
Abstract
An estimated 20 million chemical transformations occur in an average human cell every second; however, the vast majority of nature’s small-molecule “dark chemical matter” remains uncharted, limiting our understanding of basic biological processes and throttling progress in human health. High-throughput metabolomics coupled with artificial intelligence and machine learning can decode life’s chemistry, illuminating novel metabolites involved in giving rise to living systems and transforming human health.
The sequencing of the human genome marked a monumental moment in scientific history, unlocking the blueprint of life and fueling hopes for transformative advances in medicine and industry. 1 Life-changing medicines have resulted from genomics research, including gene therapies and nucleic acid-based therapeutics, bringing new hope to thousands of patients by targeting the genetic blueprint of diseases to alter disease progression.2,3
We are all comfortable with the familiar framing of proteins being the workhorse of the cell but rarely ask, “What work do proteins do?” To quote the biophysicist Harold Morowitz, every protein, indirectly or directly, makes matter cycle or energy flow. 4 Viewed from another lens, the point of genes, transcripts, and proteins is to transform carbon, that is, metabolism. This chemical layer of biology, perhaps its very point, is largely ignored by science.
The human genome comprises roughly 20,000 genes, a finite and well-mapped terrain compared with the chemical universe. 5 The human metabolome is so unknown that we do not yet have a confident estimate of its size, but experts suggest it is likely in the millions of compounds. Nature, by contrast, is an almost inexhaustible library of molecular diversity. An estimated 99% of natural compounds remain unknown. 6 Their structures, functions, and potential applications are hidden in the shadows of what researchers call “dark chemical matter.” These molecules are the products of billions of years of evolutionary experimentation, and they hold the keys to life’s resilience, adaptation, and complexity.
This chemical richness has already proven to be an unparalleled source of innovation. Nearly two-thirds of all approved drugs originate from natural products or their derivatives, including antibiotics, anticancer agents, and immunosuppressants. 7 Nature’s compounds, shaped by evolutionary pressures, exhibit bioactivity unmatched by synthetic chemistry. And yet, despite their success in medicine and industry, we’ve barely scratched the surface of this chemical treasure trove.
A Slow and Iterative Process
Why is this chemical dark matter so elusive? Unlike genomic sequencing, the task of identifying and characterizing natural chemicals remains a laborious and complex endeavor. While mass spectrometry, the primary tool for analyzing molecular identities, predates genomics, it is still incomplete. Chemical compounds are often found in complex mixtures, without annotation of structure or biological function. Traditionally, activity in complex extracts is chased down to individual bioactive compounds using a process called bioactivity-guided fractionation. This technique identifies bioactive compounds from a natural extract by repeatedly separating the extract into smaller fractions, testing each fraction for biological activity, and then further isolating the most active fractions until the pure bioactive compound is obtained; essentially, it is a method to systematically identify the active components within a complex mixture based on their biological effects.
The main challenge of bioactivity-guided fractionation is that often the bioactivity is lost in the process of fractionation before the molecule is whittled down. This could be due to several issues, including potential degradation of bioactive compounds during purification, low concentrations of active molecules, which make isolation and purification challenging, and potentially synergistic effects between multiple compounds.8,9 When compounded by the need for time-consuming and laborious assays to isolate and identify the active principle from a complex mixture, and the iterative nature of the process becomes unscalable. 10
If one were fortunate to be able to track molecules using bioactivity-guided fractionation, the current structural elucidation methods limit the annotation of chemical dark matter. These methods include isolating the desired compound with high purity and resolving its structure using nuclear magnetic resonance (NMR), and bioactivity-guided fractionation to guide elucidation of the biological activity or function of a target compound. This process is time-consuming and expensive, and therefore impractical to apply to the millions of compounds found within complex biological mixtures. 11
In short, our conventional wet lab pipeline for discovering biochemicals—from extraction and bioassay to isolation, NMR, and functional testing—is inherently low-throughput. It is no wonder that chemical space remains the last great frontier; we simply have not had the tools to efficiently survey it.
Technological Bottlenecks in Metabolomics
The advent of modern metabolomics—using high-performance analytical chemistry to profile metabolites on a large scale—promised to accelerate the exploration of chemical diversity. Indeed, mass spectrometry can detect hundreds or thousands of metabolites in a single sample. 12
In mass spectrometry, each molecule produces a unique spectral “fingerprint” as it fragments, offering clues to its identity. In principle, these spectra allow us to cast a much wider net for unknown compounds. In practice, however, metabolomics has revealed an ironic truth: the more data we collect, the more unknowns we encounter. Several bottlenecks have prevented us from converting the deluge of spectral data into chemical knowledge, including:
Everything outside this narrow library remains invisible to database matching. In untargeted metabolomics experiments, only a small fraction of detected spectra can be matched to known molecules.
12
The rest—often >80% of the spectral features in a sample—have no reference spectrum and thus remain unannotated. In other words, most peaks in a metabolomics dataset represent molecules that have never been seen before in any database.
14
Chemists can sometimes propose partial structures or substructures from fragmentation patterns, but doing this for tens of thousands of unknowns in a study is impossible for most labs. This leads to a paradox: We can measure countless metabolite signals, but we cannot identify or use most of them. The result is a growing pile of spectral data rich in potential insights but effectively opaque—the dark matter of the metabolome.
These limitations have real consequences. In metabolomics studies of human health, for instance, researchers often observe biomarker signals correlated with disease—but if those metabolites cannot be identified, their biological interpretation remains a mystery. In natural product discovery, chemists may keep rediscovering the same familiar compounds because unknown ones can’t be recognized and prioritized from complex extracts. And in drug discovery, vast chemical diversity is going unmined: pharmaceutical libraries end up biased toward known scaffolds, while the long tail of rare or novel chemotypes remains untouched, possibly containing cures or chemistries we desperately need.7,16 In short, the pace of biomedical innovation is throttled by our inability to read the molecular lexicon that nature has written.
Artificial Intelligence and Machine Learning: Navigating the Chemical Dark Matter
This is where cutting-edge innovations in artificial intelligence (AI) and machine learning come into play. Just as AI has revolutionized proteomics (think of how AlphaFold solves protein structures), it is now beginning to tackle the interpretation of complex chemical data.17–24 The idea is straightforward: can a computer be taught to decode mass spectra of unknown molecules, effectively translating the cryptic signals into likely chemical structures? Recent advances suggest the answer is yes.17–24 In fact, researchers are now deploying the same technologies behind modern language translation and image recognition to “learn” the language of mass spectrometry. One promising approach uses deep learning models—in particular, transformer neural networks akin to those in large language models—to recognize patterns in mass spectra. These models excel at handling high-dimensional, sequential data with intricate contextual relationships.
Mass spectrometry generates a wealth of data that reflects the fundamental chemical and physical properties of a molecule, including its mass-to-charge ratio, fragmentation patterns, and isotopic distributions. Unlike molecular sequences, which are a linear representation of atomic connectivity, mass spectra provide a high-dimensional fingerprint capturing both connectivity and fragmentation behaviors under defined conditions. These inherently rich data lend themselves to deciphering complex chemical structures that may involve nonlinear or cyclic arrangements, which are often obscured in sequence-based representations.
Transformer models, including attention-based architectures like Bidirectional Encoder Representations from Transformers (BERT) and Generative Pre-trained Transformer (GPT), excel at processing this type of high-dimensional, nonlinear data with intricate relationships. 25 Transformers, which have revolutionized natural language processing, can ingest a spectrum and tease out the correlations between fragment peaks that correspond to specific substructures, stereochemistry, or functional groups. BERT uses a training technique known as masked language modeling, where parts of a sentence are “masked” to teach the model the structure of natural language by predicting the missing elements based on the remaining context. For instance, in the sentence “The dog tried to follow the person,” the word “follow” could be masked, presenting the model with “The dog tried to [MASK] the person.” The model then learns to predict the hidden word using the contextual clues provided by the rest of the sentence (Fig. 1). 19
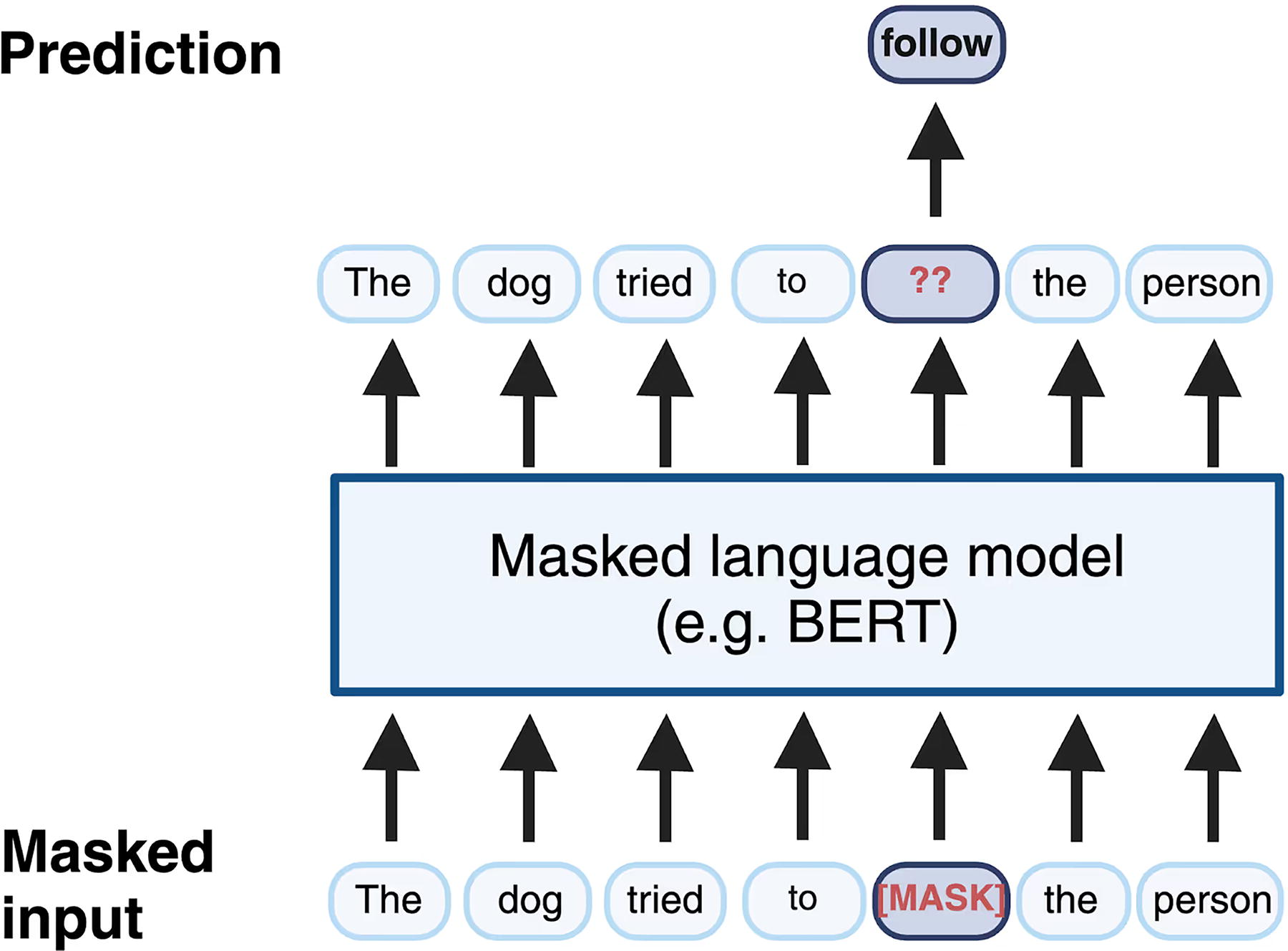
Transformers that have been tailored for tandem mass spectra essentially learn the grammar of chemistry. Early studies have demonstrated that such models can predict molecular features more accurately and comprehensively than traditional rule-based methods. 19
Different research groups have taken slightly different approaches. Some have developed encoder–decoder networks that directly generate a molecule’s structure (for instance, a Simplified Molecular Input Line Entry System [SMILES] string) from its spectrum, analogous to speech-to-text translation. One such model, appropriately named Spec2Mol, uses an encoder to create a learned representation of the spectrum and a decoder to output plausible molecular structures, drawing from the knowledge of chemical syntax. 26 Other efforts (such as CSI:FingerID, MS2DeepScore, and MSNovelist) combine machine learning with clever heuristic or rule-based steps to expand the search for candidate structures beyond known libraries.20,27,28
AI is enabling a leap from relying solely on known spectra to predicting the unknown. The application of these advances is profound. Imagine being able to rapidly identify the metabolites in a person’s blood sample that have no entries in any database—AI could flag a novel molecule, providing the first clues to its identity. In drug discovery, instead of randomly testing fractions, one could prioritize specific spectra that the AI predicts to have “drug-like” substructures or interesting novelty. AI models can also learn from a vast corpus of uncharacterized spectra, finding patterns across experiments and labs that humans might never notice. In essence, machine learning offers a scalable way to illuminate the chemical dark matter, turning masses of raw data into actionable hypotheses about molecules.
Bridging the Gap: A New Era of Scalable Chemical Discovery
Realizing this vision requires not just clever algorithms but also massive amounts of data and computational muscle. This is leading to the rise of foundation models for chemistry—large AI models trained on extremely broad datasets of molecular data, analogous to how GPT-4 was trained on the internet’s text (see Box 1).
Box 1. Pioneering Foundation Models for Decoding the Dark Chemical Space
A recent example is work by our company, Enveda, an AI-powered metabolomics company seeking to unlock nature’s chemistry. Enveda is leveraging AI, particularly transformer models such as those used by large language models like ChatGPT, to bridge the gap between mass spectral data and chemical knowledge.17–19 By training AI on large datasets, Enveda’s approach essentially translates the cryptic language of mass spectra into the chemical structures they represent. This approach is akin to teaching a machine to read a foreign language, enabling it to uncover the identities and properties of previously unknown molecules.
Enveda’s most recent model is a foundation model trained on an unprecedented 1.2 billion small-molecule mass spectra. 19 This model, called PRISM (Pretrained Representations Informed by Spectral Masking), represents (to our knowledge) the largest training set of tandem MS data assembled to date. Importantly, the vast majority of those 1.2 billion spectra were unannotated—no one knows what compounds produced them. By training in a self-supervised manner (learning the patterns within spectra themselves), the model effectively imbibes the latent grammar of fragmentation across an almost astronomical number of molecules.
Enveda’s automated pipeline for mass spectral profiling has already generated hundreds of millions of MS/MS spectra, and as we scale our search for new molecules, we plan to scale the size and diversity of experimental data. Large amounts of raw data for training translates into better predictive models, which in turn will enable scientists to decode the chemistry of life to find interesting new biomarkers.
AI models can be further trained on chemical and biological properties which would be of interest specifically to target disease etiology. Furthermore, the self-supervised learning approaches used for PRISM are well-suited for large language models that allow the model to learn from abundant unlabeled mass spectrometry data. 29
Crucially, approaches like this break from the past reliance on only labeled data. Traditionally, machine learning in metabolomics was limited to using spectra of known compounds (e.g., to classify a spectrum or to compare against candidates). But annotated spectra represent only a tiny fraction of all available data—on the order of maybe 50,000–60,000 unique molecules in public libraries. In contrast, repositories of raw experimental data (such as Global Natural Products Social Molecular Networking [GNPS], MetaboLights, and the Metabolomics Workbench) contain hundreds of millions of spectra, reflecting a far broader swath of chemical space. By harnessing this mountain of dark data, next-generation AI models do not need to wait for a molecule to be isolated and cataloged to learn its signature—they learn directly from the uncharacterized spectra, effectively mapping the wilderness without a predefined guidebook.
Conclusion
The implications of closing the metabolomic knowledge gap are enormous. In biotechnology and pharmaceutical research, a more complete understanding of nature’s chemical playbook could open up avenues to new drug leads, enzymes, and biomolecules that humanity has never encountered. In diagnostics, comprehensively mapping the human metabolome (and how it changes in disease) could reveal novel biomarkers—potentially early warning signals or mechanistic clues for conditions like cancer, neurodegeneration, inflammation or metabolic disorders. In agriculture and ecology, identifying the myriad chemical signals used in plant–microbe interactions or animal communication could lead to sustainable innovations (natural pest control agents, growth promoters, etc.). And in basic science, illuminating the structures and pathways of unknown metabolites will fill glaring gaps in our understanding of physiology and biochemistry, much like sequencing the human genome revealed previously “hidden” genes and regulatory elements. Recall that not long ago, vast stretches of genomics were dubbed “junk DNA” and thought to be irrelevant. 30 We now know those noncoding regions hold crucial regulatory roles—a testament to how dark matter in science can surprise us once illumination is possible.
Today’s chemical dark matter may likewise harbor treasures we can scarcely imagine—from new therapeutics to fundamental biological insights. The convergence of high-throughput metabolomics and AI is finally creating a path to explore this last frontier. By systematically identifying the metabolites that have eluded us, science is poised to complete the map of life’s molecular landscape.
Chemistry is steadily yielding its secrets. As we learn to read the chemical alphabet at scale, we move closer to a future where no biologically relevant molecule will lurk in the shadows, and where understanding life in its totality is a genuine possibility.
The genome may be life’s blueprint, but chemistry is its language. To understand life—and to harness its full potential—we must learn to read the messages hidden in nature’s molecules. In doing so, we will open a new chapter in science, one where the unknown chemistry of nature takes center stage in shaping the future of human health.
Footnotes
Acknowledgements
Author Disclosure Statement
The authors are affiliated with Enveda Therapeutics, Inc.
Funding Information
No funding was received for this article.