
Abstract
Traumatic brain injury resulting from an explosive blast is one of the most serious wounds suffered by warfighters, yet the effects of explosive blast overpressure directly impacting the head are poorly understood. We developed a rodent model of direct cranial blast injury (dcBI), in which a blast overpressure could be delivered exclusively to the head, precluding indirect brain injury via thoracic transmission of the blast wave. We constructed and validated a Cranium Only Blast Injury Apparatus (COBIA) to deliver blast overpressures generated by detonating .22 caliber cartridges of smokeless powder. Blast waveforms generated by COBIA replicated those recorded within armored vehicles penetrated by munitions. Lethal dcBI (LD50 ∼ 515 kPa) was associated with: (1) apparent brainstem failure, characterized by immediate opisthotonus and apnea leading to cardiac arrest that could not be overcome by cardiopulmonary resuscitation; (2) widespread subarachnoid hemorrhages without cortical contusions or intracerebral or intraventricular hemorrhages; and (3) no pulmonary abnormalities. Sub-lethal dcBI was associated with: (1) apnea lasting up to 15 sec, with transient abnormalities in oxygen saturation; (2) very few delayed deaths; (3) subarachnoid hemorrhages, especially in the path of the blast wave; (4) abnormal immunolabeling for IgG, cleaved caspase-3, and β-amyloid precursor protein (β-APP), and staining for Fluoro-Jade C, all in deep brain regions away from the subarachnoid hemorrhages, but in the path of the blast wave; and (5) abnormalities on the accelerating Rotarod that persisted for the 1 week period of observation. We conclude that exposure of the head alone to severe explosive blast predisposes to significant neurological dysfunction.
Introduction
E
Tremendous progress has been made in understanding the pathophysiology of blast injury (BI) to the brain that is caused by the intense transient overpressure of the blast wave generated by an explosion. Importantly, the so-called “thoracic mechanism” has come to be recognized, whereby the brain is believed to be injured indirectly by a blast wave that impacts the gas-filled thoracic cavity and is transmitted to the brain by way of major blood vessels in the neck (Cernak et al., 1996, 2001; Courtney and Courtney, 2009; Long et al., 2009). Preclinical work has confirmed the importance of protecting against the thoracic mechanism of BI to the brain (Long et al., 2009; Svetlov et al., 2010).
The transmission of a blast wave through the skull to the brain has long been postulated (Clemedson, 1956), but this “direct” mechanism of injury has garnered less attention than the “indirect” thoracic mechanism, owing perhaps to the impression that the brain is protected from direct injury by the relatively rigid skull. However, it has been shown that a blast wave can traverse the cranium of the rat almost unchanged (Chavko et al., 2007). Numerical simulations that take into account the physical properties of the skull and other tissues of the head exposed to an explosive blast predict that a nonlethal blast can interact with CNS tissues to cause brain injury (Moore et al., 2009; Moss et al., 2009). In principle, a blast wave may induce sufficient flexure of the skull to generate damaging loads within the brain, or it may deposit kinetic energy at boundaries between tissues of different density. Despite such observations and considerations, the effects of an explosive blast overpressure directly impacting the head are poorly understood.
In this study, we sought to investigate the effects of a blast overpressure delivered exclusively to the head, with the aim of determining the effect of direct cranial blast injury (dcBI) uncomplicated by indirect blast injury to the brain mediated by thoracic transmission of the blast wave. We describe and validate a Cranium Only Blast Injury Apparatus (COBIA), which we used to develop a novel rodent model of dcBI in which a blast overpressure is delivered locally to the head, with no exposure of the remainder of the body to the overpressure. In this model, pulmonary injury was absent. Many, but not all, of the neuropathological and neurobehavioral abnormalities reportedly associated with bTBI in humans were observed in this model of dcBI, including prominent subarachnoid hemorrhages, axonal and neuronal injury, and persistent vestibulomotor abnormalities. Notably absent were the brain swelling and delayed death from malignant cerebral edema that have been frequently reported in humans.
Methods
The Cranium Only Blast Injury Apparatus
The COBIA is constructed of several elements (Fig. 1). The central component of the COBIA is a .22 caliber, single-shot, powder-actuated tool (Ramset RS22; ITW Ramset, Glendale Heights, IL). The tool was modified by removing the piston that normally drives the fastener, making the tool function like a firearm and allowing the blast wave to propagate undamped through the barrel. The tool is held vertically using a custom-fabricated stand, which also serves to allow safe use of the tool. The blast is directed downwards into a blast dissipation chamber (BDC), which interfaces snugly with the muzzle of the tool. BDCs of different sizes were used to adjust the magnitude of the blast overpressure that was delivered to the head. The BDCs were fabricated from various lengths of polyvinyl chloride (PVC) pipe (ID 2.05 and 2.54 cm), with the shortest chamber delivering the largest blast overpressure to the head (Table 1). The BDC terminates at the blast dissipation chamber-cranium interface (BDCCI). The BDC-cranium interface is a circular section of PVC pipe (cut from a tee connector; ID 2.54 cm) that holds the rat's head in place. The interface with the scalp is lined with an O-ring made of synthetic foam rubber. The blast wave was generated by firing a .22 caliber crimped brass cartridge (power hammer loads, power level 2 or 4, brown and yellow color coding, with 128±4 and 179±5 mg of smokeless powder, respectively).

Cranium Only Blast Injury Apparatus (COBIA). (
The values shown are the means of 5–10 test firings.
Measurements of blast waveforms
Pressure measuring system
The overpressures generated by blasts from the COBIA and from the pressure-step generator used for validation (see below) were recorded using a precision dynamic piezoelectric pressure transducer (150 pC/psi; 0.002–12 kHz; Model 100-P; Columbia Research Laboratories, Inc., Woodlyn, PA), and charge amplifier (Model 4601; Columbia Research Laboratories). The transducer was connected to the charge amplifier using a 2-m cable with a 100-pF capacitor in parallel with the input. The output of the charge amplifier was digitized (DigiData 1200, sampling frequency 333 kHz; Axon Instruments, Foster City, CA), and waveforms were stored for offline analysis using pClamp software (version 7; Axon Instruments). The output of the charge amplifier is very sensitive to the setting of the transducer sensitivity control, which adjusts for the total input capacitance, including that for the specific piezoelectric transducer (nominally 150 pF), the input cable (∼ 100 pF/m), and the input capacitor (100 pF).
Pressure-step generator
Although the charge amplifier was factory calibrated, we sought to independently verify the calibration for the range of pressures that we would be studying. We constructed a device to generate pressure steps of known magnitude that could be recorded by the pressure measuring system. The pressure-step generator consisted of two chambers, a source chamber and a test chamber, separated by a solenoid-operated valve (#8262G202 120/60, normally closed; Asco, Florham Park, NJ; Fig. 2A). The source chamber (22,712 mL) was pressurized using an air compressor (6 gallon, Model #C2002-WK; Porter-Cable, Jackson, TN), with the exact pressure adjusted to the desired value using a bleed valve, and measured using a factory-calibrated, precision static pressure gauge (4001 heavy duty 4-inch; 0.5% accuracy full scale; Precision Instrument Co., Kennesaw, GA). The test chamber (15 mL) included a valve to allow decompression of the test chamber after each test cycle, and an end-cap to interface with the piezoelectric pressure transducer being tested. The apparatus was capable of generating pressure steps from 0 to 600 kPa with a rise time (from 10–90%) of ∼ 9 msec. Generating a pressure step did not result in any measureable drop in pressure in the source chamber, due to the large difference in volume between the chambers.
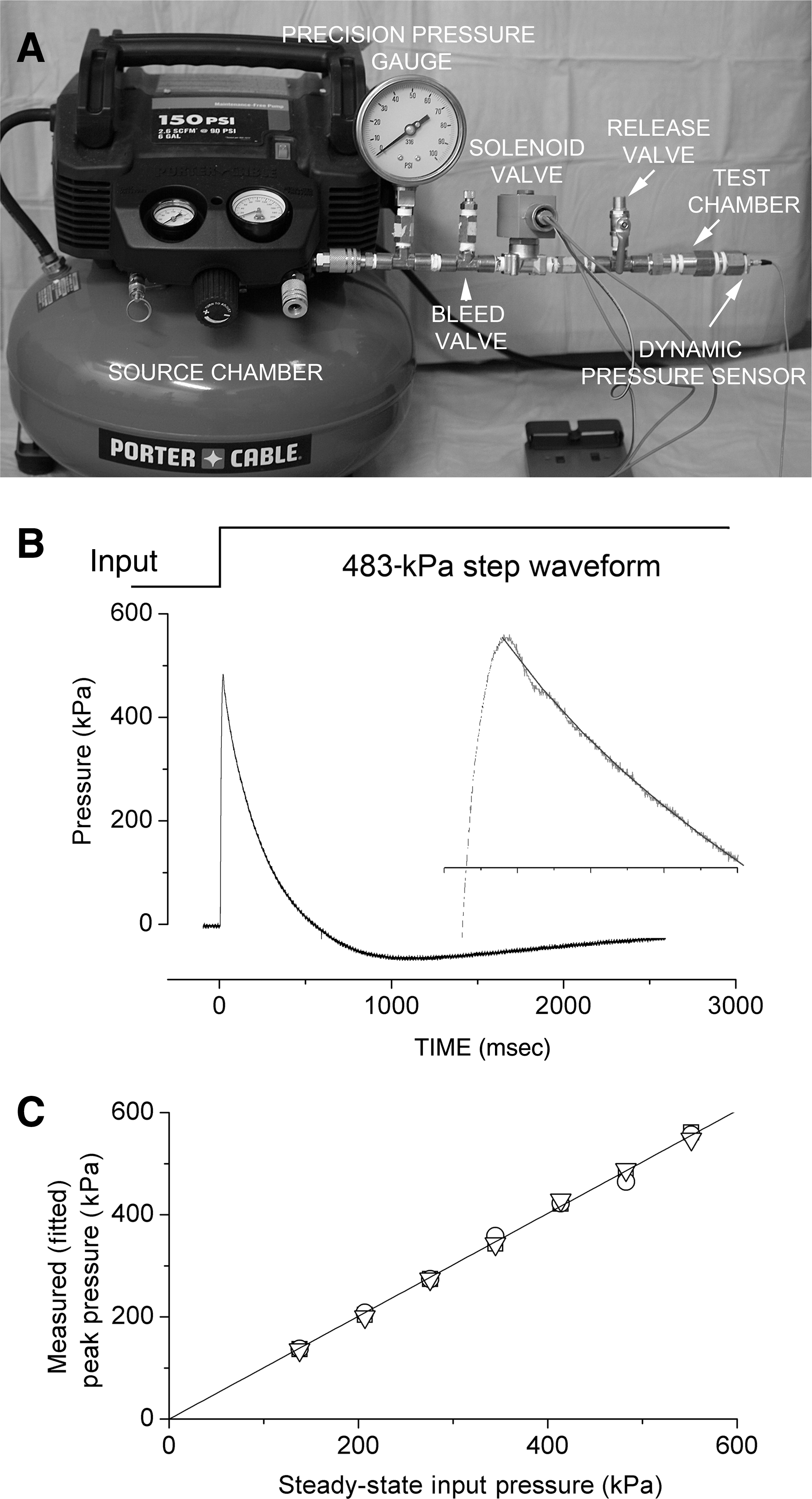
Validation of the pressure measuring system. (
Validating the pressure measuring system
Pressure steps from 0 to 137.9–551.6 kPa, in increments of 68.9 kPa, were generated using the pressure-step generator, and were recorded using the pressure measuring system. All measurements were repeated in triplicate. Because the pressure measuring system acts like a second-order bandpass filter, measurements of steady-state pressures were not possible and some ringing of the peak was observed (Fig. 2B). Therefore the decay phases of the recorded waveforms (from the peak out to 1000 msec) were fit with a second-order exponential equation (τ1, 230–300 msec; τ2, 50–90 msec) to estimate the peak pressure. Values from the fits were plotted against the steady-state input pressure, showing excellent agreement; a least-squares fit of the data yielded a slope of 1.025 and an intercept of −7.1 kPa (Fig. 2C), confirming proper calibration of the pressure measuring system.
Blast injury procedure
All procedures were approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine. This research was conducted in compliance with the Animal Welfare Act Regulations and other federal statutes relating to animals and experiments involving animals, and adheres to the principles set forth in the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996. Male Long-Evans rats (250–320 g; Harlan, Indianapolis, IN) were anesthetized (60 mg/kg ketamine plus 7.5 mg/kg xylazine, IP), intubated with an endotracheal tube, and were allowed to breathe air spontaneously. Core temperature was maintained at 37°C using an isothermal pad (Deltaphase; Braintree Scientific, Braintree, MA). The hair was clipped from the dorsum of the head.
The rat was positioned prone with the vertex carefully placed against the O-ring of the BDCCI (Fig. 1D). The head was positioned so that when viewed from above, the external occipital crest was ∼ 1/3 of the way inside the inner circumference of the BDCCI (Fig. 3A–C). Just prior to detonation, a small balloon under the rat's lower jaw was inflated to 10 kPa to promote contact between the scalp and the O-ring, resulting in reproducible positioning of the head inside the COBIA before the blast. The balloon beneath the jaw also served to cushion (but not prevent) the head from downward acceleration during the blast.
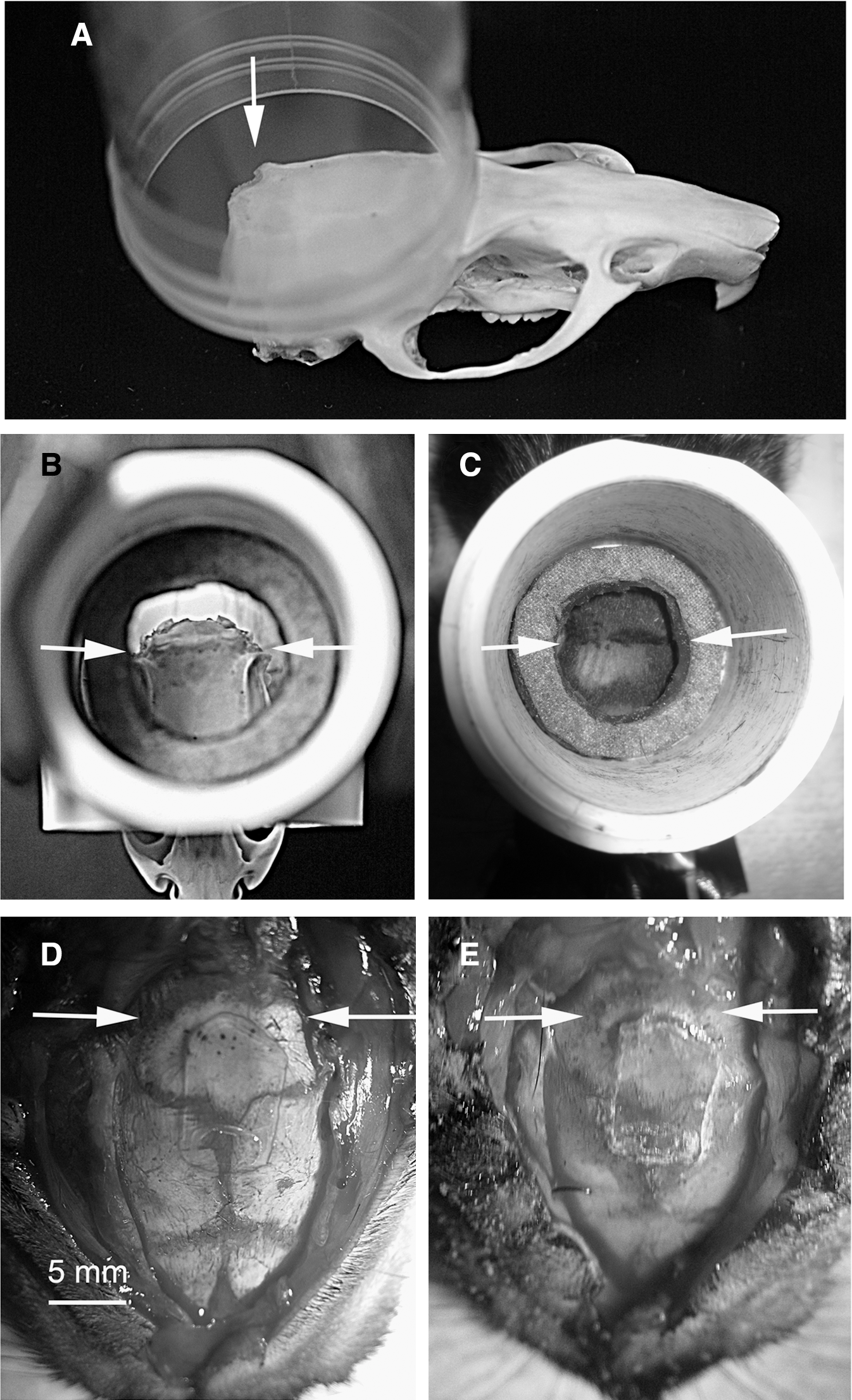
Positioning of the head inside the Cranium Only Blast Injury Apparatus (COBIA), and absence of significant inward flexure of the skull by the blast. (
Positioning the head as described above reduced the exposure to blast of the thin parietal and frontal bones, and of the weakest structures, the sutures between these bones, and allowed the external occipital crest to act as a buttress to prevent inward flexure of the skull during the blast. We assessed for possible inward flexure of the skull, which has been reported in other models (Svetlov et al., 2010), using a thin piece of glass as a strain gauge spanning the sutures exposed to the blast (Fig. 3D). In separate measurements, the glass strain gauge (a cover-slip 0.15 mm thick) was found to fracture by deflecting 0.2 mm midway over a span of 10 mm (n=6). After exposing the dorsum of the skull of an anesthetized rat, the glass strain gauge was glued to the skull, centered over lambda, using alpha cyanoacrylate adhesive (Fig. 3D). The scalp incision was sutured and the rat was exposed to a blast overpressure (517 kPa), as detailed above. After the blast, examination of the glass strain gauge revealed no fracture (Fig. 3E), indicating that if inward flexure occurred, it was <200 μm at the lambda.
In the usual blast injury procedure, after the blast, the rat was removed from the apparatus and was observed for apnea. If spontaneous respiration did not resume within 10 sec, the endotracheal tube was connected to a ventilator (MicroVent gas, compressed air; Hallowell, Pittsfield, MA), and ventilatory support as well as chest massage were instituted and maintained until either spontaneous cardiopulmonary function resumed or until cardiac arrest was deemed to be irreversible. Survivors were nursed on a heating pad until they recovered spontaneous movements. For sham injury, all of the above was performed except for detonating the cartridge.
Experimental series
Overall we studied 160 rats in two series of experiments: series 1 (79 rats) with BDCs having an internal diameter of 2.05 cm, and series 2 (81 rats) with BDCs having an internal diameter of 2.54 cm. The experiments in series 1 were conducted using BDCs of various lengths and cartridges of power level 2 and 4, and were designed to determine the best combination of BDC size and cartridge power to obtain the desired biological response. Experiments in series 2 were all conducted using cartridges of power level 4, and BDCs of various lengths (all internal diameter 2.54 cm). The 79 rats in series 1 included 62 with dcBI that provided usable data, 4 intended for dcBI that were excluded for various technical reasons (abnormal baseline variables, anesthesia-related death before injury, or improper positioning in the BDCCI), and 13 with sham injuries. Of the 62 with usable dcBI data, 9 died following injury, 11 were euthanized at 24 h, 32 were euthanized at 7 days, and 10 were euthanized at later times. The 81 rats in series 2 included 65 with dcBI that provided usable data, 8 intended for dcBI that were excluded for various technical reasons (as above), and 8 with sham injuries. Of the 65 with usable dcBI data, 25 died following injury, 2 were euthanized at 24 h, and 38 were euthanized at 10 days or beyond.
Physiological measurements
All rats underwent continuous pulse oximetry and heart rate recordings with the sensor placed on the hindlimb (Mouse Ox™; STARR Life Sciences Corp., Oakmont, PA). Readings were taken at baseline, immediately after intubation, and then continuously for 30 min after the injury or sham procedure. A group of 33 rats (7 that died shortly after dcBI, plus 26 rats that survived), underwent cannulation of the tail artery, which was performed after induction of anesthesia and prior to injury; arterial access was used to monitor arterial blood pressure (CyQ; CyberSense Inc., Nicholasville, KY), and in some rats, arterial blood gases (iSTAT; Heska Inc., Loveland, CO).
Neurofunctional testing
The accelerating Rotarod test was used to assess coerced locomotor activity (Hamm, 2001). The rats were placed on the drum of the accelerating Rotarod (starting at 4 rpm, accelerating at a rate of 2 rpm every 5 sec up to a maximum of 45 rpm; IITC, Life Science, Woodland Hills, CA), with 3 trials separated by 20 min administered on each day of testing. We report the average latency to falling off of the drum.
Pathology
Necropsies (gross examination of brains and lungs) were performed on all rats that died following dcBI; microscopic examinations (hematoxylin and eosin [H&E] sections) of brains and lungs were performed on 10 rats that died of dcBI, and on 13 rats with sub-lethal dcBI that were euthanized at 24 h. Rats that died shortly after injury were rapidly perfused with saline to remove intravascular contents, and were perfusion-fixed using 4% paraformaldehyde. Rats that survived were euthanized using a lethal dose of pentobarbital IP, after which they were perfused with saline followed by paraformaldehyde fixative, as above. The brains and lungs were harvested and photographed to document subarachnoid hemorrhages and other abnormalities. The fixed brains were cut sagittally and scanned at high resolution to assess for contusions and intraparenchymal or intraventricular hemorrhages. The tissues were then processed for paraffin embedding, sectioned, and stained with H&E for microscopic examination.
Immunohistochemistry
For immunolabeling, perfusion-fixed brains were cryoprotected with 30% sucrose. Cryosections (10 μm) were mounted on slides, blocked in 5% goat serum with 0.2% Triton X-100 in PBS for 1 h, then incubated overnight with primary antibody directed against rat IgG (1:200, catalogue #SC-2011; Santa Cruz Biotechnology, Santa Cruz, CA), β-amyloid precursor protein (β-APP, 1:200, catalogue #51-2700; Invitrogen, Camarillo, CA), or activated (cleaved) caspase-3 (1:200, catalogue #9661; Cell Signaling Technology, Danvers, MA) at 4°C. For fluorescent secondary labeling, the sections were washed three times in phosphate-buffered saline, then incubated in the dark with fluorescent-labeled secondary antibody (1:500, goat anti-rabbit Alexa-Fluor 555; Invitrogen). After 1 h, the slides were washed and cover-slipped with Prolong Antifade reagent with 4,6-diamino-2-phenylindole (DAPI, P36931; Invitrogen). Alternatively, for examining β-APP associated with axonal injury, we used the protocol with diaminobenzidine described previously in this laboratory (Kilbourne et al., 2009). Control experiments included the omission of primary antibody. Low- and high-power photomicrographs were taken using a CoolSNAP camera (Photometrics, Tucson, AZ), and images were adjusted for brightness and contrast using IPLab Software.
Histochemistry
Fluoro-Jade C histo-fluorescence staining was carried out as previously described (Schmued et al., 2005). Briefly, cryosections were first immersed in a solution containing 1% NaOH in 80% EtOH for 5 min. The sections were then rinsed for 2 min each in 70% EtOH and distilled water before being oxidized for 10 min in 0.06% potassium permanganate solution. Following a rinse in distilled water, the sections were stained in 0.0001% Fluoro-Jade C (Chemicon, Temecula, CA), and 0.0001% DAPI (Sigma-Aldrich, St. Louis, MO) solution for 10 min. Finally, the slides were washed 3×with distilled water for 1 min, and dried on a slide warmer at 50°C for 30 min. After clearing in xylene and cover-slipping with non-polar mounting medium (Cytoseal XYL; Richard-Allan Scientific, Kalamazoo, MI), the sections were examined with an epifluorescence microscope under a fluorescein isothiocyanate (FITC) filter for Fluoro-Jade C signal, with care being taken to limit the time of exposure in order to reduce the likelihood of photobleaching.
Statistical analysis
Unless otherwise noted, values are given as mean±standard error (SE). Data were analyzed using a repeated-measures one-way analysis of variance (ANOVA) with Bonferroni comparisons. Effects were judged to be statistically significant if p<0.05.
Results
Blast overpressures generated by COBIA
Blast waves generated by the COBIA were measured using a dynamic pressure sensor embedded in a model of the rat head and positioned where the rat's cranium would be in the BDCCI. Recordings of the blast waves showed a large brief transient overpressure, followed by smaller slower transient under- and overpressures that were fully damped within 2 sec (Fig. 4A). At high temporal resolution, the initial overpressure was found to be complex, and included an initial transient lasting 50–100 μsec, followed by a “quasi-static” pressure component. Notably, the initial complex overpressures generated by COBIA resembled published recordings of blast waveforms obtained in an armored vehicle penetrated by a shaped-charge munition (Fig. 4B).
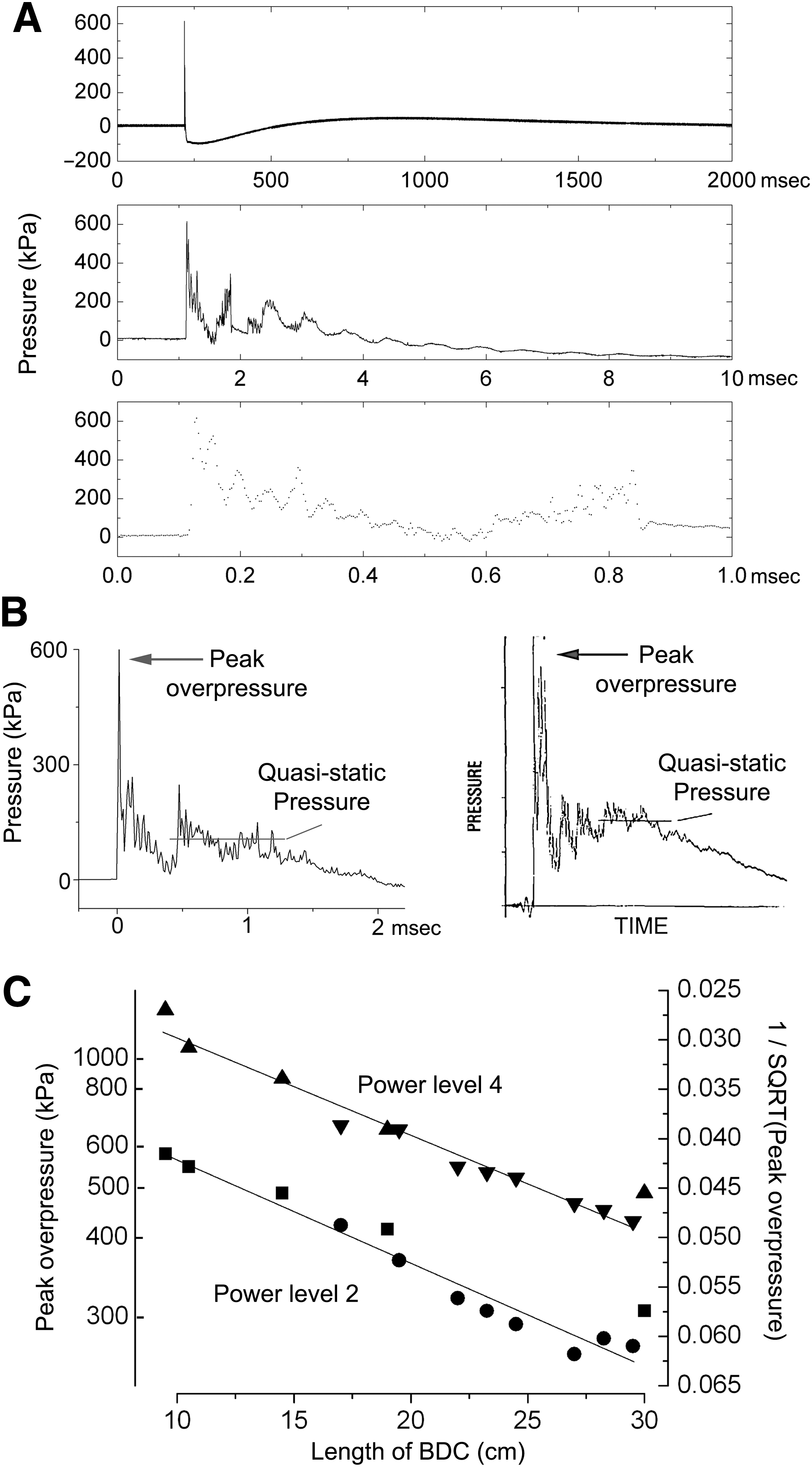
Pressure waveforms generated by the Cranium Only Blast Injury Apparatus (COBIA). (
COBIA was designed so that BDCs of different lengths would generate blast overpressures of different magnitude. The average peak overpressures recorded using a variety of BDCs and two different power level cartridges ranged from 250 to >1000 kPa (Table 1). As a first approximation, the magnitudes of the peak overpressures generated using BDCs of different lengths followed the inverse square law (Fig. 4C).
The velocities of the blast waves generated by COBIA were not measured, but could be estimated from fluid dynamic considerations. The relationship between peak overpressure and velocity in air is given by (Harvey et al., 1984; Zel'dovich and Raizer, 1966):
where P is the shock overpressure (kPa); Po is the atmospheric pressure (101.3 kPa); γ is the specific heat ratio (for air, γ=1.4); U2 S is the shock velocity (m/sec); and C is the velocity of sound in air (343 m/sec). Thus an overpressure of 500 kPa is estimated to have a supersonic velocity of 784 m/sec.
Lethal blast injury to the cranium
At the time of dcBI, the rats were intubated endotracheally and breathed room air spontaneously. Detonating the cartridge in the COBIA resulted in a blast wind that caused slight downward displacement of the head, which was partially cushioned by the balloon beneath the lower jaw, and a 1–2 cm backward displacement of the head and body. Immediately after lethal dcBI, the rats exhibited opisthotonus (i.e., an extreme, dorsally hyperextended posture of the spine with strong extension of the tail). All rats subjected to dcBI experienced apnea, but with lethal dcBI, apnea was persistent and was followed within 30–45 sec by cardiac arrest. In 7 rats, continuous monitoring of blood pressure and heart rate using an intra-arterial catheter failed to show any rise in pressure or fall in heart rate during the first 15–30 sec, suggesting the absence of a surge in circulating catecholamines or in vagal tone; systolic blood pressure fell only after cardiac arrest. Mortality was related to the magnitude of the blast overpressure to which the head was exposed. Exposures below 450 kPa were never fatal, whereas exposures to 650 kPa and higher were associated with >85% mortality; the LD50 was ∼ 515 kPa (Fig. 5A). Fatality was invariably associated with apnea that could not be reversed; mechanical ventilation and cardiac massage never succeeded in resuscitating rats with prolonged apnea (>15 sec). The rapid onset and profound nature of cardiopulmonary arrest following dcBI was consistent with a central mechanism involving the brainstem (i.e., apparent brainstem failure).
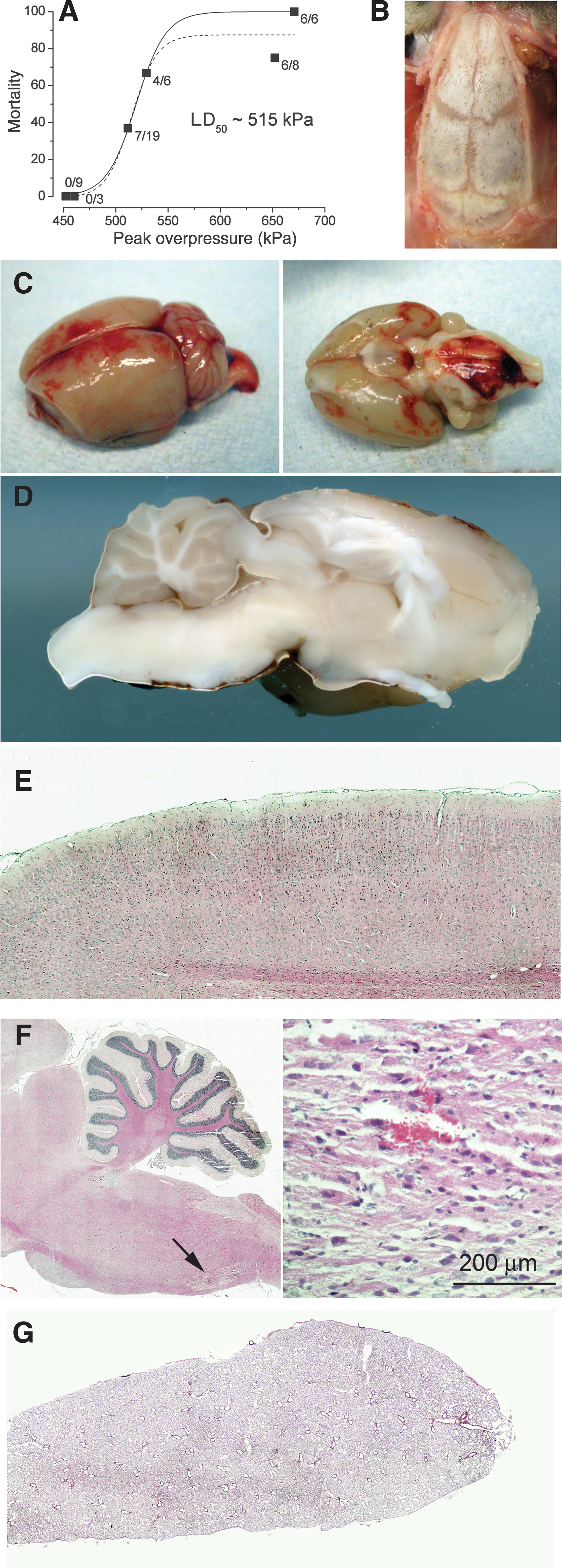
Lethal direct cranial blast injury (dcBI). (
Necropsies were performed on all rats with lethal dcBI to further investigate the cause of death. The scalp at the site of maximum blast exposure was always slightly discolored, but showed no evidence of hemorrhage, contusion, or laceration. Subgaleal hemorrhages or contusions of the temporalis muscles were not observed. Examination of the skull, including the coronal, lambdoidal, and sagittal sutures, showed no evidence of injury or weakening of the bones or sutures (Fig. 5B), suggesting the absence of significant inward flexure of the dorsal cranium. All brains excised following lethal dcBI invariably showed extensive subarachnoid hemorrhages, including dorsally over the cerebrum and cerebellum, and ventrally over the entorhinal cortex and brainstem (Fig. 5C). In 10 rats, unprocessed sagittal sections of perfused brains confirmed the presence of subarachnoid hemorrhages, but revealed no apparent cortical contusions or intraparenchymal or intraventricular hemorrhages, and no overt damage to the brainstem (Fig. 5D). Paraffin sections stained with H&E confirmed these findings, including the absence of contusions in the parietal cortex beneath the site of impact of the blast wave and blast wind (Fig. 5E). In five rats, one or two microscopic hemorrhages <200 μm were identified in the brainstem (Fig. 5F). H&E-stained sections of the lungs from 10 rats showed no evidence of overt damage or hemorrhage (Fig. 5G). Thus death was best attributed to brainstem failure due to neural dysfunction in the absence of overt tissue damage.
Notably, rats that survived the initial apneic event almost invariably recovered and survived; delayed deaths were rare. Of the 34 rats that died as a result of dcBI (9 in series 1 and 25 in series 2), all but one in each series died within 1 min of dcBI. The other 2 rats died at ∼ 30 min and 12 h, with the specific reason for the delayed death not determined.
Sub-lethal blast injury to the cranium
As noted above, all rats with dcBI became apneic. For survivors, the duration of apnea was generally related to the magnitude of the peak overpressure, and was typically less than 12–15 sec (Fig. 6A).
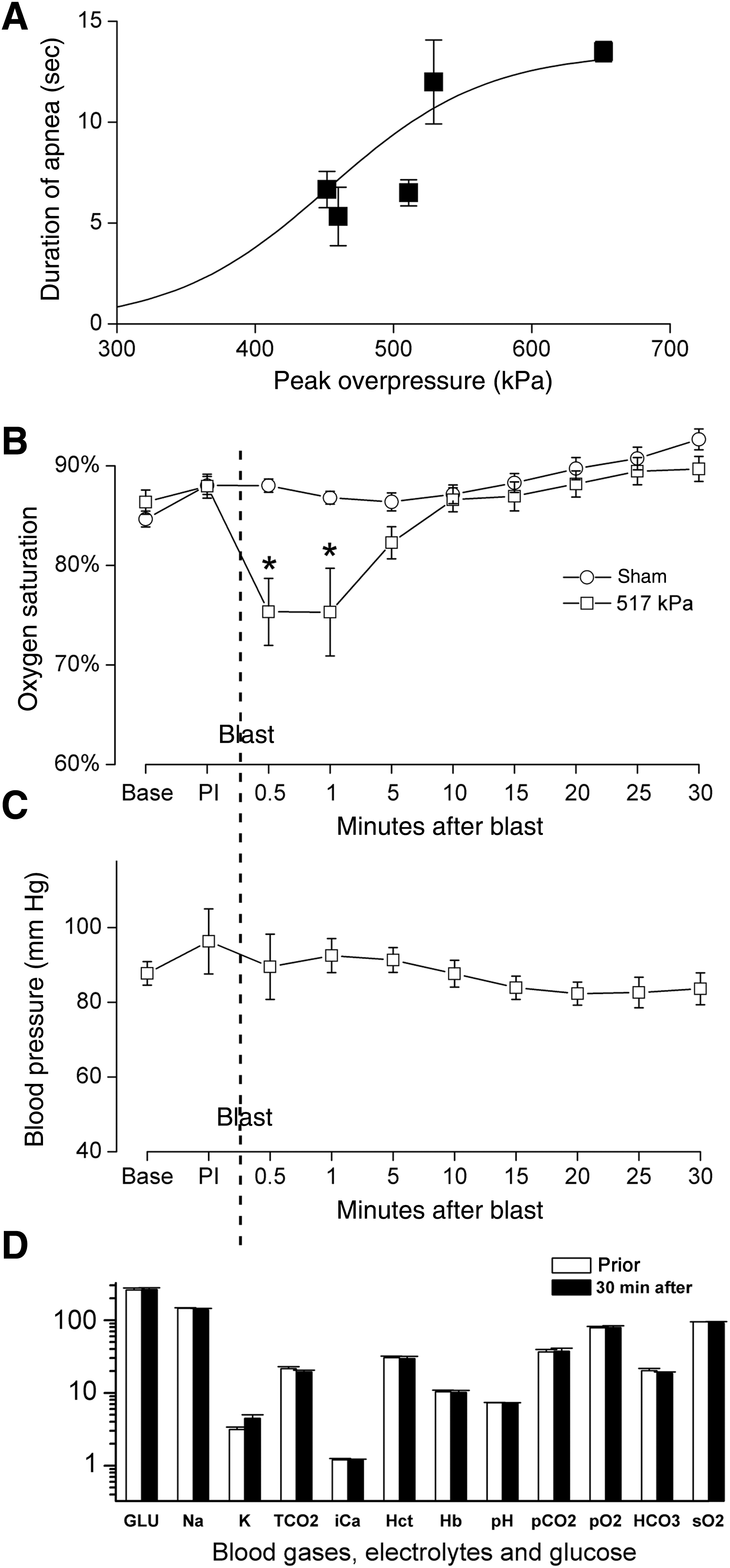
Sub-lethal direct cranial blast injury (dcBI). (
We monitored standard physiological parameters during the 30 min following sub-lethal blast injury. Sub-lethal dcBI resulted in a significant but brief decline in oxygen saturation attributable to apnea, but no significant changes in blood pressure, heart rate, blood gases, serum electrolytes, or serum glucose (Fig. 6B, C, and D). The absence of any appreciable changes in these variables suggested minimal if any pulmonary effects of the direct cranial blast.
Necropsies were performed at 24 h on 13 rats that survived dcBI (414–655 kPa). Examination of the skull, including the coronal, lambdoidal, and sagittal sutures, showed no evidence of injury or weakening of the bones or sutures, again suggesting the absence of significant inward flexure of the dorsal cranium. Subarachnoid hemorrhages were an invariant finding, but the amount of blood and the extent of the hemorrhages was less than with lethal dcBI (Fig. 7A). Notably, subarachnoid hemorrhages were evident in the path of the blast wave, including dorsally in mid- and posterior parietal regions of the cerebrum and over the anterior cerebellum, but subarachnoid hemorrhages were not present more rostrally or caudally outside the path of the blast wave (Fig. 7A). As with lethal dcBI, unprocessed sagittal sections revealed no parenchymal hemorrhages, and paraffin sections stained with H&E showed no hemorrhages or contusions of the parietal cortex.
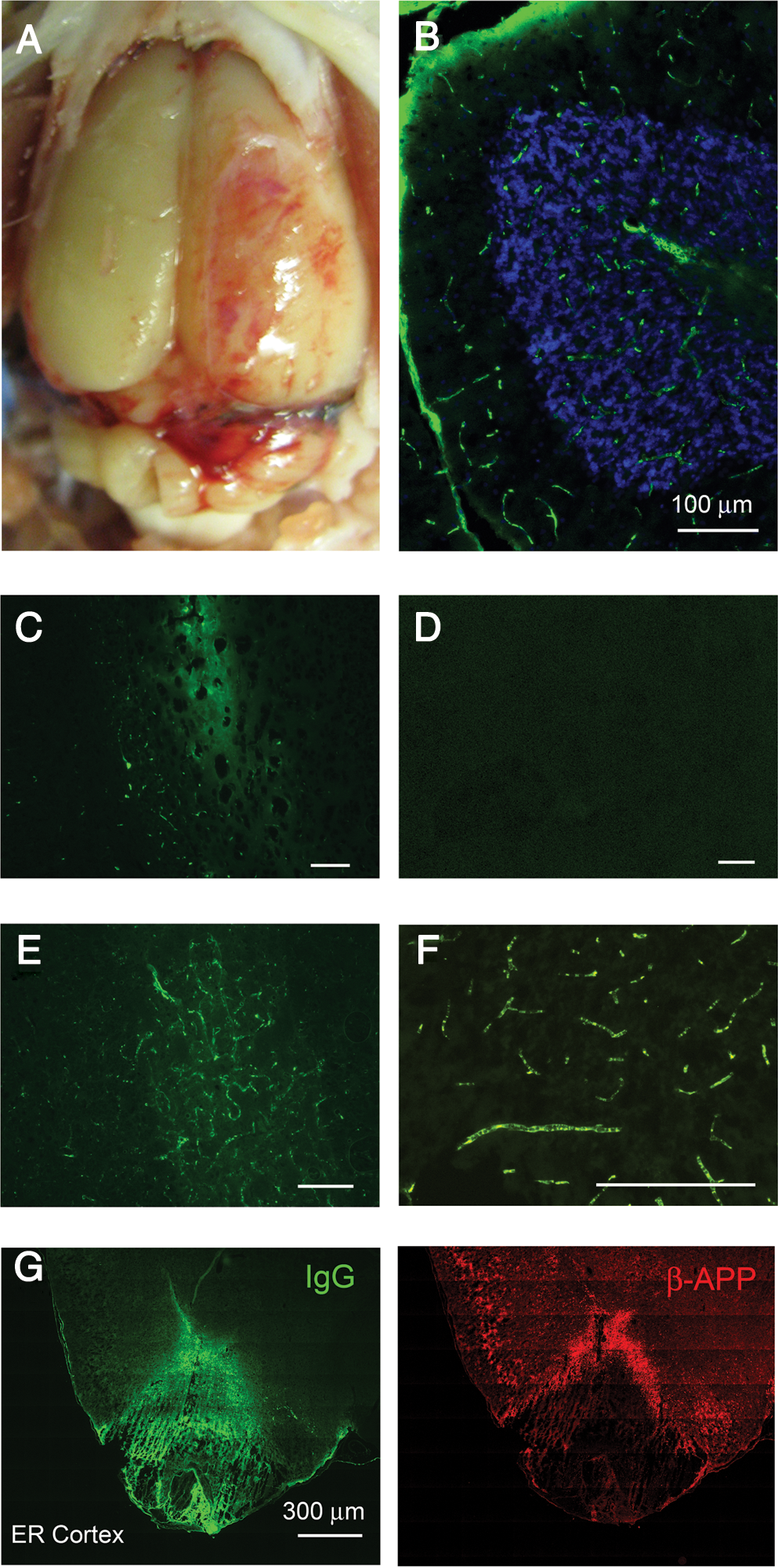
Sub-lethal direct cranial blast injury (dcBI): microvascular abnormalities and contrecoup injury. (
We used IgG immunolabeling as an indicator of microvascular dysfunction. In perfused brains, IgG immunolabeling is normally absent (Fig. 7D). However, IgG immunolabeling may be found under pathological conditions, including: (1) microvascular stasis or thrombosis that prevents intravascular clearance by perfusion; (2) endothelial injury that results in abnormal cellular uptake; and (3) extravasation associated with the formation of vasogenic edema due to breakdown of the blood–brain barrier (Simard et al., 2009a, 2009b). In 8 rats examined at 1–7 days after sub-lethal dcBI (427 and 517 kPa), abnormal IgG immunolabeling was observed in the cerebellum (Fig. 7B and C), and in the thalamus (Fig. 7E and F). In some areas, diffuse labeling was consistent with small regions of vasogenic edema (Fig. 7C and E). Most striking, however, was the unexpected paucity of evidence of vasogenic edema in these brains. In most instances, distinct microvascular labeling without extravasation appeared to be attributable to microvascular stasis or thrombosis, or possibly abnormal endothelial uptake, although post-mortem perfusion artifact could not be completely discounted (Fig. 7B and F).
Immunolabeling for IgG showed a consistent abnormality in the entorhinal cortex. Typically, the ventral-most tip of both temporal lobes exhibited a distinct region of abnormal IgG extravasation, with the superior aspect of this region marked by a zone of excess labeling for β-APP (Fig. 7G). These findings correlated with the gross examination of these brains, which consistently showed a ring-like pattern of subarachnoid hemorrhage surrounding the ventral tip of the temporal lobes (see Fig. 5C, right panel). This pattern of injury was suggestive of a contrecoup injury, which could have resulted from downward acceleration of the head by the blast wind impacting the dorsal cranium, but a direct effect of the blast wave itself could not be excluded.
Several deep brain regions that were distant from the subarachnoid hemorrhages showed consistent elevations in nuclear cleaved caspase-3, including cerebellum, hippocampus, brainstem, and corpus callosum (Fig. 8A–D). Quantification of the number of nuclei with cleaved caspase-3 in these regions showed significant elevations compared to uninjured controls (Fig. 8E).
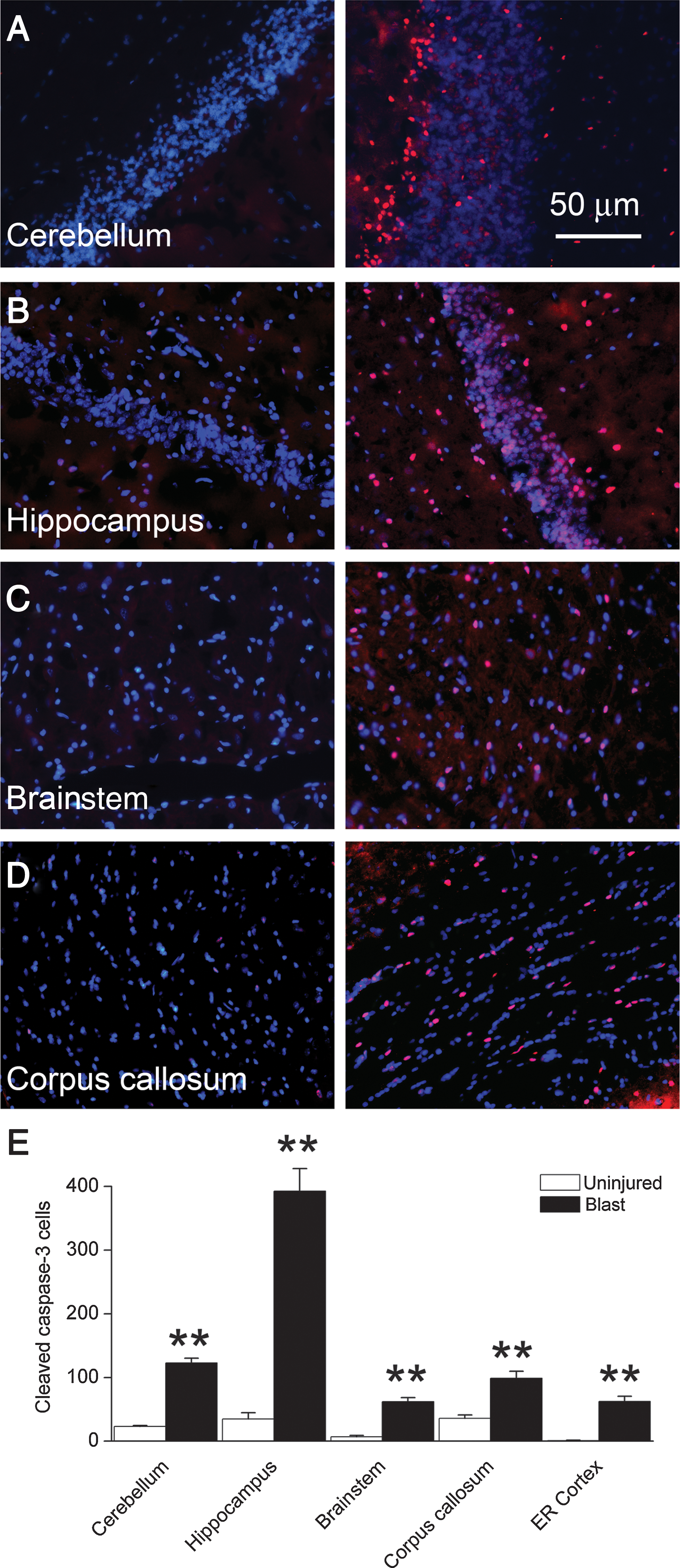
Sub-lethal direct cranial blast injury (dcBI): cleaved caspase-3. (
After axonal injury, persistent axonal transport upstream of the injury leads to bulbous accumulations of β-APP in damaged axons (Blumbergs et al., 1995; Bramlett et al., 1997; Pierce et al., 1996; Stone et al., 2000). In addition, injured neurons may exhibit an increase in cytoplasmic β-APP in their perikarya (Itoh et al., 2009; Kilbourne et al., 2009). We examined β-APP immunolabeling in 8 rats 7 days after sub-lethal dcBI (517 kPa). In only 2/8 rats examined we found abnormal beaded accumulations of β-APP consistent with axonal damage in deep white matter in the path of the blast wave (Fig. 9A), suggesting that axonal injury is not a prominent feature in this model. In all rats, upregulation of β-APP was evident in cells deep to the surface, away from subarachnoid hemorrhages, including cerebellar Purkinje cells and hippocampal neurons (Fig. 9B). Notably, regions with cellular upregulation of β-APP coincided with regions that stained for Fluoro-Jade C (Fig. 9C and D), a marker of cell degeneration. These findings also correlated with regions that labeled strongly for nuclear cleaved caspase-3 (Fig. 8A and B).
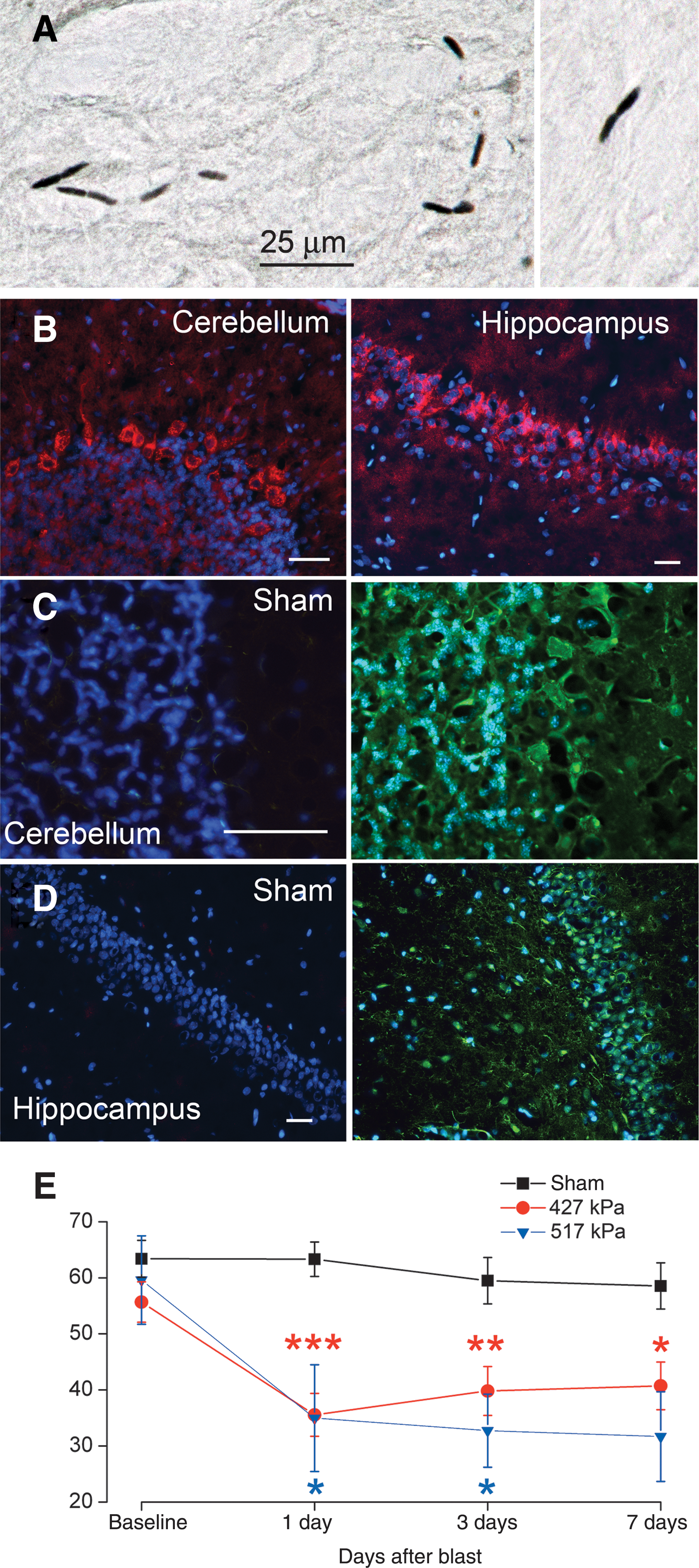
Sub-lethal direct cranial blast injury (dcBI): axonal and neuronal abnormalities and neurological dysfunction. (
We used the accelerating Rotarod task to gauge neurological function. Following sub-lethal dcBI (427 and 517 kPa), performance was significantly degraded compared to controls, with the abnormality persisting for the entire week of observation (Fig. 9E).
Discussion
Blast injury is classified into four phases, each of which can result in brain injury (
In general, it is likely that most models of bTBI that utilize shock tubes or open field exposures to explosive blasts (Long et al., 2009; Svetlov et al., 2010) result in combinations of direct PBI, indirect PBI, and tertiary blast injury to the brain, the latter due to acceleration of the head by the blast wind. The model that we developed and validated here using the COBIA was designed specifically to eliminate indirect PBI to the brain mediated by thoracic transmission of the blast wave. The COBIA likely caused bTBI due to direct PBI plus possible tertiary blast injury to the brain. With the COBIA, tertiary injury was suggested by the observation that the head was displaced by the blast, and by evidence of possible contrecoup injury involving the ventral tips of the temporal lobes, which are located directly opposite the dorsal impact site of the blast wind. On the other hand, the observation that subarachnoid hemorrhages were always present and were located predominantly in the path of the blast wave, especially after sub-lethal dcBI (Fig. 7A), combined with the observation that intraparenchymal hemorrhages were extremely rare, suggests that subarachnoid hemorrhages may have been due to direct PBI.
The physical mechanism by which a blast wave injures tissues is not well understood, but one mechanism believed to be involved is the deposition of kinetic energy at density boundaries. The air-water interface in the lung, the organ most sensitive to blast waves, is an important density boundary. The water-lipid interface where cerebrospinal fluid (CSF) touches the pial surface also forms a density boundary, albeit one with a lesser gradient than an air-water interface. Deposition of kinetic energy at the CSF-pial interface could rupture delicate pial venules, giving rise to widespread subarachnoid hemorrhages. Favoring this interpretation is the observation that parenchymal hemorrhages were almost invariably absent, arguing against a more generalized mechanism for hemorrhage such as transient arterial or venous hypertension. Also favoring this interpretation is that cortical contusions beneath the site of blast impact were never observed, arguing against coup injury from acceleration or from inward flexure of the skull by the blast wind as the cause of subarachnoid hemorrhage. By contrast, impact/acceleration models such as the closed head injury model of Marmarou invariably result in cortical contusion beneath the site of impact, due to coup injury or to inward flexure of the skull (Foda and Marmarou, 1994; Marmarou et al., 1994). Our direct measurements of the strain induced in the skull by the blast indicated that if inward flexure occurred, it was <200 μm at the lambda, consistent with the observation that skull injuries were invariably absent at necropsy. At present, the best explanation for the widespread subarachnoid hemorrhages observed in our model is that kinetic energy from the blast wave was deposited at the density boundary between brain and CSF, but additional study of this hypothesis is warranted.
Blast waves generated by the COBIA
We generated blast waves by detonating smokeless powder inside of crimped brass .22 caliber cartridges. The .22 caliber cartridges were detonated inside of a commercial .22 caliber powder-actuated tool, with the tool modified by removing its piston. This modification made the tool resemble a firearm, and allowed undamped propagation of the blast wave within the barrel of the tool and into the BDC. The blast waves generated by the COBIA bore features that are relevant to real-world explosions. At low temporal resolution, the blast waveforms from COBIA resembled simple Friedlander waves. However, at higher temporal resolution, reflections from the barrel of the tool and from the BDC resulted in complex waveforms with post-peak overpressures that resembled those recorded inside combat vehicles penetrated by munitions (Fig. 4B). The peak overpressures generated by the COBIA ranged from 250 to >1000 kPa (Table 1), with specific values determined using BDCs of different sizes and cartridges of different power levels. The peak overpressures generated by the COBIA may be compared with those generated in shock tube experiments, which typically utilize peak overpressures of 40–350 kPa for whole-body exposures (Bauman et al., 1997; Cernak et al., 2001; Chavko et al., 2007; Long et al., 2009; Petras et al., 1997; Saljo et al., 2000). Calculations based on liquid dynamic theory indicated that the velocities of the blast waves generated by the COBIA were on the order of 750 m/sec, comparing favorably to velocities calculated in simulations of humans experiencing sub-lethal air-blasts, wherein the blast wave is propagating at a velocity of 450 m/sec at the time it reaches the body (Moss et al., 2009).
The COBIA and pulmonary effects
In our experiments with sub-lethal dcBI, we observed only a small reversible effect on oxygen saturation, with all other indicators of pulmonary and other vital physiological functions remaining unaffected. In addition, histopathological examination of the lungs was unremarkable. Together, these findings indicate that the COBIA had minimal if any pulmonary effects. By contrast, whole-body as well as thorax-alone exposure of rats to lesser blast insults leads to unmistakable abnormalities involving the lungs. With whole-body exposure, one study (129 kPa) found diffuse, moderate-to-marked hemorrhage, congestion, and edema in the lungs (Bauman et al., 1997); another study (339 kPa) reported petechiae, ecchymoses, blebs, and isolated or confluent hemorrhages in the trachea and lungs (Cernak et al., 2001). With thorax-alone exposure, one study (69 kPa) found widespread areas of hemorrhage that could be confluent and involve up to 30% of the lungs; another study (304 kPa) reported a fall in mean arterial pressure, a decrease of systolic and diastolic pressures, bradycardia, and tachypnea, and a significant decrease of pH in both arterial and venous blood (Cernak et al., 1996). Given the well-documented sensitivity of the lungs to PBI, our data clearly establish that in our model, spillover of the primary insult to the lungs was minimal, and if it occurred, did not have any important physiological consequence.
The COBIA and CNS effects
Lethal blast
We found that exposures of the cranium to peak overpressures below 450 kPa were never fatal, whereas exposures to 650 kPa and higher were associated with high mortality; the LD50 was ∼ 515 kPa. Animals with lethal dcBI died rapidly of cardiopulmonary arrest following prolonged apnea. By comparison, whole-body exposure of rats to blast is associated with an LD50 that is 2 times smaller, 250 kPa (Richmond et al., 1961, 1962). The reason for the apparent twofold difference in sensitivity associated with cranium-only exposure compared to whole-body exposure is not known, but numerous factors could potentially contribute. First, it must be borne in mind that pressure measurements may not be standardized between laboratories. Although the Richmond data cited above indicated an LD50 of 250 kPa, data from another laboratory indicate that whole-body exposure to 240 kPa peak overpressure is non-lethal, causes no visible damage to the brain, and shows no evidence of damage on routine histological evaluation (Saljo et al., 2000). Additional reasons include the possibility that: (1) a certain degree of protection is afforded by the scalp and skull; (2) amplification of the blast wave occurs after it enters the torso as it propagates to the head; (3) there is added injury due to systemic effects of blast that are absent with cranium-only exposure; and (4) the cause of death with whole-body exposure does not involve the CNS exclusively. Determining the causes for the apparent differences in sensitivity will require additional investigations, including repeating experiments such as ours on rats after craniectomy.
Death following dcBI almost invariably occurred very rapidly (<1 min), and appeared to be due to primary brainstem failure without overt contusive or similar tissue-damaging injury. The rats exhibited opisthotonic posturing and apnea, both of which reflect major brainstem dysfunction. We did not attempt any pharmacological intervention, but intubation, mechanical ventilation, and cardiac massage failed to prevent cardiac arrest. Apnea is also reported with shock-tube experiments (Long et al., 2009). When examined at necropsy, the lungs and other organs except the brain appeared to be normal. Gross examination of the brain showed diffuse subarachnoid hemorrhages, but no internal brain injury. Almost invariably, animals that survived the first several minutes after blast injury did not die later; only 2 of 35 deaths were delayed.
Our findings regarding rapidity of death and pathology from fatal blast injury may be compared to reports on rats after whole-body exposure to blast. With whole-body exposure, animals that die are found to have multifocal hemorrhages in the meninges, cerebrum, and cerebellum, although whether these hemorrhages were intraparenchymal or not was unclear (Bauman et al., 1997). Also, 50% of deaths occur within 2 min, and all occur within 30 min of the insult (Richmond et al., 1962). It is not clear from available reports what the specific cause of death is following whole-body exposure to blast, but the rapid times to death (2 min) argue against entities such as malignant cerebral edema and neurogenic pulmonary edema, and favor instead primary brainstem failure involving cardiac and respiratory centers.
In humans, severe blast injury often results in delayed death that is commonly attributed to cerebral edema and brain swelling (Bauman et al., 2009; Ling et al., 2009). To our knowledge, delayed death has not been reported following blast exposure of rats, suggesting that cerebral edema and brain swelling may not be prominent effects of blast, at least not in this species. Transient edema and a modest rise in intracranial pressure have been reported in rats exposed to repeated whole-body blast, but the effects observed were small and non-lethal (Saljo et al., 2009). It appears that the threshold for blast-induced fatal brainstem injury is lower than that for microvascular dysfunction, at least in the rat. If this is also true in humans, it would suggest that cases of brain edema and swelling may actually reflect quaternary blast injury to the brain (i.e., may be due to PBI to the brain complicated by some other insult such as hemorrhage or hypotension) (DeWitt and Prough, 2009). Understanding this complex relationship is of paramount importance because of the critical therapeutic implications.
Sub-lethal effects on neuropathology
Rats with exposure of the head alone to peak overpressures of 500 kPa or less were likely to survive. Animals that survived blast awoke from anesthesia without undue delay and typically could right themselves and sit; spontaneous activity was blunted, but they never lapsed into coma or unresponsiveness.
When examined 24 h after injury, all animals that survived the blast exhibited subarachnoid hemorrhages over the cerebrum, cerebellum, and brainstem. Subarachnoid hemorrhages were especially prominent in the path of the blast wave, and as discussed above, may have been due to physical disruption of surface venules by the blast wave itself. Subarachnoid hemorrhage is a frequent observation in humans with bTBI and is associated with worse clinical outcomes (Armonda et al., 2006), making this aspect of our model particularly relevant to the human condition. Insofar as the subarachnoid hemorrhages are diffuse, one expects that the resultant inflammatory response to the blood and blood products in the cortex would result in widespread abnormalities predisposing to delayed cognitive dysfunction.
Subarachnoid hemorrhages were observed in rats that survived exposures of 414–655 kPa. The literature suggests a rather steep pressure-response relationship for neuropathological injury in rats with whole-body exposure. Though 147 kPa overpresssure results in subarachnoid hemorrhage, necrosis, cortical cell loss, gliosis, and infiltration, exposure to 22% smaller overpressures (114 kPa) results in no detectable neuropathological changes (Long et al., 2009).
Sub-lethal dcBI was associated with abnormal immunolabeling of perfused brains for IgG, consistent with microvascular injury. The abnormalities included scattered regions of microvascular stasis and patches suggestive of vasogenic edema. However, widespread or generalized extravasation of IgG was not observed, consistent with the absence of malignant cerebral edema, the absence of brain swelling, and the near absence of delayed death. A modest transient increase in intracranial pressure to 16 mm Hg was reported after 3 successive blasts (60 kPa) over 20 min that was attributed to edema formation (Saljo et al., 2009), but overall, brain swelling and malignant edema are not prominent features in any of the rat models reported.
One important abnormality found with sub-lethal dcBI involved immunolabeling for β-APP. Both axonal and neuronal abnormalities were identified that were broadly similar to findings reported in other forms of TBI (Blumbergs et al., 1995; Bramlett et al., 1997; Itoh et al., 2009; Kilbourne et al., 2009; Pierce et al., 1996; Stone et al., 2000). Abnormalities in β-APP were widespread, and involved deep brain regions away from subarachnoid hemorrhages. The abnormalities in axonal β-APP that we observed, although quantitatively not a prominent feature of our model, likely reflect an injury similar to that identified in other studies of bTBI using silver stains (Long et al., 2009; Svetlov et al., 2010). Pronounced changes in the neuronal cytoskeleton have been reported after blast exposure, with a redistribution of neurofilament proteins in neuronal perikarya that is believed to predispose to delayed nerve cell loss (Saljo et al., 2000). In addition, the perikaryal abnormalities in β-APP that we observed correlated with Fluoro-Jade staining and abnormal nuclear cleaved caspase-3, two other markers of cell injury. Neurons in the rat brain may be injured by exposure to overpressures as low as 20 kPa (Moochhala et al., 2004), with exposure to higher levels (100–300 kPa) causing significant injury to both neuronal and glial cells (Cernak et al., 2001; Kaur et al., 1995; Saljo et al., 2010).
In general, the pathological, cellular, and axonal abnormalities reported here following dcBI may account for the persistent vestibulomotor abnormalities that we observed in the accelerated Rotarod test. Previous studies with whole-body exposure to blast with body protection in rodents found that vestibulomotor abnormalities normalized within 1 week of injury (Long et al., 2009), although in other studies, abnormalities in wheel running (Bauman et al., 1997), and in an active avoidance task (Cernak et al., 2001) persisted for 5 days. Models in which functional deficits endure are particularly important because bTBI in humans is well known to be associated with long-lasting neurofunctional abnormalities (Finkel, 2006; Ling et al., 2009).
Some shortcomings of the present study should be noted. Because our primary purpose was to describe the COBIA and to use it to establish a novel model of dcBI, our characterization of the many brain abnormalities induced by the COBIA was necessarily limited. A more complete description of the neuropathological findings reported here is needed. Additional studies will be required for more comprehensive characterization of the functional, cognitive, and behavioral abnormalities induced in this model. Also, the hypothesis that subarachnoid hemorrhage is due to deposition of kinetic energy at the brain-CSF boundary calls for additional study.
In summary, we developed and validated a novel model of bTBI that is unique in allowing study of the direct effects of explosive blast on the brain, uncomplicated by indirect effects mediated via the thoracic mechanism. There is no doubt that in combat and other real-life situations, both direct and indirect mechanisms of PBI to the brain, as well as secondary, tertiary, and quarternary mechanisms, all play crucial roles in determining the overall outcome from bTBI. However, we postulate that understanding the specific contribution of each of these separate mechanisms will lead to important insights that will improve the eventual protection and treatment of the unfortunate warfighters and civilians who are victims of bTBI.
Footnotes
Acknowledgments
This work was supported by a grant to J.M.S. from the Department of the Army (PT074766; the U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick, MD 21702-5014 is the awarding and administering acquisition office). The information in this article does not necessarily reflect the position or the policy of the United States Government, and no official endorsement should be inferred.
Author Disclosure Statement
No competing financial interests exist.