
Abstract
Despite a Cambrian explosion in therapeutic modalities, small-molecule drugs remain a prominent and advantageous medical intervention. The universe of synthesizable, drug-like small molecules is astronomical. Given this scale, efficiently narrowing in on therapeutic candidates that are potent, selective, and tolerable cannot occur by happenstance. Over the past several decades, computational tools have become commonplace among pharma companies seeking to discover new small-molecule drugs. For example, molecular mechanics force fields are used to power molecular dynamics simulations—an effective approach for virtually screening and optimizing candidate molecules. In parallel, data-driven methods such as machine learning have supercharged the field’s ability to design potentially bioactive compounds. Despite these advances, established computational methods still suffer from issues relating to throughput, accuracy, generalizability, or combinations thereof. We argue that a merger of these technologies is inevitable and desirable, allowing the strengths of each to address the weaknesses of the other. This fusion—in the form of neural network potentials (NNPs)—is an exciting frontier for small-molecule discovery and design. Ostensibly, NNPs enable a swift, accurate, and generalizable solution for researchers developing the next generation of small-molecule drugs.
Introduction
Small molecules are the dominant therapeutic modality. 1 They are the therapies of the past and are likely to be the therapies of the future. Defined as any compound with a low molecular weight (often <500 Daltons), small molecules can be designed for oral administration, penetrate cell membranes to reach intracellular proteins, and engage their targets through multiple mechanisms of action.1,2 Critically, small-molecule drugs can be readily manufacturable and are frequently formulated as tablets that do not require cold chain storage or other complex logistics.
Biologics and other emerging modalities (e.g., gene therapies) have some proven advantages over small molecules—such as the ability to deliver or express functional copies of proteins sidelined by genetic mutations—but also disadvantages presenting significant new challenges.3,4 For these reasons, small-molecule drugs will retain a central position in the future pharmacopoeia.
Most small-molecule drugs bind to proteins—often protein active sites. Binding can inhibit or activate a protein’s function, altering an ostensibly dysregulated gene pathway and potentially rescuing a patient’s phenotype. Some small molecules target protein allosteric sites—regulatory pockets on a protein’s surface. 5 These so-called allosteric modulators are harder to develop, but are often more selective for their targets, leading to safer medicines. 6
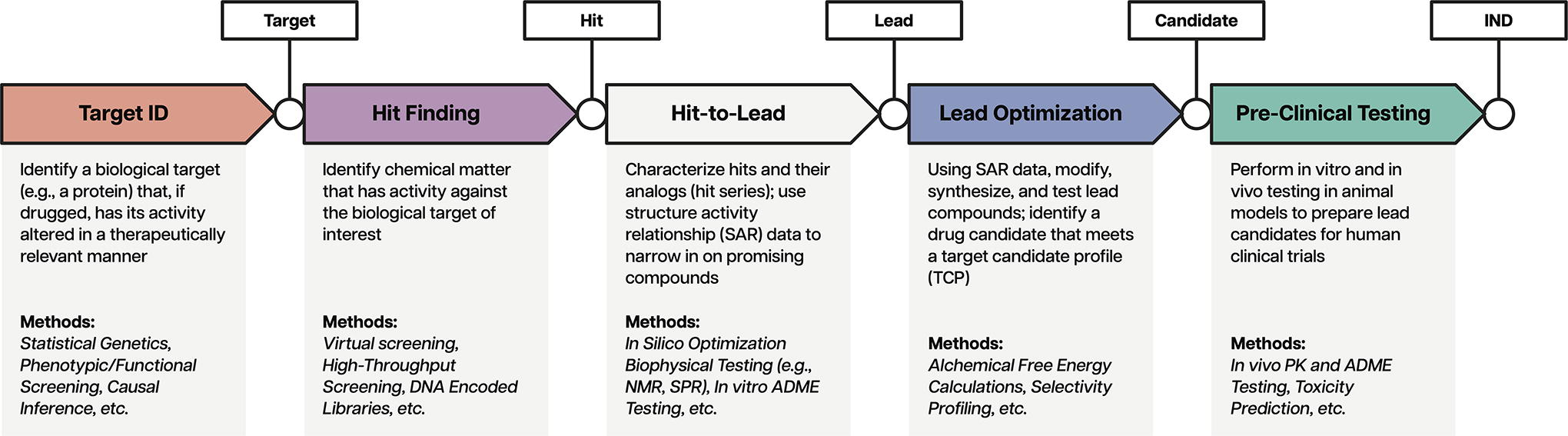
Transforming random small molecules into drugs cannot happen by serendipity. Scientists estimate there are roughly 1024 synthesizable, drug-like compounds—that is equivalent to the number of stars in the known universe.7–9 Given this astronomical search space, the pharmaceutical industry increasingly has turned to computer-aided drug design (CADD) over the past decades to assist with rapidly narrowing in on the complex objectives that define a drug discovery program.
Virtually every component of a traditional small-molecule discovery and development campaign benefits from CADD tools. As outlined in Figure 1, pharma companies can use an inexorably expanding array of computational methods across target validation, hit finding, and lead optimization. Molecular mechanics (MM) force fields (FFs) are a key technological foundation upon which many of these tools have been built.10,11 As such, MM FFs have proven their utility through dozens, if not hundreds, of successful discovery campaigns.
Force Fields Are Critical Components of Widely Used CADD Tools
Chemistry underwent a quantitative revolution in the early 20th century. Although scientists understood the relationship between a molecule’s 3D structure and its physiochemical properties, they lacked a rigorous, mathematical bridge linking the two. MM seemed to satisfy this need in a manner within reach of contemporary computing hardware.
MM is rooted in the Born–Oppenheimer approximation, which exploits the separation of timescales between fast electron relaxation and slow nuclear motion to describe a molecule’s potential energy as a function of its atoms’ 3D nuclear coordinates. 12 The mathematical framework connecting potential energy and structure is called a force field (FF), which can be used to compute useful equilibrium properties relevant to chemistry and drug discovery.
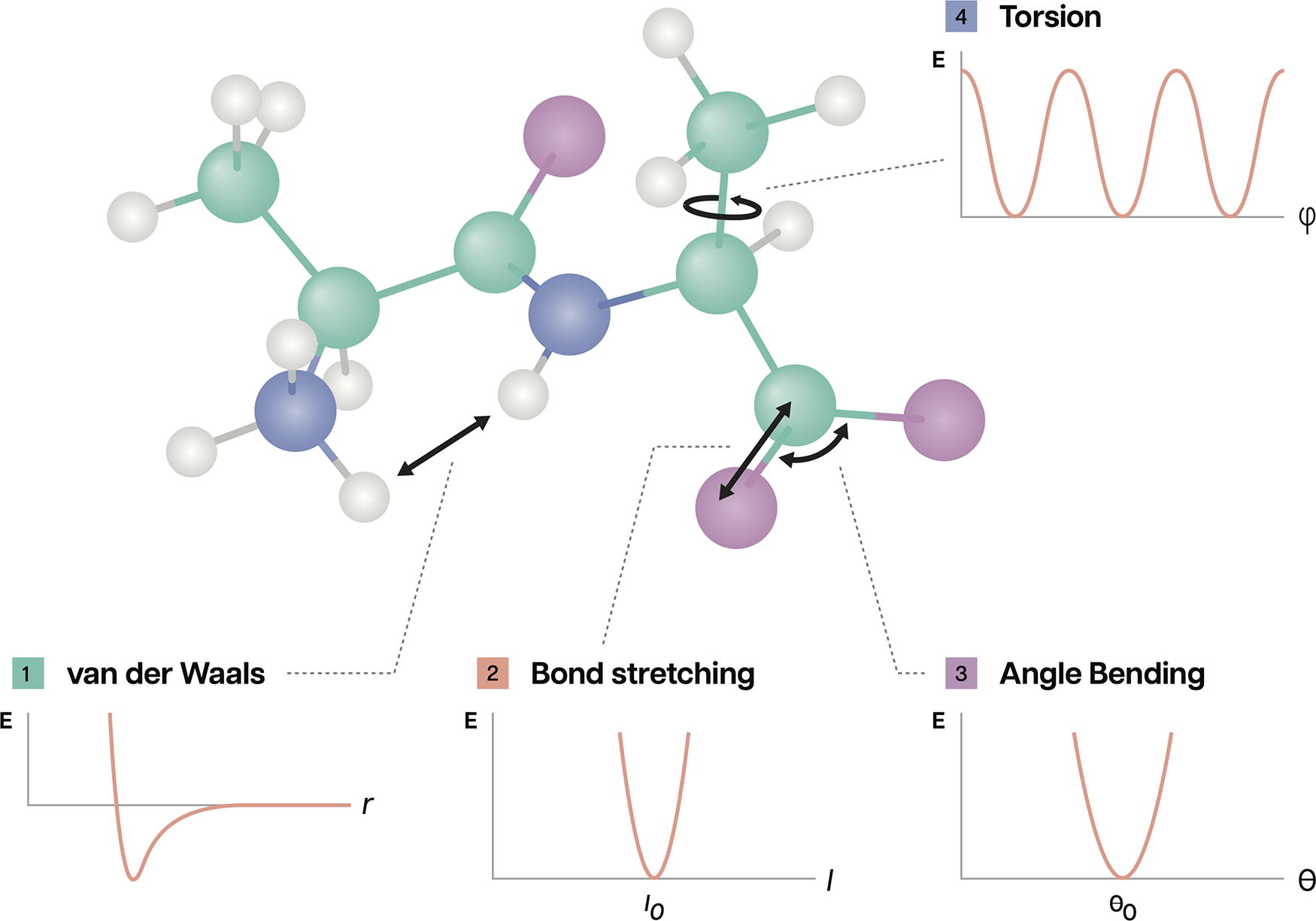
Many FFs calculate a system’s potential energy by summing terms for bonded and nonbonded interactions, as shown in Figure 2. The former considers quantum chemical valence interactions (e.g., harmonic bond stretching and angle bending), while the latter may include Van der Waals forces and electrostatics, for example. 13 These terms are expressed using a variety of functional forms—equations that yield the energy contribution(s) of a particular interaction using a set of learned parameters that must be fit to data such as quantum chemical calculations or physical property measurements. 14 Until the early 1960s, chemists carried out these laborious calculations by hand. 10
In 1961, James Hendrickson used MM to compute the conformational dynamics of hydrocarbon rings, catalyzing a new era of computational chemistry. 10 Hendrickson used an IBM 709, a mainframe capable of just 4,000 multiplication operations per second—that is roughly 1012 fewer operations per second than modern graphics processing units (GPUs).15,16 With Pandora’s box now opened, scientists raced to accelerate FFs using computational methods. The ensuing decade would see the rise of the first open-source, scientific FFs.
The Allinger laboratory published some of the first open FF software packages (e.g., MM2) optimized for small organic molecules in the 1970s. 17 These made use of minicomputers such as the PDP-11, capable of a more impressive 700,000 operations per second. 18 A decade later, the Karplus and Kollman laboratories released CHARMM and AMBER, FFs specializing in larger biomolecules such as proteins and nucleic acids.19,20
Despite matching the fastest minicomputers of the era, these frameworks did not advance the underlying functional forms. In fact, CHARMM and AMBER used simplified functional forms compared with the earlier FF models, trading accuracy for speed and simplicity. GROMOS and OPLS, which shared these simplified functional forms, became popular in the 1990s for simulating compounds in aqueous environments. 21
Schrödinger, a pioneer in molecular simulation and software development, has driven the commercialization of FFs. In the early 2000s, Schrödinger began offering DESMOND through its platform, a molecular dynamics (MD) simulation engine developed by D.E. Shaw Research, which supported Schrödinger’s commercial FF, OPLS3.22–24 As of 2024, Schrödinger’s software products are used by all the top 20 established pharma companies for small-molecule drug discovery. 25 While not required for program success, Schrödinger’s ubiquity is suggestive of the broad utility of commercial FF packages. Its success has spawned numerous other modern FF efforts, such as the open-source Open Force Field Consortium supported by an industry consortium funding model.
Molecular Dynamics Simulations Are Moving Pictures
MD simulations breathe life into static representations of molecules. 26 During an MD simulation, a FF acts on the structure of a small molecule (a ligand) or a protein–ligand complex, causing it to bend, twist, wiggle, and contort through time. Pressing “Go” on an MD simulation generates a trajectory—a movie of how the complex explores the conformational landscape imposed by the molecules’ constituent atoms. 27
Provided there are sufficiently long trajectories, MD simulations will describe the equilibrium behavior of a system. This enables chemists to gain a quantitative understanding of the system’s microscopic conformational preferences and macroscopic equilibrium physical properties (e.g., thermodynamics). Comprehensively sampling the conformational landscape is crucial for accurately capturing the behavior of complex biological systems. This is because both energetically favorable (frequently sampled) and unfavorable (rarely visited) conformations can play significant roles.
A practical example is that of DDR1—a tyrosine kinase protein whose dysregulation has been attributed to myriad cancers. Kinases are one of the largest enzyme families in humans generally involved in cell signaling. Adenosine triphosphate (ATP)-competitive inhibitors are classified by the conformation of the activation loop containing the highly conserved Asp-Phe-Gly (DFG) motif upon ligand binding. 28 Type I inhibitors stabilize the DFG-in state (Asp facing inward), while type II inhibitors stabilize the DFG-out state (Asp facing bulk solvent). Understanding the full equilibrium behavior between these states of the wild-type DDR1 protein is a critical first step for a rational inhibitor design against mutant versions of DDR1, as shown in Figure 3.
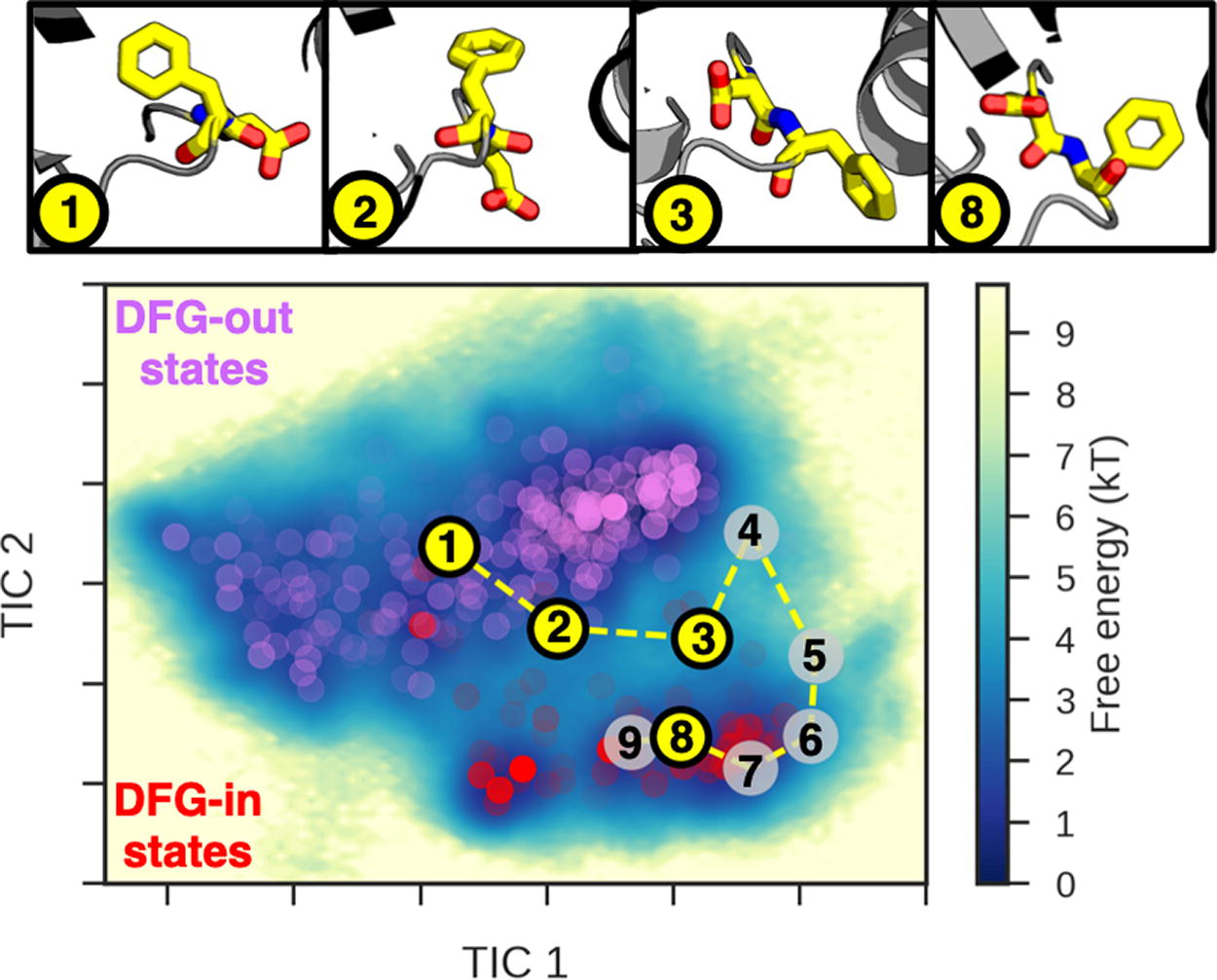
MM FFs and MD simulations are ubiquitous in industry owing to their usefulness throughout the small-molecule discovery process, from early hit finding through lead optimization. Common as they are, these techniques are replete with limitations that constrain their domain of applicability.
The central challenge is that the accuracy of a FF is generally inversely proportional to its speed. 29 Long timescale accurate simulations confer the most commercial value, but can be computationally intractable. Brief low-resolution, fast-but-inaccurate simulations are not as predictive, but can be executed at high-throughput. This trade-off governs how FFs are used in small-molecule drug discovery.
At the top of the discovery funnel, virtual screening of ultralarge chemical libraries involves checking many millions of candidate ligands against a rigid or semiflexible representation of a protein target. 30 Pruning this vast universe involves docking and scoring compounds, oftentimes using methods powered by FFs. 31 The scale of the problem necessitates speed and throughput. Therefore, the FFs used during screening are lightweight—they drop terms, ignore protein dynamics, and make a plethora of assumptions. 32 These shortcuts all contribute to the sobering fact that docking scores are not generally predictive of experimentally determined binding affinities, instead leading practitioners to focus on enrichment (biasing the selection of molecules toward hits more than random chance) and ultralarge library docking (where a small, top-scoring fraction of the docked library has a substantially higher hit rate).33–35
Since screening shrinks the number of candidate molecules drastically, scientists apply more rigorous MD methods during lead optimization. Directly predicting the relative binding affinities (Kd) of candidate ligands is a prime MD use case at this stage. 36
Protein–ligand association kinetics (kon) is often diffusion-limited and fast—once a small molecule bumps into its target, they snap together rapidly. However, it takes on the order of hours for some compounds to dissociate (koff). 37 This means that a one-hour MD trajectory might observe a single dissociation event—if one is lucky. Because binding affinity is proportional to the ratio koff/kon, comparing the affinities of any two candidate small molecules with useful statistical precision might require simulating 102–103 binding/unbinding cycles. Even with modern hardware, this MD workload may take 108–109 years of compute time.
Sidestepping this Herculean computational task required the development of alchemical free energy methods. 38 Alchemical methods decompose the lengthy, continuous process of protein–ligand binding into a series of more easily computable alchemical intermediates (e.g., steric decoupling). As the name might suggest, alchemical intermediates are oftentimes not real—they are imaginary, interpolated snapshots of molecules as they transmutate between different molecules or environments, as shown in Figure 4.
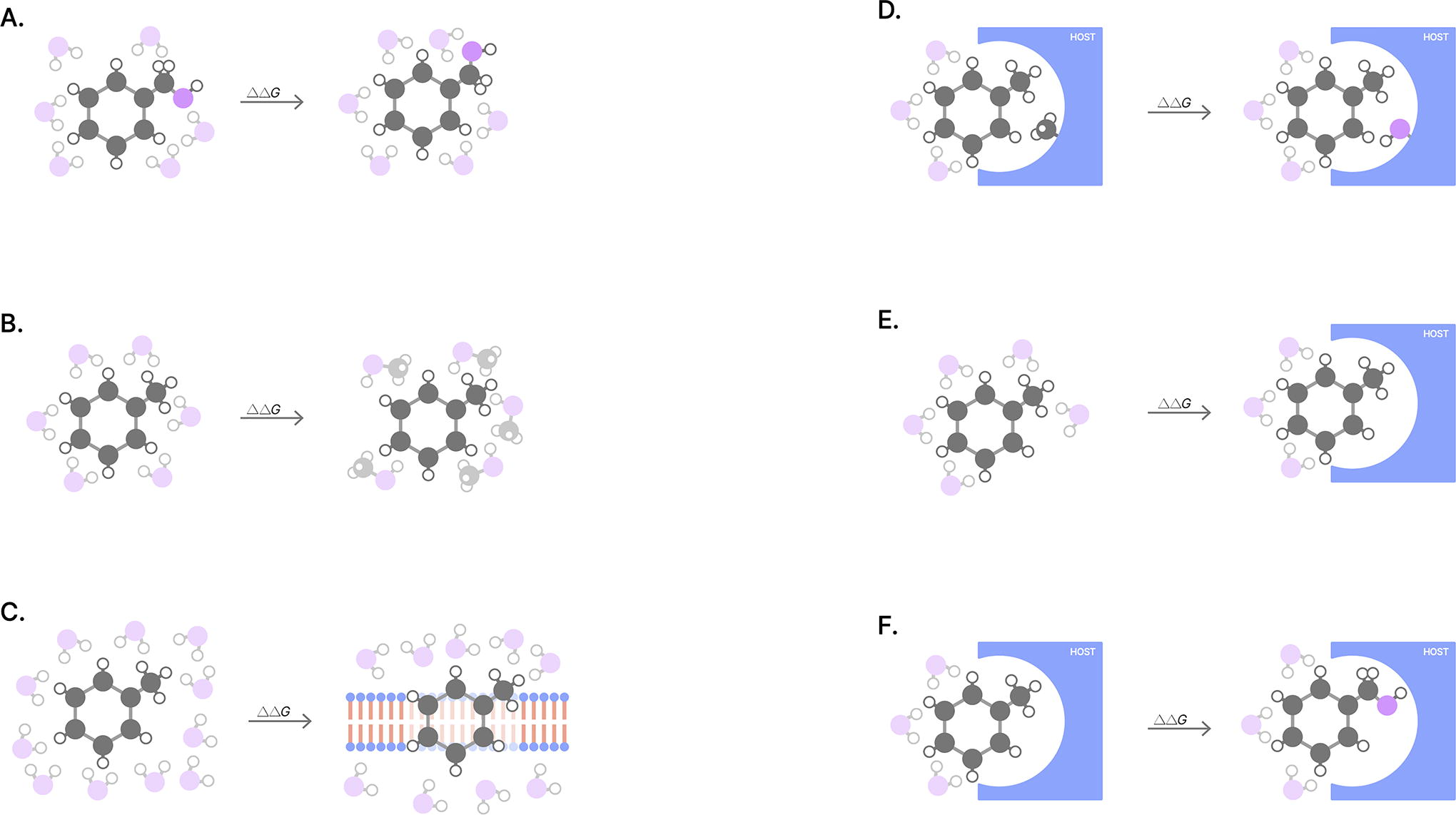
By breaking the binding affinity prediction problem into discrete, solvable pieces, alchemical free energy calculations require orders of magnitude less effort while still retaining all entropic and enthalpic contributions to binding free energy.
Unlike docking techniques, alchemical methods can produce results that more closely match experimental data, although only in well-behaved instances. 40 Many contemporary methods perform more poorly for complexes with side-chain motion, multiple ligand binding modes, conformationally dynamic proteins, dynamic protonation states, bound metals, and more. Unfortunately, these obstacles are not uncommon in industry. 41
In essence, binding affinity predictions fail for the following three main reasons: (1) the FF does a poor job modeling the physics of the system, (2) the simulation omits relevant chemical effects that modulate the chemical components in the system (e.g., protonation and tautomerization of the ligand or binding site residues), and (3) the simulation is not sampling all the relevant protein conformations—or combinations of all three.42,43
Accurately predicting binding affinity is critically valuable because it helps chemists prioritize compounds to synthesize and test—saving time and money by reducing the number of design–make–test–analyze cycles required in a discovery program.
Generative Molecular Modeling Is an Efficient Frontier
Data-driven techniques such as machine learning (ML) are becoming increasingly transformative to the life sciences. 44 ML excels at learning complex, nonlinear functions [f(x)], provided there are sufficient training data from the distribution of (x) one wishes to predict on or generate from. Chemistry is replete with nonlinear mappings. For example, medicinal chemists seek to connect a molecule’s structure to its bioactivity and other physiochemical properties—which is not straightforward.
Property prediction and de novo generative molecular design both are means to bridge the structure–activity chasm. The latter aims to build a potent molecule from scratch. While ML techniques are commonplace across both regimes, generative de novo design has become the field’s aspirational North Star.
In the ideal scenario, one could express their design objectives in a target candidate profile and directly generate a small number of candidate molecules ready for preclinical development. While this goal may seem far off, it is vital to acknowledge the breathtaking progress the field has made in recent decades.
CADD tools for generative design date back to the early 2000s, before the advent of deep learning techniques. By the early 2010s, medicinal chemists used rules-based algorithms for enumerating chemical libraries and conducting reaction-driven design—enabling the synthesis of plausible, bioactive compounds for high-throughput screening.45,46
Around the same time, researchers began leveraging early neural architectures (e.g., recurrent neural networks) to generate bioactive ligands in silico. 47 Many of these early models used text-like representations of molecules termed Simplified Molecular-Input Line-Entry System (SMILES). 48 ChEMBL is a popular, open-access repository of matched molecule–activity data containing experimental measurements for ∼2.4 million compounds, with compound identities expressed in machine-readable formats such as SMILES (Fig. 5). 50
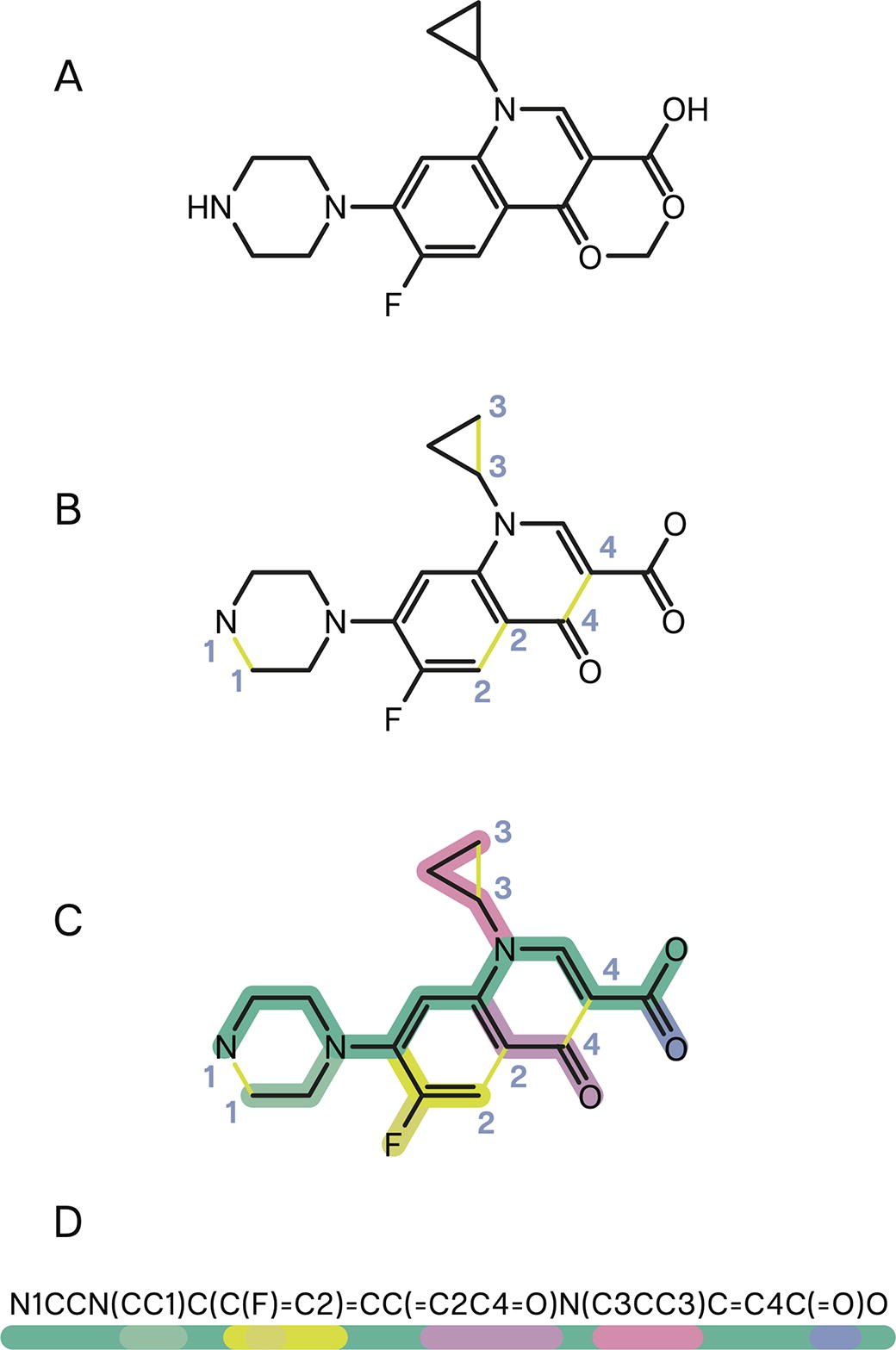
The current set of generative small-molecule ML models often incorporates structural data (e.g., co-crystal structures) in some manner. Many generative ML models are structure-implicit, meaning they are not trained on structural data. 51 Instead, these models are exposed to protein structure during inference time. Several structure-implicit models leverage goal-directed optimization, a common strategy through which the output of a generative model is docked and scored using a conventional MM software package. Recently, the number of structure-explicit models trained on structure data has increased. Unfortunately, very few of these models have been prospectively validated in the physical world. 51
A total of 50 years of systematic data curation powered by roughly $20 billion of investment created the Protein Data Bank (PDB). 52 The PDB contains >200,000 experimentally determined crystal structures with roughly 10% of these containing bound ligands—an excellent resource for training ML models, although small compared with typical ML corpora. 53 Since 1994, the Critical Assessment of Structure Prediction (CASP) competition has honed metrics of structure prediction accuracy. 54 Abundant training data and consensus performance benchmarks laid the foundation for the breathtaking results generated by Deepmind’s AlphaFold2 (AF2) in 2021. 55
Early commenters (including the organizers of CASP) hailed AF2 as the solution to the century-old protein structure prediction problem.56,57 Although not directly related to generative chemistry, AF2 fomented enormous excitement within the small-molecule discovery community—and the life sciences writ large. Would computational biologists be able to dock (or generate) molecules within digitally folded protein pockets, would this eliminate the need to obtain expensive crystal structures, and would structure become the best data representation for ML models within biology?
A few things seem clear in 2024. While AF2’s scientific and social impacts cannot be understated, AF2 was not a panacea for replacing crystal structures in small-molecule discovery programs. 58 Instead, AF2 can be considered a continuation of the trend exploiting structural data representations in biology already set in motion by the rise of geometric deep learning models—models that make use of the spatial relationships between parts of each training example. 59
Although AF2 was powered by one of the few highly curated data sets in the biological sciences (the PDB), more than data curation is needed to succeed in modeling biochemistry. To make the most out of relatively small data sets, ML models can leverage simplifying rules based on the laws of physics. For example, E(3) equivariant models understand that rotating and translating molecules in 3D space do not change their identity, properties, or conformation. 60 E(3) refers to the Euclidean group of translations, rotations, and reflections in 3D space. By understanding these physical invariances, models may make better guesses in situations where biochemical data are particularly scarce. 61
Indeed, most current state-of-the-art models for protein–ligand complex structure prediction train on structure use equivariant networks. These include NeuralPLexer2, AlphaFold-latest, and RoseTTAFold All-Atom, among others, with AlphaFold3 being a notable departure.62–65 While often orders of magnitude slower than docking, the potential for these methods to predict protein conformational changes necessary for ligand binding is tantalizing. 66
Within the traditional small-molecule discovery lexicon, generative ML models enumerate digital chemical libraries that are “virtually screened” using ML docking algorithms (e.g., DiffDock) or complex structure prediction models. 67 Recently, benchmark articles such as PoseBusters and PoseCheck have given upbeat critiques over ML’s current ability to generate physically viable ligands.68,69 PoseCheck shows how several generative algorithms propose ligand poses with unfavorable energetics, steric clashes, missing key interactions, and high strain energy.
Regardless of the model architecture or training objective, what is certain is that data are the central deciding factor for model performance and generalizability. Unlike proteins, where sequence data are abundant and inexpensive thanks to the shrinking sequencing prices, experimental small-molecule data are scarce and expensive to generate. 70 Once filtered for sequence and structural similarity, the PDB contains only a few hundred unique protein–ligand complex structures, posing challenges for ML training. 71 Real proteins are conformationally dynamic (with kinases being one of the few well-understood examples), a characteristic unrepresented in single static 3D crystal structures. 72
Directly predicting ligand binding affinity using deep learning methods also is rife with pitfalls. Binding affinity data sets such as ChEMBL and PDBbind contain significant interlaboratory variation, making simple pooling of all IC50 and K i data ill-suited for training, even when separated by measurement class.73,74 For this reason, some ML-focused companies (e.g., Terray) have begun generating enormous batches of new affinity data in a consistent, controlled manner. 75
Altogether, the generative de novo design of small molecules is an exciting frontier. Data-driven methods such as ML are blossoming, opening new opportunities within drug discovery and development. Unfortunately, extant open-access data sets of small molecules are noisy, sparse, and often incompletely annotated, highlighting the need for internal data generation.73,76,77 Physics-informed techniques and generative modeling are not diametrically opposed. In fact, emerging ML architectures are deeply connected with physical phenomena. Diffusion models, for example, are rooted in equilibrium statistical mechanics—specifically, Boltzmann distributions.78,79
The present–future intersection of ML and physics beyond MM, one that could reshape what is possible in small-molecule discovery, is the neural network potential (NNP). 80
NNPs Combine Advantages from Both Worlds
MM-based methods for affinity prediction have a generalizability advantage over current data-driven approaches. As mentioned previously, however, current MM FFs have a somewhat constrained domain of applicability where their accuracy is diminished. Previously, there seemed to be two paths for improving FF generalizability and accuracy.
The first approach involves tacking on a combinatorial explosion of mathematical terms to existing MM equations to account for specific scenarios. Generally, these are Taylor or Fourier series terms that account for complex quantum chemical multibody valence interactions. Ostensibly, this avenue could expand MM methods’ domain of applicability where currently there are limitations. Early efforts such as MMFF and other “Class II” MM FFs attempted this, but ran into difficulties with increasingly burdensome parameterization due to a combinatorial explosion of coupling terms.10,81
The second approach involves switching to quantum mechanical (QM) FFs to power MD simulations. QM frameworks are exquisitely accurate owing to their inclusion of chemical features such as electronic polarization, charge transfer, and orbital hybridization. However, QM is computationally intractable at the scales necessary to appropriately capture the statistical mechanics of protein–ligand binding in industry settings. 82 An emerging third path forward is to use NNPs.
NNPs are ML models designed to directly compute the potential energy of a molecular system as a function of atomic coordinates. 80 NNPs learn to describe physical interactions between atoms, and implicitly, electrons. Unlike MM potentials that rely on lower order functional forms, NNPs use flexible (and fast) neural forms that can capture higher order multibody interactions with greater fidelity. 80
Typically, NNPs are trained on quantum chemical calculations (e.g., forces, energies) of clusters of molecules. 83 Similar to MM FFs, NNPs may be further fit to experimental physical property data (e.g., liquid densities, heats of vaporization, and other condensed-phase properties). 84 NNPs are differentiable similar to other neural networks, meaning that their energy predictions are continuous functions of atomic position and model parameters. Why is this so important?
Much of the complexity of MM simulation engines extends from the onerous and manual process of programming efficient routines for calculating position and parameter gradients—how changes in atomic coordinates or model settings influence the system’s potential energy. Owing to their automatic differentiability and just-in-time compilation, NNPs within ML frameworks (e.g., PyTorch, JAX, and TensorFlow) can precisely and efficiently calculate gradients—accelerating simulation without compromising accuracy.
Recall that MM simulations sometimes fail to predict binding affinity due to inaccurate FFs, omission of certain chemical components (e.g., protonation states), and poor conformational sampling. NNPs promise to improve on all these aspects:
Several concurrent technological advances are driving innovation within NNPs. These models benefit from the explosion of fit-for-purpose model architectures (e.g., ANI-1). 94 Combined with ML methods to reduce the cost for accurate QM calculations, cheap CPU compute ($0.01/core-hour) makes generating large QM training sets more feasible. 95 Moreover, pressure on GPU vendors to deliver better hardware as well as the porting over of efficient software algorithms from MM codes should galvanize rapid advances in NNP performance, rapidly bringing it within reach of MM for small systems. 96
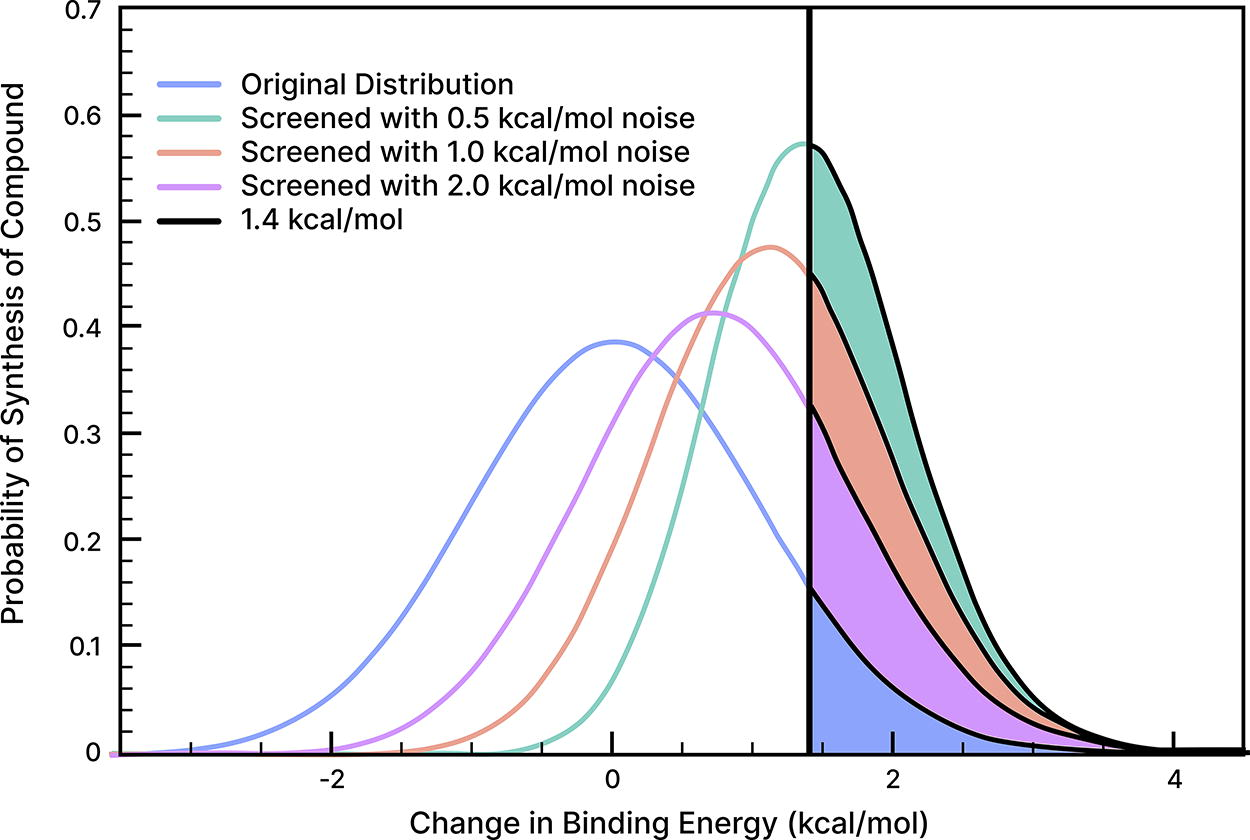
For a given discovery program, an NNP that confers near-experimental predictive accuracy on binding affinity could have profound implications. As shown in Figure 6, models capable of predicting ligand binding affinities to chemical accuracy (∼0.5 kcal/mol) could accelerate lead optimization by a factor of 8× and greatly reduce the number of compounds required for synthesis. 97
NNPs should coexist beautifully with de novo generative modeling techniques. For example, NNPs could serve as accurate simulators to provide synthetic training data for generative ML models. Alternatively, generative models that share the same fundamental architectures as NNPs could be trained on (1) the same QM data powering the NNP and (2) the data that generative models typically are trained on (e.g., crystal structures), opening the front end of the funnel to a broader universe of data.
There still are risks and unknowns associated with NNPs. As with any ML technique, overfitting is an immediate concern. The training corpus could omit relevant chemical features or be biased toward specific scenarios, creating generalizability issues. Specific models are well-known to struggle with common drug moieties such as sulfonamides, for example. Adequately mapping entire conformational landscapes during training is critical as well for NNP generalizability. There are several active learning and adversarial approaches to remedy this issue.98,99 Finally, simulations are not always stable. 100 Improper simulations can explode—rapidly deteriorating into nonphysical scenarios.
Although there is more work ahead, NNPs are a powerful evolution of the CADD toolkit for small-molecule discovery efforts. By combining the inference speed and throughput of data-driven modeling and the physical constraints conferred by rich QM and experimental data, NNPs could become the best of both worlds.
What Will the Future Look Like?
The shift to NNPs should be rapid over the next several years. At first, costly yet accurate NNPs should begin replacing expensive QM methods for many applications in small-molecule drug discovery since they are more efficient and well-suited to GPUs.85,101–103
We are already seeing the emergence of NNP/MM hybrid simulations, in which a part of the system (e.g., the ligand) is treated with more accurate NNPs—which appears to provide substantial accuracy boosts while only causing an ∼5× drag on simulation speed compared with the pure MM counterpart.96,104
At the same time, we are nearing the limit of what GPU-accelerated hardware can deliver for small biomolecular atomistic MM systems. There are certainly still efficiency gains to be made for larger systems (e.g., large membrane proteins) on current-generation hardware. For small drug-like molecules, however, there is no reason to not utilize more accurate NNPs if there is no speed penalty over MM.
We have barely scratched the surface of what the clever melding of NNP MD simulations and generative ML algorithms could do. These technologies have not been practically intertwined for long enough. There will be enormous creativity brought to bear on this problem in the coming years. Researchers will continue improving NNP inference speed through improved algorithmic innovation that exploits the simplicity of NNP atomic potentials and the rapid iteration cycles commonly found with ML. 105
The future should follow the paradigm—simulate, emulate, generate. Accurate simulations will serve as the foundation. Fast ML methods will be trained to emulate accurate simulations at a greatly reduced cost. Finally, generative ML models will gain the ability to generate physically valid compounds that meet the complex design objectives required by the small-molecule discovery teams of tomorrow.
Footnotes
Acknowledgments
JDC thanks Justin Smith (NVIDIA), Gianni de Fabritiis (Universitat Pompeu Fabra), Marcus Wieder (Open Molecular Software Foundation), Antonia S. J. S. Mey (University of Edinburgh), Peter Eastman (Stanford University), Michael Shirts (University of Colorado at Boulder), and Davis Lm Mobley (University of California at Irvine) for helpful discussions.
SB thanks Eric Dai, Zavain Dar, Adam Goulburn, and Nan Li from Dimension as well as Henri Palacci (DESRES), Nathan Frey (Prescient Design), Sam Stanton (Prescient Design), Charlie Harris (University of Cambridge), and Eric Atkins for helpful conversations and inspiration.
Author Disclosure Statement
JDC is a current member of the Scientific Advisory Board of OpenEye Scientific Software, Redesign Science, Ventus Therapeutics, and Interline Therapeutics, and has equity interests in Redesign Science and Interline Therapeutics. The Chodera laboratory receives or has received funding from multiple sources, including the National Institutes of Health, the National Science Foundation, the Parker Institute for Cancer Immunotherapy, Relay Therapeutics, Entasis Therapeutics, Silicon Therapeutics, EMD Serono (Merck KGaA), AstraZeneca, Vir Biotechnology, Bayer, XtalPi, Interline Therapeutics, the Molecular Sciences Software Institute, the Starr Cancer Consortium, the Open Force Field Consortium, Cycle for Survival, a Louis V. Gerstner Young Investigator Award, and the Sloan Kettering Institute. A complete funding history for the Chodera lab can be found at http://choderalab.org/funding.
SB is a current member of Dimension, a venture capital firm that invests at the intersection of technology and the life sciences. Dimension has financial interests in companies developing small molecule therapeutics including Enveda Biosciences, Kimia Therapeutics, and Monte Rosa Therapeutics.
Funding Information
JDC acknowledges financial support from the Sloan Kettering Institute.