the OGD/R group (p < 0.05, Fig. 10D). We then examined the effects of NKCC1 or NHE1 inhibition on cellular swelling, as gauged by the cell volume, in cultured spinal cord astrocytes after OGD/R. The inhibition of NKCC1 or NHE1 significantly prevented the increase in the astrocyte volume at 12 h of reoxygenation compared with that of the OGD/R group (p < 0.05, Fig. 10E).
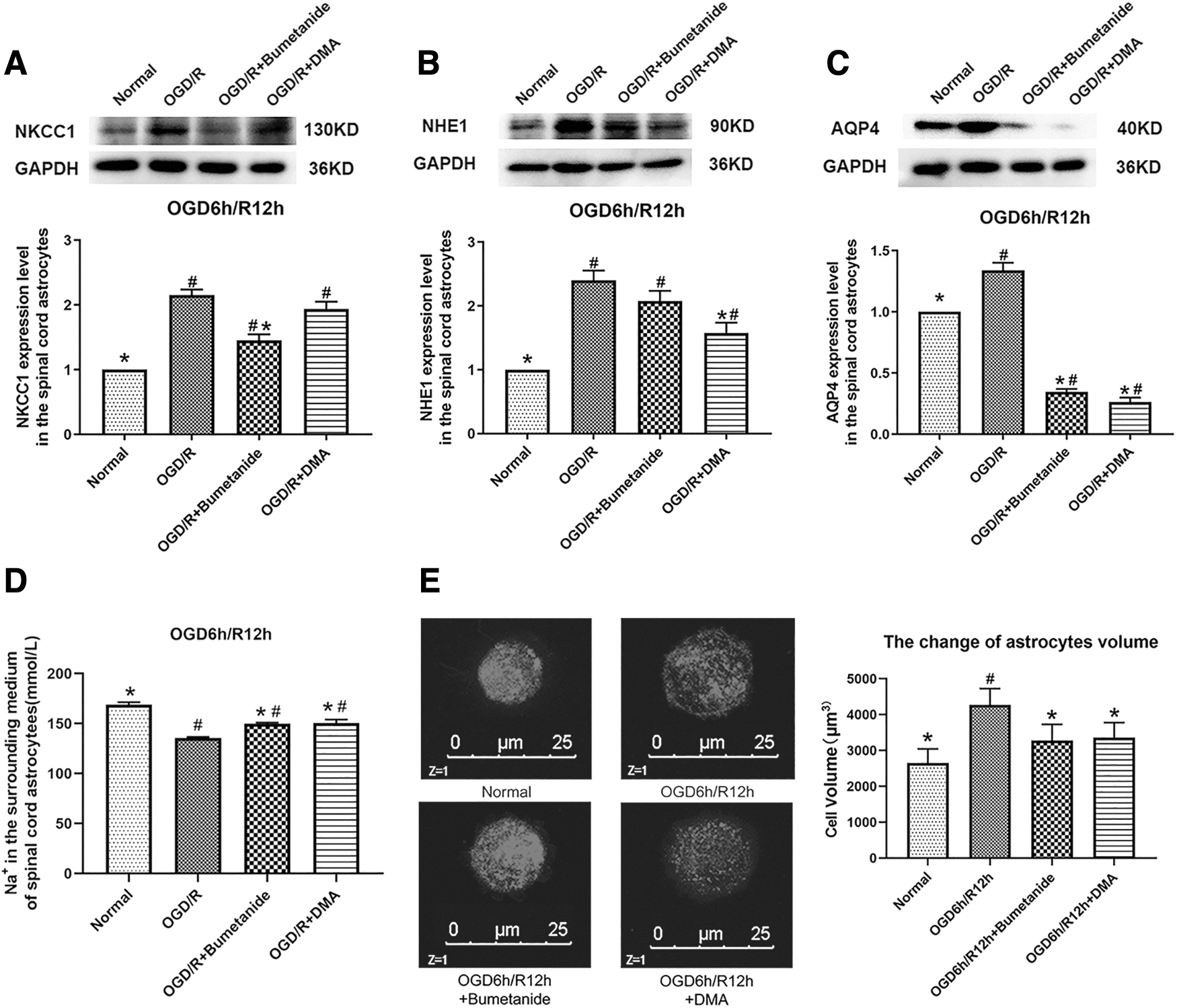
The effect of NKCC1 and NHE1 regulation on cellular swelling and AQP4 expression in spinal cord astrocytes and the Na+ concentration in the surrounding medium after OGD/R in vitro.
Discussion
This study demonstrated that inhibiting HMGB1 reduced NKCC1 and NHE1 expression in the spinal cord of an SCI rat model, in cultured spinal cord astrocytes after OGD/R, and in microvascular endothelial cells of the CNS after OGD/R in vitro. This reduction in NKCC1 and NHE1 expression through HMGB1 inhibition in rat spinal cords, cultured astrocytes, and cultured endothelial cells might be related to the HMGB1–TLR4–TRIF–NF-κB pathway. Further experiments demonstrated that a reduction in NKCC1 or NHE1 decreased the water content of the spinal cord after injury, reduced astrocytic cellular swelling and AQP4 expression after OGD/R in vitro, and increased the Na+ concentration in the surrounding medium.
HMGB1, a non-histone DNA-binding protein, plays an important role in transcriptional regulation in cells and as a damage-associated molecular pattern (DAMP) outside cells. 15 Increased HMGB1 in the CNS, which is actively secreted from reactive astrocytes and microglia and passively released from necrotic cells following pathogenic and/or tissue damage, mediates the inflammatory response, disrupts the endothelial barrier, impairs extracellular glutamate clearance, and creates edema. 16,17,24,25,31 –33 As previously reported, the maximum plasma HMGB1 expression in patients with acute SCI was significantly higher than that in uninjured individuals. 22 In our previous studies, we found higher HMGB1 expression in the spinal cord and serum of SCI rats, and expression could be inhibited by the intraperitoneal injection of either EP or GL. 24 The HMGB1 levels in spinal cord astrocytes cultured in vitro and HMGB1 expression in the surrounding medium were noticeably increased in OGD/R, and these changes could also be inhibited by either HMGB1-specific shRNA or EP. 25
Spinal cord edema following SCI has been associated with increased intrathecal pressure, reduced blood flow, increased tissue damage, and greater functional deficits. 1 –4 Cytotoxic edema, particularly astrocytic swelling, predominates in the initial phase of spinal cord edema after SCI. 4 In astrocytes, water fluxes into the cell passively follow Na+ influxes through NKCC1, NHE1, and so on, which are usually triggered by exposure to endogenous toxins (extracellular K+, intracellular H+, etc.) in the CNS after pathogenic and/or tissue injury. 4,7,10
For astrocytic intracellular Na+ overload and extracellular Na+ reduction, the potential energy contained in the transendothelial Na+ gradient drives the extravasation of sodium and water into interstitial tissue in the CNS through Na+ transporters in microvascular endothelial cells, which results in the formation of ionic edema. 4,11 Further damage to the blood–spinal cord barrier induces formation of vasogenic edema (plasma protein leakage and transvascular edema). 4 –7 Our previous studies demonstrated that the inhibition of HMGB1 significantly reduces spinal cord edema in SCI rats and decreases astrocytic swelling in vitro after OGD/R. 24,25 This study examined the role of HMGB1 on NKCC1 and NHE1 expression in the spinal cord of SCI rats in cultured astrocytes and endothelial cells, which are crucial Na+ transporters in astrocytes and endothelial cells in the CNS. 10,12,13,34 We also studied the role of NKCC1 and NHE1 regulation in the decrease in spinal cord edema observed after the inhibition of HMGB1 in SCI rats and in sodium/water transport in spinal cord astrocytes in vitro.
NKCC1 is a member of the Na+/K+/2Cl– transporter family and is broadly distributed in astrocytes, neurons, oligodendrocytes, and endothelial cells; it is activated by high extracellular K+, a salient feature of ischemia. 10,12,35 Following CNS injury, intracellular adenosine triphosphate (ATP) becomes depleted, Na+/K+-ATPase fails, and thus, NKCC1, a mechanism that is independent of intracellular ATP, is more likely to be particularly important to extracellular K+ accumulation. 4 During early phases after injury, astrocyte swelling is driven by elevated extracellular K+ and potassium upregulation of NKCC1, which transports K+ and Na+ into astrocytes. 10,36 –38 NKCC1 in endothelial cells is also involved when ionic edema occurs for new ionic and osmotic gradients across the endothelium after sodium and water influxes into astrocytes. 4,7,13 In our study, we found that HMGB1 inhibition reduced spinal cord NKCC1 expression in SCI rats and in both spinal cord astrocytes and endothelial cells in vitro after OGD/R. NKCC1 reduction attenuated SCI-induced spinal cord edema in rats, decreased cultured astrocytic swelling and AQP4 expression, and increased the Na+ concentration in the surrounding medium in astrocytes after OGD/R in vitro.
NHE1 is the primary regulator of pH in the CNS after pathogenic and/or tissue injury, which is activated in response to decreased pH and facilitates a 1:1 exchange of intracellular H+ for extracellular Na+ in astrocytes and endothelial cells. 4,7,14,34,39 NHE1 is involved in pH homeostasis and acidosis-induced astrocyte swelling, and during ionic edema, NHE1 contributes to Na+ influx across the luminal membrane. 4,34,39 As reported, blocking NHE1 activation in astrocytes may reduce astrogliosis and blood–brain barrier damage and provide neuroprotection in the CNS after ischemia. 40,41 Nishioka and colleagues 42 showed that treadmill exercise ameliorates ischemia-induced brain edema while suppressing NHE1 expression. In our study, NHE1 expression was reduced by HMGB1 inhibition in the spinal cords of SCI rats, cultured spinal cord astrocytes, and cultured endothelial cells. The inhibition of NHE1 reduced SCI-induced spinal cord edema in rats. In cultured spinal cord astrocytes, NHE1 reduction reduced astrocytic swelling and AQP4 expression and increased the Na+ concentration in the surrounding medium in vitro after OGD/R.
The fluxion of Na+ could result in the flux of water, which moves to maintain osmotic and electrical neutrality. Although water transport can occur to some extent by simple diffusion across the plasma membrane, water channel proteins display high capacity and selectivity for transporting water molecules across cell membranes. 4,5,7,43 AQP4 is a water channel protein found in astrocytes, and the overexpression of AQP4 might contribute to increased water entry into astrocytes. 5,25,43 Our previous studies demonstrated that inhibiting HMGB1 significantly decreased astrocytic swelling and AQP4 expression in vitro after OGD/R. 25 In this study, we cultured rat spinal cord astrocytes and then subjected them to OGD/R. The inhibition of HMGB1 reduced NKCC1 and NHE1 expression. Both NKCC1 and NHE1 inhibition markedly decreased the cellular volume and AQP4 protein expression in astrocytes and increased the Na+ concentration in the surrounding medium.
We further investigated the possible mechanisms involved in the decreases in NKCC1 and NHE1 overexpression induced by inhibiting HMGB1 in the spinal cord, astrocytes, and endothelial cells. Our previous studies demonstrated that inhibiting HMGB1 significantly reduced TLR4 expression both in the spinal cord of rats with SCI and in astrocytes in vitro after OGD/R. 24,25 In this study, inhibiting HMGB1 also significantly reduced TLR4 expression in cultured endothelial cells after OGD/R. Our results showed that both NKCC1 and NHE1 expression were decreased by the inhibition of TLR4, TRIF, and NF-κB in cultured spinal cord astrocytes and CNS endothelial cells after OGD/R. In the rat spinal cord following SCI, activating TLR4 increased NKCC1 and NHE1 expression, and inhibiting TRIF and NF-κB decreased NKCC1 and NHE1 expression.
These findings demonstrate that a possible mechanism within a complex, interconnected network of pathways, the HMGB1/TLR4–TRIF–NF-κB signaling pathway. TLR4 is the key CNS receptor on which HMGB1 signaling depends, and the interaction between HMGB1 and TLR4 can activate TRIF, leading to the activation and translocation of NF-κB. 27,44 As reported, TLR4 and/or NF-κB could regulate the expression of NKCC1 in the choroid plexus epithelium in post-hemorrhagic hydrocephalus as well as in rats following subarachnoid hemorrhage, 45,46 and NHE1 expression in cerebral microvascular endothelial cells was also regulated by the cell signaling pathway. 34
Limitations
This study has a number of limitations. First, there are several different ion transporters in astrocytes and endothelial cells in the spinal cord, all of which exert distinct functions and interact with each other during both cytotoxic edema and ionic edema in SCI. In this study, we examined only the effects of the inhibition of HMGB1 on NKCC1 and NHE1 expression and the underlying roles in cytotoxic edema and ionic edema after SCI. Further in vitro and in vivo studies are needed to explore other ion transporters. Second, other receptors of HMGB1 and signaling pathways that are involved in HMGB1 intracellular signaling were not investigated and need to be targeted in future studies. Third, vasogenic edema is also important for the development of spinal cord edema following SCI. In previous studies and the present study, we observed the effects of HMGB1 inhibition on cytotoxic edema and ionic edema. We believe that inhibiting HMGB1 may have some effects on the blood spinal cord barrier (BSCB), which might regulate vasogenic edema in the spinal cord following SCI. Further studies should be performed to evaluate the effects of HMGB1 inhibition on vasogenic edema following SCI.
Conclusions
In summary, we demonstrated that HMGB1 upregulated NKCC1 and NHE1 expression and that this regulation induced spinal cord edema after SCI in rats. HMGB1 inhibition resulted in a reduction in NKCC1 and NHE1 expression in both spinal cord astrocytes and microvascular endothelial cells in vitro, and these reductions in NKCC1 and NHE1 expression decreased sodium/water influx in spinal cord astrocytes after OGD/R. These effects of HMGB1 on NKCC1 and NHE1 expression occur through HMGB1/TLR4–TRIF–NF-κB signaling. HMGB1 is a promising, new target for future studies as well as treatment plans for spinal cord edema after SCI.
Transparency, Rigor, and Reproducibility Summary
We sought to identify both the role of HMGB1 and the mechanism of its effect on NKCC1 and NHE1 expression in astrocytes and endothelial cells, as well as the role of the regulation of spinal cord edema after SCI. An SCI model was generated in adult female rats using a heavy falling object, and an in vitro oxygen-glucose deprivation/reoxygenation (OGD/R) model was generated in rat spinal cord astrocytes and microvascular endothelial cells. These results imply that HMGB1 inhibition reduces NKCC1 and NHE1 expression in both astrocytes and microvascular endothelial cells and thus decreases spinal cord edema after SCI in rats and that these effects occur through the HMGB1–TLR4–TRIF–NF-κB signaling pathway.
Footnotes
Authors' Contributions
LS conceived and designed the work that led to the submission, played an important role in interpreting the results, and was a major contributor to the writing of the manuscript. ML and JQL contributed significantly to the data acquisition, data analysis, and manuscript preparation. ZQW performed the experiments and contributed to the data acquisition. LPW performed the experiments, helped perform the analysis, and engaged in constructive discussions. ZXQ performed the experiments and helped perform the literature search. CD performed the experiments and contributed to the data acquisition. JML performed the experiments and contributed to the data acquisition.
Funding Information
This study was supported by a grant from the National Natural Science Foundation of China (no. 81870976) and by the Shanxi Provincial Health Commission Key Projects to Tackle Key Problems (no. 2020XM27).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Appendix S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.