
Abstract
Design of guide RNA (gRNA) with high efficiency and specificity is vital for successful application of the CRISPR gene editing technology. Although many machine learning (ML) and deep learning (DL)-based tools have been developed to predict gRNA activities, a systematic and unbiased evaluation of their predictive performance is still needed. Here, we provide a brief overview of in silico tools for CRISPR design and assess the CRISPR datasets and statistical metrics used for evaluating model performance. We benchmark seven ML and DL-based CRISPR-Cas9 editing efficiency prediction tools across nine CRISPR datasets covering six cell types and three species. The DL models CRISPRon and DeepHF outperform the other models exhibiting greater accuracy and higher Spearman correlation coefficient across multiple datasets. We compile all CRISPR datasets and in silico prediction tools into a GuideNet resource web portal, aiming to facilitate and streamline the sharing of CRISPR datasets. Furthermore, we summarize features affecting CRISPR gene editing activity, providing important insights into model performance and the further development of more accurate CRISPR prediction models.
Introduction
The CRISPR-Cas9 system, following the advent of transcription activator-like effector nucleases and zinc-finger nucleases, is the third-generation programmable gene-editing technique. Using a single guide RNA (gRNA) and the Cas9 protein, the CRISPR system is capable of editing virtually all genetic loci. Since its discovery for programmable gene editing, the CRISPR-Cas9 from Streptococcus pyogenes Cas9 ([SpCas9], hereafter referred to Cas9) has been the most broadly studied and used system. 1 The gRNA is an engineered chimeric RNA comprising the CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). The Cas9 protein forms a ribonucleoprotein complex with the gRNA and is transported from the cytoplasm to the cell nucleus, directed by the nuclear localization signal of the Cas9 protein (Fig. 1). The Cas9-gRNA complex binds to the target DNA through specific interactions with the protospacer adjacent motif (PAM) sequence (NGG for SpCas9) and gRNA/DNA complementary base-pairing. 2 The formation of an R-loop activates Cas9 DNA endonuclease activity, which induces a DNA double-strand break (DSB) at the targeted genomic locus, 3 and triggers the cellular DSB repair mechanisms. In mammalian cells, DSBs are repaired via two major DNA repair pathways: homology-directed repair (HDR) and classical or alternative nonhomologous end joining (NHEJ).4–6 Repaired DNA, characterized by indels or desired mutations, leads to the generation of modified gene products. Due to its simplicity, this technique has been rapidly and widely adopted by the scientific community, with the potential to decipher genetic functions and most importantly provide hope for curing many genetic disorders.7,8 However, an undeniable challenge in the effective and safe application of the CRISPR system is the necessity to design gRNAs with high-efficiency, as well as minimizing off-target effects (specificity). 9 To overcome this, computational tools have been developed to facilitate gRNA design, especially those based on machine learning (ML) and deep learning (DL)-based methods.10–12 Although many CRISPR design tools have been developed and exhibit good predictive performance for specific cell types or species during claimed model testing, their performance and suitable experimental scenarios in practical applications remain unclear. Therefore, a systematic and unbiased evaluation of prediction tools in the context of different cell types is still needed. Here, we thoroughly evaluate and establish a benchmarking pipeline used for CRISPR activity prediction evaluations. Using this tailored process, we summarized nine CRISPR gene editing datasets to perform objective, reliable comparisons across seven representative tools. We compile all of these into an online web portal (GuideNet) to streamline the access to CRISPR datasets and design tools (https://dream.au.dk/tools-and-resources/guidenet). Finally, we investigate the significant factors affecting gRNA efficiency, providing valuable insights for further improvement and development of CRISPR on-target prediction tools.
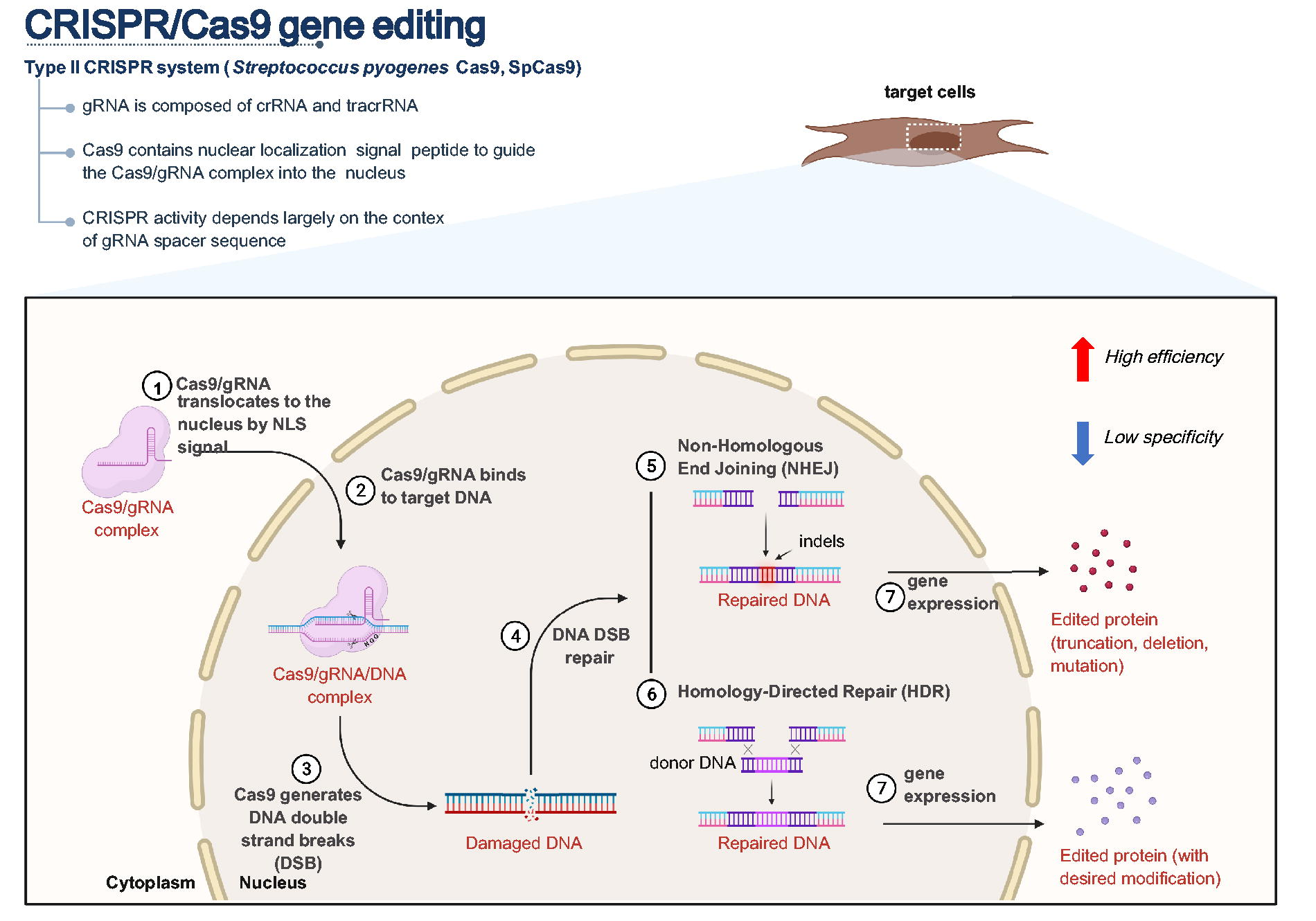
Schematic diagram of Type II CRISPR-SpCas9 genome editing system. The illustrative figure shows the CRISPR-SpCas9 system-mediated gene editing in mammalian cells. Seven key steps are highlighted. The guide RNA (gRNA) is a chimeric gRNA consisting of CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA). In the gene editing process, the Cas9-gRNA complex is translocated from the cytoplasm into the nucleus (step 01), followed by binding to the on-target DNA site (step 02). The presence of an NGG protospacer adjacent motif (PAM) is critical for the target search and the formation of a stable R-loop structure composing Cas9, gRNA, and target DNA. Double strand break (DSB) is then introduced at the target site (step 03), which will be repaired by the endogenous DNA repair mechanisms (step 04). Two major DSB repair pathways are illustrated: non-homologous end joining ([NHEJ], step 05) and homology-directed repair (HDR), step 06), which will introduce indels or intended modifications at the DSB respectively. Upon gene expression, the impact of CRISPR-introduced edits will be seen e.g., at the protein level (step 07). This figure is generated with BioRender.com with a license for publication.
An Overview of Tools for in Silico CRISPR Design
Many tools for pre-experimental CRISPR gRNA design have been developed, which can be further categorized into those aimed at enhancing on-target activity, 13 and minimizing off-target effects. 14 A non-exhaustive list of CRISPR design tools that take these roles into account is summarized and provided in the integrated GuideNet web portal.
Here we provide a brief overview of these tools, specifically for on-target prediction, according to a conventional categorization system 9 : (1) Alignment-based approach, where gRNAs are aligned within the provided genome exclusively by identifying a PAM; (2) Hypothesis-driven approach, where gRNAs are aligned and evaluated based on a hypothesis that took into account specific genome context factors contributing to CRISPR on-target activity; and (3) Learning-based approach (including ML- and DL-based tools), where gRNAs undergo scoring and prediction through a pre-trained model.
The conventional selection of CRISPR gRNAs for a target DNA or gene is based on PAM alignment. Cas-Designer, 15 is one such representative tool, previously recommended by Liu et al. 16 This tool, which also integrates the off-target tool Cas-OFFinder, 17 provides a rapid retrieval of gRNA spacer sequences and potential off-targets information. Since the algorithm depends on only sequences, the tool provides users the flexibility to submit the sequence of a target locus (e.g., target exon, maximum length = 1,000-nt), and to select one of 44 PAM types covering different Cas enzymes/variants and span over 350 genomes for in silico gRNA design. The design outcomes are conveniently redirected to the Ensemble genome browser for further inspection.
For the hypothesis-driven approach, these tools utilize empirically derived rules to rank gRNAs based on their predicted gRNA features. E-CRISP, 18 a tool recommended by a previous evaluative study, 19 enables rapid iterations through gRNA design and parameter selection. It facilitates the design of gRNAs targets, ranging from single exons to entire genomes, across multiple species and applications. The output includes a novel ‘SAE (Specificity, Annotation, Efficacy) score,’ where the ‘efficacy score’ is determined by gRNA sequence features such as the GC content, the presence of a G preceding the sequence, micro-homology characteristics, as well as efficiency scoring schemes developed by Doench et al. (‘Rule Set 1’) 20 and Xu et al. (‘Spacer Scoring for CRISPR’). 13 Several other integrated gRNA design platforms such as CHOPCHOP, 21 and CRISPick, 20 that also incorporate existing on-target and off-target prediction algorithms were not included in our comparative analysis but were considered in our tool recommendation (discussed below).
Here we focus on the third category of CRISPR design tools, learning-based tools, since the first category of tools merely provided alignment results without scores to rank them, and the optimal model in the second category (E-CRISP) 18 has been confirmed to have weaker performance than learning-based models in previous research. 11 These tools can be further classified into two categories, ML or DL-based strategies. ML tools typically need the pre-generation of multiple features from a data set originated from experiments. A highly correlated, non-redundant feature set determined by empirical methods has crucial impacts on model prediction accuracy and contributes to the variance between different tools.22–24 These features can be used as input to a (supervised) learning algorithm using the efficiencies on known gRNAs as the training set. Subsequently, the trained model can be applied to the efficiencies for selecting gRNAs in new experiments.
Several CRISPR on-target prediction tools with tailored features have been developed. Among these tools, we selected four ML-based tools for further benchmarking analysis, including Azimuth, 25 sgDesigner, 26 Rule set3, 27 and CRISPRedict. 28 The Azimuth, 25 (referring to the latest version, Azimuth 2.0) is a state-of-the-art tool developed by an ensemble learning method, specially trained by the gradient boosting regression tree algorithm. The training data was based on gRNA efficiency readouts from lentivirus library screens targeting human and murine genes, which were generated through flow cytometry and resistance-based experiments, respectively. The sgDesigner tool, 26 developed by Hiranniramol et al., utilized a quantitative gRNA experimental dataset consisting of 12,472 oligonucleotide sequences delivered by lentivirus transduction. The study distinguished efficient and inefficient gRNA groups based on a χ2 test and employed sequence and structural features, such as the gRNA secondary structure, for logistic regression model training. The sgDesigner tool served as an alternative updated version to the WU-CRISPR tool 29 (recommended in the study by Haeussler et al. 10 ). The rule set3 tool used 46,526 unique gRNA data from seven datasets, encompassing two tracrRNA variants: Hsu tracrRNA, 30 and the modified Chen tracrRNA.31,32 It trained a gradient boosting regressor to predict z-scored gRNA activity. Features were extracted from the sequence context and target site. The model was ultimately evaluated using a new in-house screening dataset. Recently, Konstantakos et al. reported a novel interpretable gRNA efficiency prediction tool along with a corresponding web named CRISPRedict, 28 that used both data from lentivirus library screens, 33 and single gRNA experiments. 10 This model included related features, e.g., both overall and site-specific nucleotide compositions, as well as variables reflecting gRNA structural characteristics. Through a multi-step feature selection analysis, they obtained a minimum subset of relevant features to train four distinct models for the corresponding four tasks. The tool CRISPRedict, despite the simplicity of their used algorithm, enabled interpretable efficiency prediction with comparable predictive performance and provides two models based on in vivo or in vitro gRNA expression systems enabling the user to tailor the gRNA design to their experimental setup.
In addition to ML-based CRISPR design tools, the DL-based approach has been successfully applied to improve CRISPR design. Here we focused on three DL-based tools and benchmarked their performance: DeepHF, 34 DeepSpcas9, 33 and CRISPRon. 24 All three DL-based tools use experimental data from lentivirus libraries consisting of gRNA-target site pairs where a surrogate target site is integrated downstream of the gRNA expression cassette. These libraries can be used to directly quantify indel frequencies arising from CRISPR-Cas9 editing. Unlike functional readout data from flow cytometry or resistance-based (phenotype) experiments, this surrogate target site approach is not biased against gRNAs that induce non-frameshift mutations. The DeepHF tool employs a bidirectional long short-term memory neural network (one type of the recurrent neural networks [RNN], Figure 2) to extract one-hot-encoded gRNA features, which are trained on a dataset of ∼50,000 gRNA and integrated with several biological features to construct the final model. They also developed a model based on data from T7 promoter transcribed gRNAs, 10 and applied fine-tuning strategies to enhance the predictive accuracy under different expression conditions (the U6 or T7 promoters). The DeepSpCas9 model used a training dataset of 12,832 target-specific gRNAs to develop the model. This tool considered the 30-nt sequential features and employed a convolutional neural network ([CNN], Fig. 2) architecture to learn gRNAs sequence information to predict gRNA efficiency. Previously, we developed the DL-based CRISPRon tool which exhibited significantly improved performance. This tool also used the 30-nt one-hot-encoding sequences as input and applied multi-channels CNN feature extraction for training with experimentally measured CRISPR activity datasets. Particularly, by incorporating in-house data and Kim et al. dataset, 33 to expand the training sample, CRISPRon adopted the gRNA similarity-based data partition method to generate fully independent test sets for a fair benchmarking of tools. Also, it introduced binding energy features, which differed from the first two tools, and finally achieved optimal predictive performance by cross-validation training of models. Collectively, these DL-based tools mainly leveraged neural networks, such as representative CNN and RNN architectures, to train prediction models, eliminating the need for a labor-intensive feature engineering process which was necessary for conventional ML-based models. The DL approach was able to recognize the hidden patterns within the data and showed significant advantages than ML models in understanding the features affecting the gRNA on-target activities.
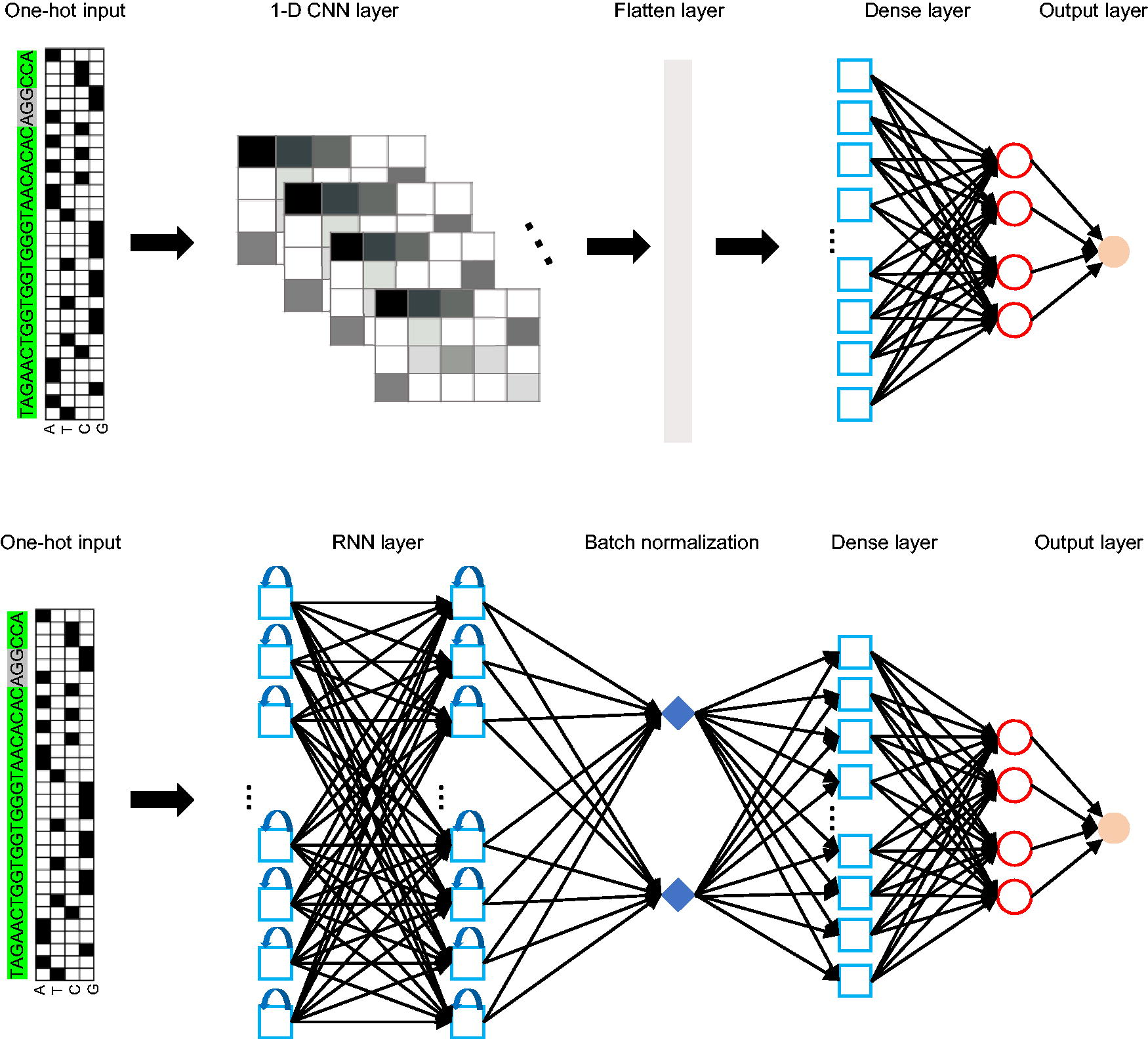
Typical deep learning network (CNN, and RNN architectures) used for CRISPR prediction modeling. For each network, the encoded matrix containing the gRNA–DNA sequence pair information is used as input. CNN, convolutional neural network; RNN, recurrent neural networks; gRNA, guide RNA.
It is important to note that several gRNA design tools, while they not providing comparable accuracy in gRNA efficiency predictions as ML/DL tools, have been developed for use in specific organisms and may prove beneficial in specific scenarios. For instance, the CRISPR-P tool, 35 (http://crispr.hzau.edu.cn/CRISPR2) was exclusively designed for gRNA prediction in plant genomes, EuPaGDT, 36 was tailored for designing CRISPR gRNAs of eukaryotic pathogens, and the flyCRISPR, 37 tool was developed for gene editing in the model system Drosophila melanogaster.
Datasets for CRISPR Modeling and Evaluation
Experimentally generated CRISPR editing datasets are vital for the generation of CRISPR prediction models and evaluation of performance. While several CRISPR activity datasets have been generated and successfully used for developing models, a challenge remains in the comprehensive and unbiased assessment of tools using identical and independent test datasets.11,38 Here we summarize nine large datasets, including seven measured with loss of gene function assays (by Doench, 25 Hart, 39 Labuhn, 40 Koike, 41 Gagnon, 42 Varshney, 43 and Telboul, 10 ) and two based on deep sequencing-based indel quantification (Xiang, 24 and Chen, 44 ). Free and direct access to these benchmarking datasets is provided in the online GuideNet database (https://dream.au.dk/tools-and-resources/guidenet).
For an overview of the CRISPR-Cas9 on-target activity prediction tools, readers are referred to the recent reviews.11,45 According to two previous comparative studies,10,11 Azimuth, 25 and WU-CRISPR, 29 were reported to exhibit the best performance among seven CRISPR on-target prediction tools for U6 promoter-based assays. Regarding DL-based tools, DeepSpCas9, 33 and DeepHF, 46 demonstrated good performance across U6 promoter-based gRNA expression data in humans and mice, and T7 promoter-based setting in zebrafish. Building upon these findings, we selected seven representative tools for benchmarking analysis using nine compiled datasets, including four ML models: Azimuth, 25 sgDesigner (an updated version of WU-CRISPR), 26 Rule set3, 27 and CRISPRedict, 28 and three DL models: DeepHF, 34 DeepSpcas9, 33 and CRISPRon. 24 CRISPRon and CRISPRedict tools were included in our benchmark due to their demonstrated predictive performance and model advantages. Moreover, certain tools were reported to demonstrate satisfactory performance in the model training stage but exhibited relatively worse performance than expected on independent test sets.10,47 To mitigate this training bias and ensure an impartial evaluation, we excluded the intersected gRNAs between the training sets of each individual tool and our benchmarking datasets. The Xiang dataset, which was employed to develop the CRISPRon model, was excluded from the comparison involving CRISPRon and other models. Similarly, the Doench dataset was excluded from the comparison involving Azimuth and other models. Both Xiang and Doench datasets were also excluded for comparisons related to the Rule set3. To ensure a fair benchmark, we further examine the overlapped gRNAs present in our test sets but trained by the evaluated tools within the same cell line. There are 270 identical gRNAs between the Xiang dataset and the DeepHF training dataset, which were removed from the benchmarking (Supplementary Fig. S1).
Evaluation of Model Performance
For an unbiased justification of model performance, the selection of comparison criteria and outcome-based statistical tests is crucial. Therefore, we summarized the three comparison principles and evaluation matrix as follows:
First, we introduced an average model, which calculates predictions by averaging the scores from all tools. Second, we selected two quantitative metrics, Spearman correlation and normalized discounted cumulative gain (nDCG, further information is shown in Supplementary Data S4), both ranging from (0, 1), to measure the disparities between the predicted gRNA ranking lists output by the various tools and the gRNA lists of the benchmark test set. The Spearman correlation was selected due to its lack of assumptions about the data distribution and its increased robustness to outliers, rendering it more suitable for quantitating the overall difference of prediction results compared with the Pearson correlation. 48 Employing the nDCG criterion allowed for a more focused examination of highly efficient gRNA prediction, rather than inefficient gRNAs. This metric is now effectively applied to evaluate the performance of methods in returning the top gRNAs per gene knockout. Similar evaluation standards have been utilized previously in other evaluative studies.11,19 Third, to determine the statistical significance of the evaluation results, we selected Steiger’s test and Friedman’s test, which were separately applied to the comparison of Spearman correlation as well as nDCG values. Collectively, the entire evaluation process is illustrated in Figure 3. The evaluation process enables a robust, unbiased, and systematic comparison between CRISPR-Cas9 on-target prediction tools, effectively avoiding the potential pitfalls of comparison results.
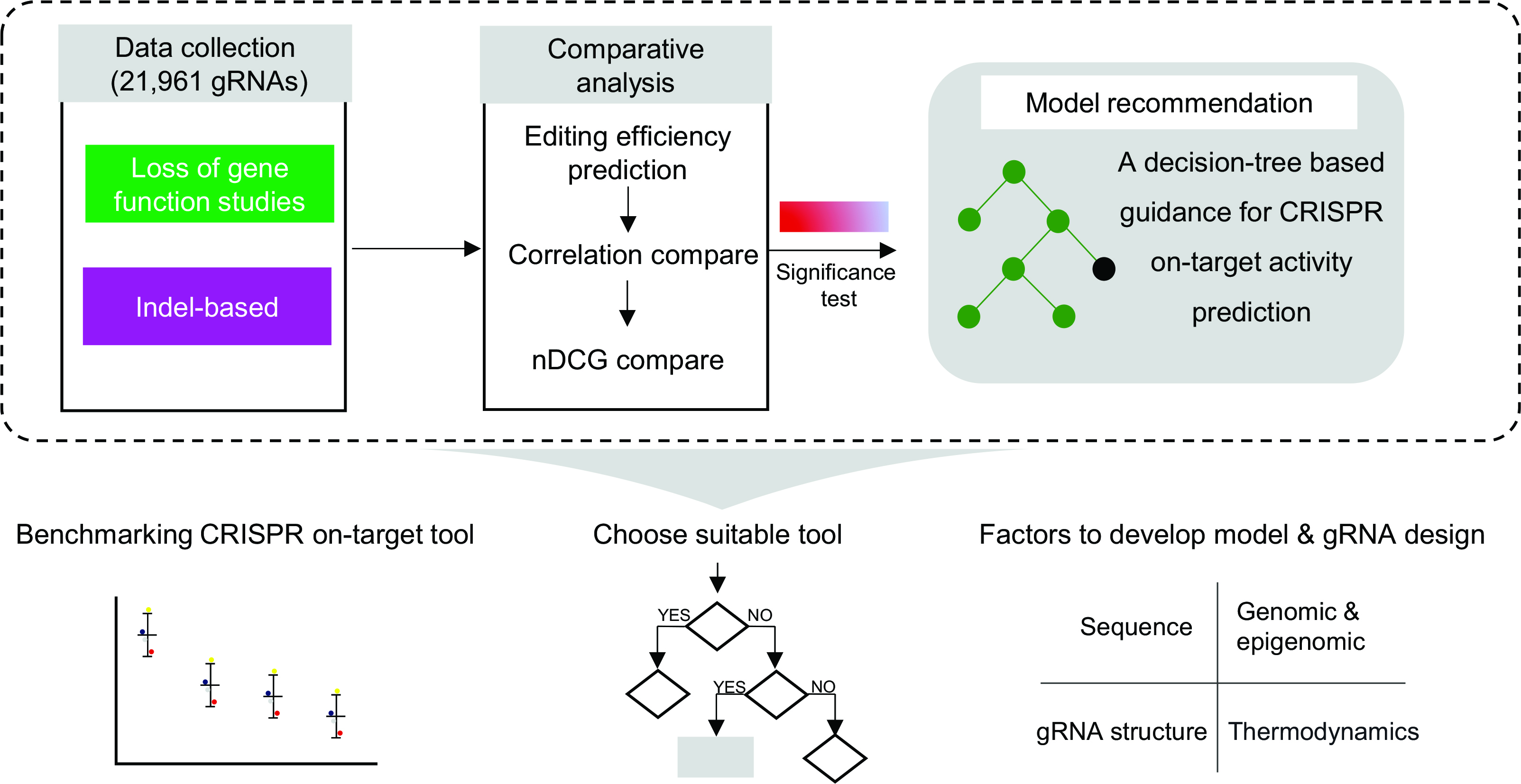
Flow chart presenting the inter-models comparison process. First, we collected benchmark test sets and established a tailored comparison pipeline. Next, following in-depth benchmarking analysis, a decision tree-based practical guide was generated for pre-experimental tool choices. Finally, a retrospective analysis of key features was performed, providing insights for the future development of tools.
Comparative Analysis of CRISPR on-Target Prediction Tools
Consistent with previous findings, considerable variations were found in the performance between the datasets for each model by correlation analysis (Fig. 4), emphasizing the necessity of considering contexts (e.g., cell types, activity determination methods, expression vectors) for CRISPR activity prediction. For instance, the DeepHF model obtained a Spearman correlation of 0.751 with the Xiang dataset, while a correlation of only 0.229 was observed with the Labuhn dataset. Despite relying on a simple linear regression model, the ML-based CRISPRedict model displayed a relatively narrow predictive range across all datasets, showing robust generalization capability. Overall, the DL-based tool CRISPRon and the ML-based tool Rule set3 exhibited better performances than the other tools in U6 gRNAs, with CRISPRon demonstrating higher average predictive accuracy, followed by the Rule set3, DeepSpcas9, CRISPRedict, and Azimuth. Due to the difference in the expressed system, DeepHF has a higher predictive performance for T7 gRNAs.
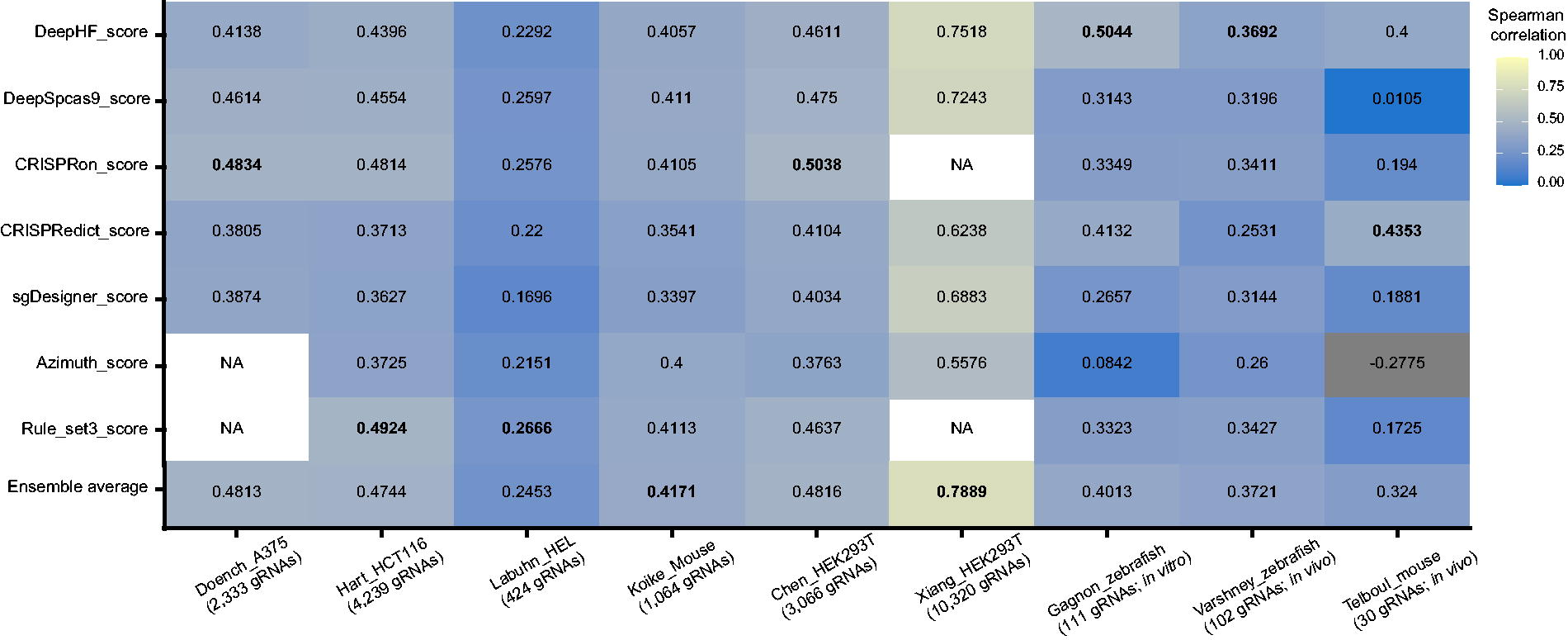
Heatmap of Spearman rank correlation coefficients between efficiency scores and data sets. The number of gRNAs, cell type, and species are shown at the bottom. Correlation scores are shown along the horizontal axis, data sets are on the vertical. Correlations of an algorithm against its own training data set were not provided, and shown in white and marked as NA. The highest scores are in bold. Corresponding results from Steiger’s significance test can be found in Supplementary Tables S1 and S2. gRNA, guide RNA.
To further refine the benchmarking analysis, we conducted additional comparative analysis focusing on CRISPRon and DeepHF, both of which provided user-friendly computational platforms. The Xiang dataset which was used for training the CRISPRon model was excluded from the benchmarking analysis. CRISPRon exhibited optimal prediction performance in two out of five independent test sets and showed a comparable performance to the Rule set3 on the Hart, Labuhn, and Koike datasets. Steiger’s test confirmed that the performance differences between CRISPRon and Rule set3 were not statistically significant. Compared to the other tools, the performance of CRISPRon was significantly better across most testing datasets (Steiger test, p < 0.05) (Fig. 4, Supplementary Table S1). The Labuhn dataset contained a limited number of gRNAs (n = 424), making it challenging to draw any convincing conclusion on model performance. On the contrary, the DeepHF model outperformed other tools in T7 gRNAs in zebrafish and demonstrated prediction performance comparable to CRISPRedict for in vivo mouse datasets. Unfortunately, Steiger’s test merely shows individual significant results, likely due to the limited sample size (Supplementary Table S2).
We also evaluated the ranking ability of these models. As measured by nDCG@10 and nDCG@20, the comparison focuses on the top 10 and top 20 high-effective gRNAs for each knock-out gene in the Doench dataset. As shown in Figure 5, our findings indicated that the top-ranking performance of all tools is exceptionally high and similar, with average nDCG values for all tools exceeding 0.79. The sgDesigner model, which is based on a stacking ensemble learning strategy, demonstrated a ‘slight advantage’ in both the top 10 and top 20 gRNAs. However, after careful examination, we found that the predicted scores by sgDesigner are highly redundant and possibly cannot reflect the real distinction of top-ranking gRNAs (see the predictions of sgDesigner in descending order in Supplementary Data S3). This limitation might be attributed to the unique data processing during model training, where the original study selected 1,319 high-effective or low-effective gRNAs to perform modeling while disregarding those with intermediate efficiency.
26
This strategy potentially emphasized the most predictive features for the training model, but obscured the distinction of top-ranking gRNAs. Consequently, these model discrepancies from nDCG@10 to nDCG@20 narrowed, due to the great generalization of three DL models (entire range from 0.052 to 0.043). Friedman’s significance test showed that there was no significantly different performance in the ranking abilities of these five tools (Supplementary Fig. S2). To further examine whether there was a significant difference between any two tools, we performed a post hoc Nemenyi test and yielded a critical difference (
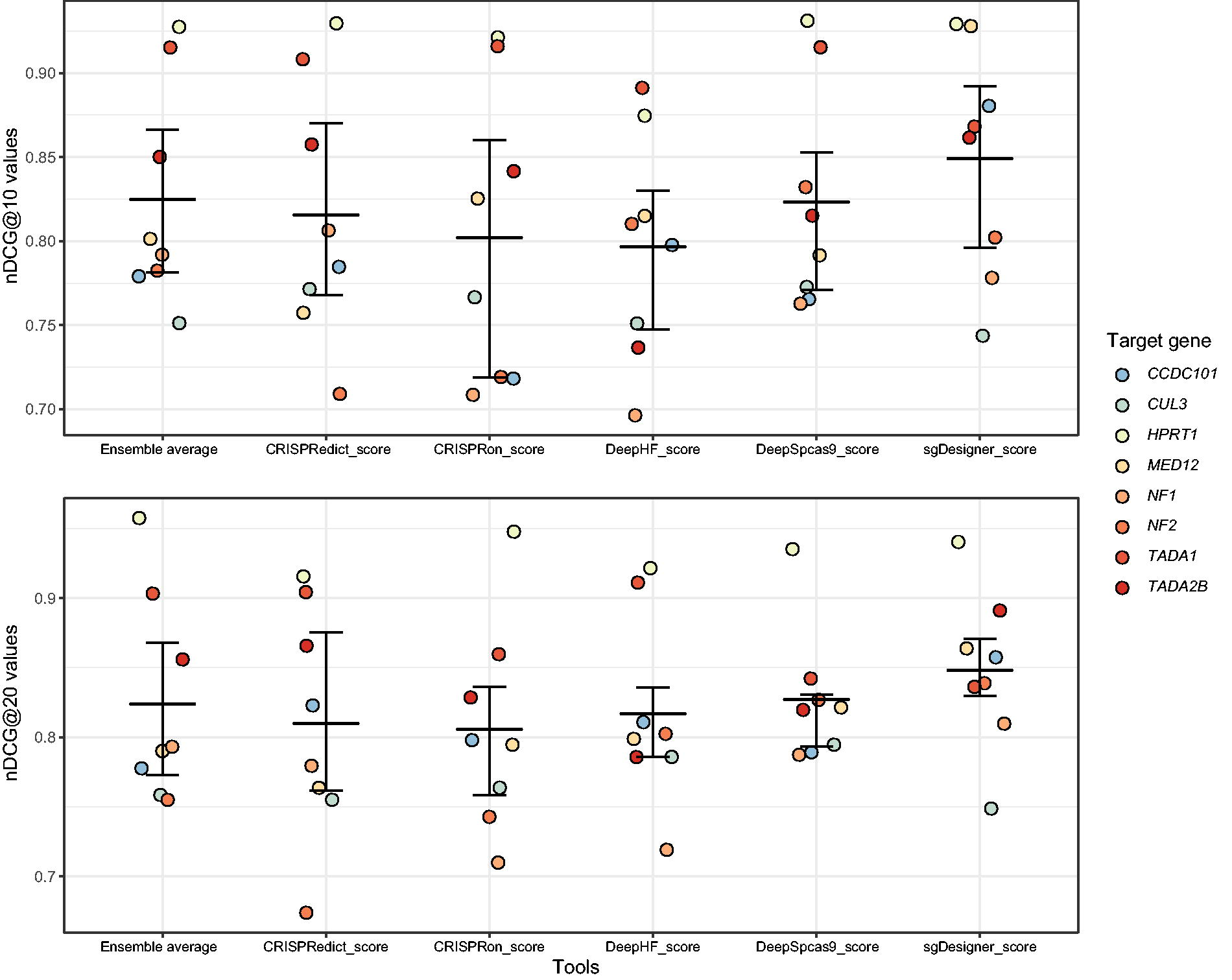
Jitter plot of the normalized discounted cumulative gain (nDCG) comparison. The nDCG values for the top 10 and 20 gRNAs per gene in the Doench knockout dataset are displayed at the top and bottom, respectively. Azimuth and Rule set3 models are excluded from this analysis due to this dataset trained by it. The line represents the 25th percentile, mean, and 75th percentile of the nDCG coefficients, from top to bottom. Each point represents an individual gene. Corresponding results from Fridman’s significance test can be found in Supplementary Figure S2.
ML tools exhibit comparable performance or even superior to DL models for some datasets, especially Rule set3 or in the nDCG comparison. This suggests that the selection of training-methods might need to be tailored to the data types. Notably, the CRISPR indel datasets generated by high-throughput sequencing, such as the datasets from Xiang and Chen, should be prioritized and generated. This format of data was found to consistently retain the robust predictive performance across all tools (Fig. 4). By utilizing massive, standardized high-throughput indel quantification data, it is possible to promptly accomplish the modeling process, bypassing the analysis of phenotypic outcomes, as exemplified by the development of tools DeepHF and CRISPRon. This data-driven manner has already been recommended by other studies,24,49 which emphasized the necessity of data integration, quantitative rules, and effective feature extraction for CRISPR modeling.
Apart from the seven ML and DL on-target prediction tools compared here, over 40 tools have been developed to streamline in silico CRISPR design. We provided a non-exhaustive list of these CRISPR design tools on the GuideNet resource website. This enables us to provide a concise and practical pre-experimental gRNA design guide by integrating our above benchmarking results and the characteristics of other design-related platforms. The Cas-Designer, 15 supported the determination of gRNAs with over 20 nucleases across various species and high processing speed. However, the model did not provide the prediction efficiency values. Regarding the species-specific CRISPR design, tools such as CRISPR-P, 35 EuPaGDT, 36 and flyCRISPR, 37 were exclusively developed for gRNA design in the genome of plants, pathogens, and drosophila, respectively. When a specific model for a given species or cell type is lacking, generalizable and relatively accurate DL-based models such as CRISPRon and DeepHF are worth recommending.
Features Affecting CRISPR Activity
As mentioned earlier, many predictive models have been developed to predict gRNA activities, which rely on learning the sequence or specific features of target sites. Understanding the impact of these features will aid the improvement and better understanding of CRISPR-Cas9 gene editing tools.13,20,24,25,34,50 Therefore, as suggested previously,16,51–53 these features can be grouped into four categories: (1) the intrinsic sequence characteristics including PAM and spacer sequence, (2) the gRNA secondary structure, (3) genetic or epigenetic factors, and (4) thermodynamic parameters in DNA-RNA binding processes. Herein, we summarize and discuss the “genomic context” in which the Cas9-gRNA complex interacts, while effects related to biochemical cleavage reactions, such as Cas9-gRNA complex concentration, 30 ion concentration, 54 delivery strategy,55,56 are not included in this review. A summary of these essential and consistently effective factors is provided below and outlined in Figure 6.
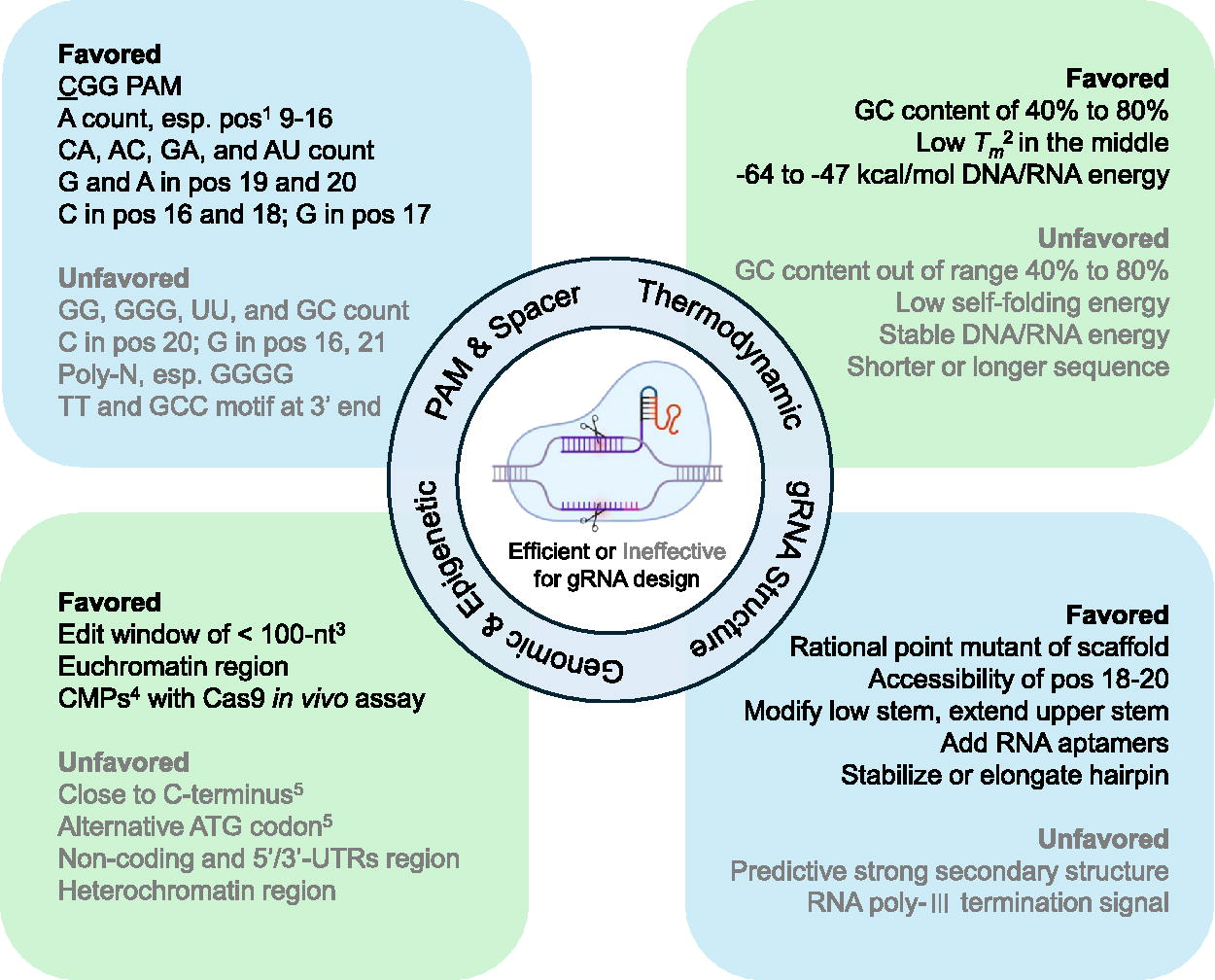
Schematic diagram of key factors that impact CRISPR on-target activity. Entire factors are categorized into four sections. Factors listed in black font indicate those that favor effective gRNA design, while factors in gray font indicate those that are less favorable. Pos., position; Tm, melting temperature; Edit window of <100-nt, applicable to CRISPR activation or CRISPR inference experiments; CMPs, chromatin-modulating peptides; Close to C-terminus, applicable to CRISPR assays related to the alteration of gene expression or gene knockout. Note that position refers to counting the distal PAM.
The PAM Sequence
CRISPR gene editing is dependent on the presence of a PAM sequence and the efficiency and specificity are strongly influenced by changes in the PAM sequence. 1 For SpCas9, the canonical PAM sequence is an NGG (N = A, C, G, or T) located at the 3′ end of the target (protospacer) site. Previous studies have found that the PAM sequence triggers Cas9 endonuclease to bind to and cleave the target DNA sequence. 57 Without the presence of a proper PAM sequence, the Cas9 fails to cleave a completely match target site, 58 a mechanism for CRISPR to distinguish self and non-self-DNA in bacteria. Although the first base of N is variable studies have found that C is favored in this position whereas T is associated with lower gRNA performance.29,40,59 In addition, “non-canonical” PAM sequences, such as NCG, NGA, and NAG, can also stimulate the activity of Cas9 but have significantly lower activity as compared with the NGG PAM. The specific order of genome editing efficiency affected by PAM for SpCas9 has now been found to be NGG > NAG > NGA.25,60 It is worth noting that for most CRISPR activity prediction tools, PAM was not included as a feature for training the prediction models, and the canonical NGG PAM sequence is generally assumed. The ability to recognize non-canonical PAM sequences is of greater importance when considering potential off-target sites.
The Guide (Protospacer) Sequence
For SpCas9, the 20-nt guide sequence is one of the most significant determinants for on-target activity prediction.22,25 In the vast majority of CRISPR applications, expression of the gRNA is driven by RNA polymerase III promoters such as U6. Therefore, it is necessary to carefully consider several potential issues pertaining to oligo synthesis and transcription. For instance, there are several well-known gRNA activity impacts about RNA polymerase preference. gRNA initiating with dinucleotide 5′ AG were observed to have a transcript bias by T7 polymerase, which is presumably due to 5′ end transcript heterogeneity from in vitro transcription.42,61 For transcription from a U6 promoter, gRNA activity is greatly improved when the transcribed sequence begins with a guanine (G). 8
Specifically, most gRNA expression vectors, including those used for generating large training datasets, are based on the U6 promoter. The presence of a UUUU motif disrupts RNA polymerase III transcription, thus lowering the editing activity of poly T containing gRNAs. 62 Cleavage efficiency is significantly decreased when the gRNA contains a poly-N motif due to the oligo synthesis, especially four contiguous Gs. 18 Furthermore, two specific types of 3′ end motifs, namely the “TT motif” and the “GCC motif,” were found to significantly reduce gRNA editing efficiency by more than 10-fold, each operating through distinct mechanistic pathways. 63 The former is characterized by a three-pyrimidine sequence containing a TT dinucleotide or a four-pyrimidine sequence with at least two Ts. This structure might be linked to the tetra-T sequence at the 5′ end of the scaffold sequence, extending the T-rich region, generating a premature transcription termination signal and consequently decreasing gene editing efficiency. The latter diminished efficiency at the Cas9 targeting stage which could potentially lead to inefficient loading of the gRNA with Cas9 into functional ribonucleoprotein complexes, nonspecific binding to off-target sites, and co-factor-dependent mechanistic issues. Notably, the impact of T-rich motifs on gRNA transcription creates a potential bias in the training sets used to develop ML and DL models that may not reflect the efficiency of chemically synthesized gRNAs used for clinical applications.
On the contrary, the gRNA overall nucleotide composition can impact gRNA activity. The gRNA with a higher abundance of adenines (As) exhibited increased editing activity. 29 This enrichment preference was predominantly found in positions 9 to 16 of the guide sequences.13,20 The presence of A in this region increases gRNA affinity to Cas9, as shown in Cas9 loading assays. 64 However, Moreno-Mateos et al. 61 found that G enrichment and A depletion contributed to the stability, binding, and on-target activity of CRISPR-Cas9 in zebrafish data. Also, the first and last positions of effective gRNAs exhibited a strong preference for G and cytosine (C) depletion, while A and uracil (U) were generally absent from the gRNA spectrum. Interestingly, this study used gRNAs transcribed from T7 promoters and delivered as short-lived RNA molecules, unlike lentivirus libraries which have an extended window of expression and activity. The differences observed in nucleotide composition may therefore reflect the characteristics of the expression systems or the kinetics of gRNA activity given the different time frames of assays using T7 and U6 transcribed gRNAs. In terms of dinucleotide and trinucleotide motifs, Wong et al. 29 reported a significant reduction of CRISPR activity due to the presence of GG and GGG for overall nucleotide composition. Malina et al. 65 observed that akin to the conclusions drawn for single nucleotides, dinucleotides UU and GC did not exhibit preferences in effective gRNAs, while those containing A, such as CA, AC, GA, and AU, showed clear cleavage preferences.
Apart from the overall nucleotide composition affecting editing outcomes, nucleotide usage at each individual sequence position of the spacer is critical for efficient editing. Nucleotides proximal to the PAM sequence tended to be the most significant, which could be interpreted by the target interrogation and Cas9 loading within this region.13,58,64 This feature-rich region was known as the core seed region and corresponds to positions 16–20 of the gRNA. 66 Within the entire region, U was consistently disfavored.20,29 At position 20, there was a pronounced preference for G and A, with a marked disfavoring of C.20,29,67 At position 19, a preference for purines was evident. C was the favored nucleotide at position 18, corresponding to the CRISPR-Cas9 cleavage site. 13 At position 16, a similar pattern emerged where C was enriched, and G was disfavored. Notably, the nucleotide preference at position 17 remained a subject of controversy, with some studies indicating a preference for G, while others suggested enrichment of C and depletion of G.13,20 Recently, a position-specific analysis based on over 2,000 high-efficiency gRNAs confirmed these base preference conclusions, showing G enrichment in position 17. Also, the presence of G downstream of the PAM, due to the DNA bulge created by ‘gRNA sliding’, was found to be ineffective. 23
The Thermodynamic Features
Several studies have indicated that thermodynamic features can influence gRNA secondary structure and impact Cas9 binding affinity, while the thermodynamic features of both the gRNA and the DNA sequence flanking the target site can influence CRISPR on-target activity.24,34,51,65 GC content, serving as a metric for measuring thermodynamic stability, is a known predictor of CRISPR activity due to the additional hydrogen bond in a GC pair compared with an AT pair. An established consensus suggested that gRNAs with extreme GC content are prone to non-functionality, emphasizing the importance of maintaining GC content within the range of 40–80%,20,29,64 with high GC sequences with the potential for creating G-quadruplexes have a greater detrimental impact on gRNA activity. 65 The impact of GC content can be further studied by analyzing the variation of GC content within sections of the gRNA. When focusing on GC content for 10-nt sub-sequences, the first ten bases could be regarded as the strongest predictor of gRNA efficiency, with higher-performing gRNAs exhibiting significantly elevated GC content (at least more than 50%) in PAM-distal region. 40 At a resolution of five nucleotides, higher GC content in positions 4–8 was most indicative of higher activity. In contrast, a preference for lower melting temperature (Tm) was observed in the middle section of the gRNA, potentially linked to the noted enrichment in A nucleotide. 52 A recent study has shown that a GC content exceeding 56% in the PAM-proximal seed region significantly reduces cleavage efficiency compared to the PAM-distal region. 68 Based on these findings, it is recommended that the GC content not exceed 56% in the PAM-proximal region and be more than 50% in the PAM-distal region. On the contrary, the metric for assessing thermodynamic stability involved the calculation of the free energy levels. The interaction between the Cas9/gRNA complex and target genome sequence can be interpreted into gRNA self-folding energy, DNA-DNA unfolding energy, and DNA-RNA hybrid energy. The self-folding free energy of gRNA could serve as an indicator of sequence stability, reflecting its tendency to form secondary RNA structures through intramolecular interactions. Non-functional gRNAs typically exhibited a more negative self-folding free energy, signifying a greater propensity for self-folding compared to their functional counterparts. 29 The stability of the DNA/RNA hybrid in the R-loop formation also influences Cas9 on-target activity with more stable DNA/RNA duplexes associated with lower editing efficiency. Interestingly, a “sweet spot” range of DNA/RNA binding energy recently was identified as a measure of optimal efficiency across different gRNAs. Through physical modeling of 11,602 Cas9 gRNAs binding to targets with 5′-N − 1GGN + 1 – 3′ PAMs, Corsi et al. defined a DNA/RNA hybrid energy interval. Compared to traditional GC-content analysis, this energy range demonstrated a more robust and distinctive ability to identify high-efficient gRNAs. Moreover, this change in energy helped explain why certain off-targets exhibited higher efficiency than on-targets. The mismatched bases facilitated energy transitions and prevalent ‘gRNA sliding’ along the target DNA sequence, particularly due to the presence of G bases flanking the NGG PAMs. 23 Finally, one addition point to note is that the total length of the gRNA could impact its editing efficiency. Shorter sequences, such as 17-nt or 18-nt have increased specificity but reduced efficiency compared to 19-nt or 20-nt spacers while gRNAs exceeding 20 nucleotides were less effective.13,61
The Impact of gRNA Structure
In addition to the influence of thermodynamic properties, the role of gRNA secondary structure is underscored in various biological processes, considering that RNA structure plays a pivotal role in determining nucleotide accessibility and ensuing interactions at each locus. 69 Conserved structural motifs within the gRNA are integral for facilitating correct secondary structure formation, proper Cas9 loading, and subsequent endonuclease activation. 70 Specifically, the secondary structure of the gRNA is determined by the scaffold as well as the spacer sequence. Some characteristics, mutants, and modulation of gRNA sequences have been found to disrupt the canonical gRNA secondary structure, thereby impacting Cas9 on-target activity. For instance, based on Gibbs energy prediction, we previously found that the formation of a strong internal secondary structure hindered Cas9-mediated DNA cleavage. 71 Yet these original low activity gRNAs can regain their efficiency through selective point mutations that disrupt alternative secondary structure formation. Moreover, by utilizing in silico RNA folding analyses, Wong et al. 29 discovered that efficient and inefficient gRNAs exhibited significant differences at positions 18–20 and positions 51–53 of the gRNA. These positions corresponded to the 3′ end of the gRNA spacer and a region of the gRNA scaffold, respectively. Importantly, a conserved motif in the gRNA generates a stable stem-loop secondary structure between nucleotides at positions 21 and 50. This conserved structure aligns positions 18–20 with 51–53 in an antiparallel configuration (Supplementary Fig. S3). Therefore, base-pairing could potentially occur between bases 19–20 and 52–53 if the sequences are complementary, resulting in an extended stem-loop structure covering positions 18–53. This alternation of high duplex stability in secondary structure might hinder either gRNA seed interactions with the target DNA or scaffold interactions with the Cas9 protein, leading to the observed decrease in efficiency. Observations on the impact of gRNA base pairings for Cas12a have also been made. 72
On the contrary, recent studies found that rational engineering of scaffold-specific regions, especially the lower stem, upper stem, and hairpin, is expected to modulate CRISPR-Cas system efficiency, given the indispensable role of bulk and nexus for gRNA activity. 73 For instance, the presence of thymidine trinucleotides or poly T sequences within the scaffold has been found to reduce gRNA activity due to RNA polymerase III transcription termination.62,66,74 Accordingly, modifying the low stem to remove termination signals, extending the upper stem, or employing the combinational strategy of both has been shown to enhance targeting efficiency. 31 Adding RNA aptamers, e.g., MS2-MCP, PP7-PCP, and com-Com binding pairs described by Jesse et al. 75 or MS2, PP7, and boxB aptamers studied by Ma et al., 76 to the upper stem or hairpin of the scaffold can further modulate gRNA activity. However, it should be noted that excessive hairpin formation may adversely affect the cleavage efficiency of CRISPR-Cas9. Generally, a divalent modification of the aptamer in the scaffold is more beneficial for gRNA activity than single fusion, whereas trivalent modification tends to perform poorly. Finally, stabilizing the hairpin structure also improves Cas9 cleavage efficiency. Modifying uridines at several positions, such as changing the 34th U of tracrRNA to A or replacing U-T bonds with G-C in the stem loop, has been shown to enhance Cas9 activity. 77 Elongating the first hairpin 3′ of the nexus part to form a locked tracrRNA (t-lock) with a super-stable loop prevents gRNA misfolding and improves cleavage efficiency by an average of 69%, especially for those difficult editing sites. 69
Despite current models primarily considering altering of spacer sequence to edit different genome sites while maintaining the gRNA scaffold sequence constant, this sequence inflexibility renders some targets intractable to CRISPR gRNA editing due to undesirable intramolecular interactions.69,78 Peter C. et al. addressed this limitation by incorporating gRNA datasets that utilize the wild-type Hsu tracrRNA, 30 and Chen tracrRNA,31,32 which modify the signal of RNA polymerase III termination and stabilize the gRNA structure. This work led to the development of Rule Set 3, 27 a novel ML model adaptable to different tracrRNA types, providing valuable insights for enhancing editing activity. Interestingly, by introducing the SELEX-based BLADE approach (a systematic evolution method for optimizing gRNA), it was found that the scaffold itself is malleable and can take different structural variations into account. 79
The Genomic and Epigenetic Features
When the aim of CRISPR gene editing is to alter gene expression, it is crucial to consider whether the cleavage site can functionally disrupt the target gene. Existing studies suggested diminished gRNA activity when targeting sites were close to the C-terminus. 20 Meanwhile, users should be aware of alternative ATG start codons when targeting sites are closer to the translational start site of the gene. In targeting genomic non-coding regions and 5′- or 3′- untranslated regions, the efficacy of these gRNAs is commonly low.20,25,64 Regarding CRISPR activation and CRISPR interference, effective gRNA’s strategies to be carried out are in an optimal targeting window of less than 100-nt, near the transcript start site of target genes.80,81
Epigenetic features, as independent factors in addition to gRNA features, contribute to differences in gRNA efficiency across different cellular contexts despite sharing the same gRNA sequence. Open chromatin status, as indicated by CCCTC-binding factor (CTCF) binding, histone modification, Cytosine-phosphate-Guanine (CpG) methylation, and Deoxyribonuclease (DNase) hypersensitivity, is associated with more efficient gene editing.59,66,82,83 The structure of chromatin has also been found to affect the balance between DNA DSB repair pathways NHEJ and HDR. 84
In silico analysis, Haeussler et al., 10 utilized a dataset where the assay was replicated in distinct cell types to compare the binding variation of gRNAs. They observed a high correlation in measured efficiencies for certain cell type combinations, but not all. The Spearman correlation between knockout efficiency for 2,076 guides in HL60 and KBM-7 cell lines was 0.752. but the correlation for the 177K library screens conducted in RPE1 and HCT116 cells was merely 0.531 and 0.585 from the two different replicates. It has been suggested that the variation was attributed to differences in chromatin state. Recently, a new study conducted a comparative analysis of gRNA efficiency based on four methylation features (including CTCF, Histone H3 lysine 4 trimethylation (H3K4me3), DNase, and reduced representation bisulfite sequencing (RRBS).11,59 They initially screened for identical gRNAs with at least one differing epigenetic feature, and then compared the efficiency differences between any two cells. According to this small-scale experiment, they observed that epigenetic features seem to have a substantial impact on CRISPR activity, rather than a subtle influence stated by Haeussler’s research. Therefore, we and others, 85 recommended performing a similar analysis across multiple cell-based CRISPR screening experiments and encouraged the future CRISPR model development to explore the prediction of gRNA efficiency under different epigenetic statuses.
Apart from cell-based CRISPR assays, Isaac et al. discovered that the editing hurdle posed by nucleosomes is more tolerant in vivo due to the dynamic nature of cell nucleosomes. This phenomenon known as ‘nucleosome breathing’ allows the temporary unwrapping of nucleosome DNA, which may shortly expose these DNA sequences to Cas9 binding. Similarly, DNA near the nucleosome edge was found to be far more permissible to Cas9 cleavage than the unexpected center of DNA when investigating a natural nucleosome positioning sequence. 86 Moreover, in actual experiments,87,88 using Cas9 nuclease in conjunction with chromatin-modulating peptides has been shown to increase editing efficiency by opening the chromatin structure. Thus, these results show that the Cas9 editing activity requires an open nucleosome status, and modulating this status can achieve higher editing efficiency.
Features Utilized in CRISPR Activity Prediction Tools
To enhance the interpretability of models and guide future tool development, we systematically analyzed feature engineering strategies across state-of-the-art CRISPR activity prediction tools. For the three DL-based frameworks, DeepSpcas9, 33 and CRISPRon, 24 employ multi-channel CNN networks to extract 30-nt sequence features. Compared to DeepSpcas9, CRISPRon incorporates the gRNA-target DNA binding free energy (ΔGB) to improve predictive performance. Biochemical and structural studies,58,89 have shown that gRNA engages in a stepwise association with DNA, suggesting that localized thermodynamic properties are critical for target recognition. Building upon this insight, the introduction of ΔGB addresses the gap by accounting for energy dynamics throughout the interaction process, including DNA-opening energy, RNA-unfolding energy, and DNA-RNA-binding energy. 23 The contribution of ΔGB to model performance has been robustly validated through Shapley Additive Explanations and Gini feature importance analyses in independent ML models. 24 Likewise, DeepHF, 34 utilizes RNN network layers for automatic feature extraction. The inclusion of additional biological features, such as secondary structure accessibility, stem—loop formations, Tm, and GC content, marginally enhances its predictive power, finally achieving better model performance.
In contrast, ML-based frameworks exhibit more diverse and individualized feature utilization, reflecting variations in data generation methods and empirical hypotheses across different studies. Azimuth, 25 sgDesigner, 26 and CRISPRedic,t 28 incorporate 582,302 and 28 features, respectively. Both Azimuth and CRISPRedict construct features based on overall nucleotide composition, position-specific nucleotide encoding (including single-nucleotide and dinucleotide features), and GC content, with the Azimuth additionally incorporating PAM-proximal “NGGN” motifs, regional Tm values, and target protein positional attributes, while the CRISPRedict augments this feature set by introducing central A counts and specific motifs (e.g., GGGG, TT, GCC). Notably, the CRISPRedict takes advantage of a multi-step feature selection strategy to refine the feature set, enabling it to achieve predictive performance comparable to DL-based models. This approach suggests the importance of feature optimization in CRISPR model development. Moreover, Rule set3, 27 an updated version of the Azimuth, utilizes the entire features from Azimuth and introduces three new sequence-based features: (1) the length of the longest consecutive stretch of each nucleotide (A, T, C, G) within the gRNA sequence, (2) Tm of the gRNA:DNA heteroduplex and (3) the free energy of the folded spacer sequence. The Rule set3 also explores certain target-site features that reflect the likelihood of disrupting gene functions. For example, the phyloP score, which assesses evolutionary conservation, and the proportion of α-helix-related amino acid near cut sites have been identified as strong predictors of gRNA activity. Consistent with previous findings, 25 the model demonstrated that targeting beyond 85% of the coding sequence results in a steep decline in gRNA activity. Instead, targeting annotated regions, defined by protein domain databases such as Smart, Pfam, PROSITE, and Gene3D,90–93 tends to yield higher activity. Finally, the sgDesigner focuses on position-specific single nucleotide features and GC content, with a particular emphasis on gRNA secondary structure features predicted by RNAfold. 94 This structural information reflects differential nucleotide accessibility within the folded gRNA, a feature incorporated in only a few tools (e.g., DeepHF). Overall, both sequence-based features and biologically relevant properties significantly contribute to model development. While an informative, discriminating, and independent feature set is integral for enhancing the performance of ML-based prediction tools, DL-based tools benefit from large, systematic, consensus-driven datasets and multi-level feature representations. A detailed overview of feature usage in our evaluated models is provided in Supplementary Data S1.
Conclusions
We present here an overview and comparative analysis of CRISPR-Cas9 on-target activity prediction. The primary take-home messages are as follows: (1) a practical decision-tree-based guide for CRISPR gRNA design can be accessed, with diverse tools listed for different scenarios; (2) DL-based tools represent the future direction of CRISPR on-target prediction, as well as off-target prediction although not discussed here. This progress relies on integrating different gRNA assays, optimizing algorithms and feature collection, and especially developing large-scale, unbiased, and varied editing context datasets for gRNA quantification, such as high-throughput sequencing-based gRNA efficiencies generation like Chen et al. and Xiang et al datasets; (3) Due to the cell type and species heterogeneity, generalizable deep learning tools such as CRISPRon is the preferred choice if the context of cell types is not available. Given the data particularly trained with, the DeepHF model is particularly suitable for predicting the activity of T7-based in vitro transcribed gRNAs. Moreover, this review mainly focuses on the on-target prediction of CRISPR experiments in humans and mice. But we do suggest that new tool development should consider the impact of epigenetic factors and thermodynamic parameters on editing efficiency, rather than relying on gRNA sequence alone.
Footnotes
Authors’ Contributions
Y.L.L.: Proposed and led this study. H.Y.: Collected the data and performed the comparative analysis. C.P.S. and H.X.X.: Drafted the supplementary material, supporting project management activities, and post-computational analysis. Y.S. and C.A.: Performed code validations. H.Y. and Y.L.L.: Drafted the original article. L.B., L.L., K.B., C.L., Y.H., and J.G.: Provided critical edits to the article. All authors have reviewed and approved it for publication.
Author Disclosure Statement
The authors declare no conflict of interest for this study.
Funding Information
This research was funded by the Novo Nordisk Foundation (NNF21OC0072031, NNF21OC0068988) and the Lundbeck Foundation (R396-2022-350). This publication is based upon work from COST Action Gene Editing for the treatment of Human Diseases, CA21113 (![]() ) supported by COST (European Cooperation of Science and Technology). K.B. held Nicolas Monardes contract from Consejería de Salud y Consumo of the Junta de Andalucía.
) supported by COST (European Cooperation of Science and Technology). K.B. held Nicolas Monardes contract from Consejería de Salud y Consumo of the Junta de Andalucía.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.