
Abstract
Inherited retinal diseases (IRDs) are a heterogeneous group of blinding genetic disorders caused by pathogenic variants in genes expressed in the retina. In this study, we sought to develop a method for rapid evaluation of IRD gene variant pathogenicity by inducing expression of retinal genes in patient-derived fibroblasts using CRISPR-activation (CRISPRa). We demonstrate CRISPRa of CRB1 expression in fibroblasts derived from patients with retinitis pigmentosa, enabling investigation of pathogenic mechanisms associated with specific variants. We show the CRB1 c.4005 + 1G>A variant caused exon 11 skipping in CRISPR-activated fibroblasts and retinal organoids (ROs) derived from the same RP12 patient. The c.652 + 5G>C variant was shown to enhance exon 2 skipping in CRISPR-activated fibroblasts and differentially affected CRB1 isoform expression in fibroblasts and ROs. Our study demonstrates an accessible platform for transcript screening of IRD gene variants in patient-derived fibroblasts, which can potentially be applied for rapid pathogenicity assessments of any gene variant.
Introduction
Inherited retinal diseases (IRDs) are the most common cause of blindness in working aged adults and the second most common cause of blindness in children in the developed world. 1 With thousands of known mutations in hundreds of IRD genes, the identification and classification of disease-causing sequence variations are a challenging process, particularly considering the large number of variants for which pathogenicity is uncertain. Moreover, in cases wherein pathogenicity can be determined from correlation with clinical phenotype, inheritance patterns, population data, and in silico analysis, the effects of the variant on gene expression remain uncertain without functional assessment and verification of predicted effects.
It is now recognized that a significant proportion of intraexonic changes can affect splicing, resulting in alternative mRNA transcripts that do not bear the predicted codon change, due to exclusion of the affected exon from the mature transcript.2,3 Understanding the precise mechanisms of variant pathogenicity, beginning with effects on mRNA and protein expression, is essential for the identification and development of appropriate emerging treatment modalities for IRDs. For example, missense or synonymous mutations affecting splicing may be amenable to treatment with splice-switching antisense oligonucleotide drugs 3 while gene replacement therapies may be required for variants that result in transcript degradation.
Functional assessments of IRD variant pathogenicity have been performed using a number of platforms, including minigene assays 4 as well as induced pluripotent stem cell (iPSC)-derived retinal organoids (ROs), 5 which provide a source of patient-derived retinal cells for assessment of retinal gene expression. Both of these platforms involve time-consuming processes, making high-throughput implementation challenging.
For more rapid assessment of IRD gene variants, we and others have utilized accessible patient cells, such as peripheral blood monocytes or leukocytes 6 and dermal fibroblasts 7 to assess IRD gene expression, however, this method is currently limited to IRD genes with detectable gene expression in these cell types. To expand the utility of this approach, we sought a method for inducing the expression of retinal genes that are not expressed in fibroblasts.
CRISPR-activation (CRISPRa) is a recently developed adaptation of CRISPR/Cas gene editing technology that is capable of inducing expression of a targeted gene. Fusion of an endonuclease-deficient dCas9 protein with a transcriptional activation domain has been shown to produce programmable transcription factors that can be targeted to any position in the genome.8–10 Targeting dCas9-transcriptional activators to the promoter region of various genes has proven to be an effective method for inducing gene expression in a variety of cells. 11 CRISPRa has been used to activate gene expression in bacteria, yeast, plants, frog, and mammalian cells.12–16
CRISPRa of transcription factors has been used to reprogram cell fate in vitro and in vivo.17–19 In the retina, CRISPRa was used to activate long-term opsin expression in mice. 20 Activation of retinal gene expression in patient-derived fibroblasts has not been previously reported.
Over 450 pathogenic variants of CRB1 have been reported to be associated with various retinal dystrophies including early-onset retinitis pigmentosa (RP), RP with preserved para-arteriolar retinal pigment epithelium, RP with intraretinal cystoid spaces, RP with Coats-like exudative vasculopathy, familial foveal retinoschisis, cystic maculopathy, fenestrated sheen macular dystrophy, and Leber congenital amaurosis. The human crumbs homologue 1 (CRB1) gene is located within a 210 kb genomic locus situated within the chromosome 1q31.3 region. 21
In the human retina, three distinct protein-coding transcripts are expressed from the CRB1 locus: CRB1-A, CRB1-B, and CRB1-C (Fig. 1A). 22 The longest transcript variant, CRB1-A, comprises 12 exons encoding a 1406 amino acid transmembrane protein expressed by Muller glial cells. In contrast, the CRB1-B transcript consists of an alternative first exon located within intron 5, followed by canonically spliced exon 6–10 sequence and an elongated terminal exon 11 incorporating additional sequence from intron 11. CRB1-B is expressed predominantly in photoreceptor cells and encodes a 1003 amino acid transmembrane protein expressed in photoreceptor cells.
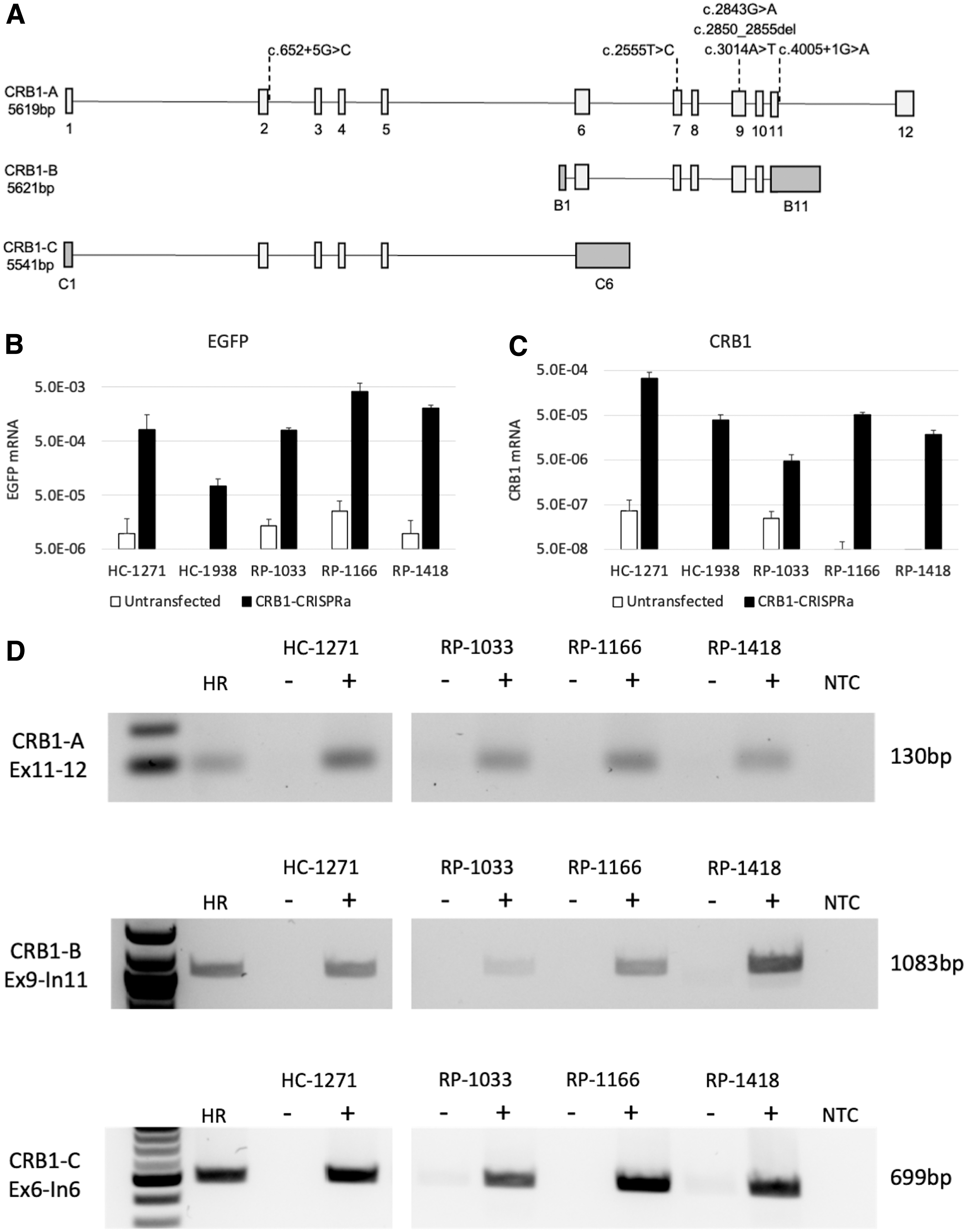
Although CRB1-B lacks the canonical CRB1-A exons 1–5, CRB1-C comprises exons 1–5, followed by an elongated terminal exon 6 incorporating additional sequence from intron 6 (Fig. 1A). 22 In this study, we utilized CRISPRa to induce expression of the CRB1 gene in fibroblasts derived from three patients with retinitis pigmentosa 12 (RP12), providing a method for rapid screening of mRNA transcripts containing CRB1 variants.
Methods
Human tissues and cell lines
The use of human tissues and cell lines in this study was approved by the University of Western Australia Human Research Ethics Committee (2021/ET000151, RA/4/1/7916 and 2019/RA/4/20/5717). Patients and healthy control subjects investigated in this study were enrolled as part of the Western Australian Retinal Degeneration (WARD) Study (Lions Eye Institute, Nedlands, Australia) and genetic diagnosis was conducted by the Australian Inherited Retinal Disease Registry and DNA Bank. 23 Dermal fibroblasts from two healthy control subjects were included in the study. Patient ID, age, gender, phenotype, mutation, and location are listed in Table 1.
Human dermal fibroblast lines
HC, healthy control; RP, retinitis pigmentosa.
Detailed clinical phenotypes of the CRB1 cases and variant pathogenicity classifications according to American College of Medical Genetics and Genomics (ACMG) guidelines have been published previously. 24 Dermal fibroblasts were cultured in T75 flasks in Dulbecco's modified Eagle's medium (DMEM, 013; Perkins Media Lab, Harry Perkins Institute of Medical Research, Nedlands, Australia) supplemented with 10% fetal bovine serum (26140079; Thermo Fisher Scientific, Waltham, MA) and antibiotic antimycotic (051; Perkins Media Lab) at 37°C and 5% carbon dioxide.
Human iPSC lines were reprogrammed from WARD patient and healthy control fibroblasts as previously described.25,26 IPSCs were cultured on Geltrex (A1413302; Thermo Fisher Scientific)-coated 6-well culture plates in mTeSR1 medium (85850; STEMCELL Technologies, Vancouver, Canada) at 37°C and 5% carbon dioxide.
Postmortem human retinal tissue was acquired from the Lions Eye Bank (Lions Eye Institute, Nedlands, Western Australia) from a single 18-year-old male donor, harvested at 4 h postmortem.
Transfection of dermal fibroblasts with dCas9-VPR-EGFP mRNA
The Edit-R CRISPRa assay (Horizon Discovery, Waterbeach, United Kingdom) was used to activate endogenous retinal gene expression in patient-derived dermal fibroblasts. Cell cultures from IRD patients and control subjects were cotransfected with mRNA encoding a dCas9-VPR-EGFP fusion protein and crRNA/tracrRNA duplexes (Horizon Discovery) using the NEON electroporation system (MPK5000; Thermo Fisher Scientific). Each crRNA consisted of four pooled sequences designed using the algorithm of Horlbeck et al. 27 to target promoter sequences within 2 kb of the transcriptional start site of the selected gene (Horizon Discovery).
crRNA/tracrRNA duplexes were formed by mixing equal volumes of crRNA (200 μM) and tracrRNA (200 μM) and incubating for 10 min at room temperature. Dermal fibroblasts were cultured in T75 flasks and harvested for electroporation at ∼70% confluency. Fibroblasts were dissociated using TrypLE Express Enzyme (12604021; Thermo Fisher Scientific), resuspended in phosphate-buffered saline, counted, and resuspended in Buffer R at 20,000 cells/μL.
Then, 0.5 μL of the crRNA/tracrRNA duplex and 0.5 μL of Cas9-VPR-EGFP RNA (500 ng) were added to 10 μL of Buffer R cell suspension (2 × 105 cells) and the resulting solution was drawn into a 10 μL NEON tip. Cells were electroporated using a single 20 ms pulse at 1700 V then plated into a 12-well plate. Culture medium was changed after 24 h, and cells were harvested 48 h post-transfection.
RO culture
RO differentiation was performed as per a previously established protocol. 28 iPSCs were dissociated using ethylenediaminetetraacetic acid (20-158; Sigma-Aldrich, St Louis, MO), then cell clusters were seeded into 6-well suspension plates containing mTeSR1 supplemented with 10 μM Y27632 (ab120129; Abcam, Cambridge, United Kingdom) for 48 h then moved into DMEM/F12 (071; Perkins Media Lab) supplemented with decreasing concentrations of knockout serum replacement (KSR, 10828028; Thermo Fisher Scientific), nonessential amino acids (099; Perkins Media Lab), antibiotic–antimycotic (051; Perkins Media Lab), B27 (089; Perkins Media Lab), and 10 ng/mL Insulin-like growth factor 1 (78022.1; STEMCELL Technologies).
After 35 days, ROs were moved into KSR-free medium and supplemented with additional N-2 Supplement (041; Perkins Media Lab).
Transcript analysis and quantitation of gene expression levels
Total mRNA was extracted from transfected and control dermal fibroblasts using TRIzol Reagent (15596026; Thermo Fisher Scientific), and cDNA was synthesized with the RT 2 First Strand Kit (330404; Qiagen, Hilden, Germany). Quantitative polymerase chain reaction (qPCR) was used to characterize gene expression. Expression values were normalized to GAPDH using the ΔCT method.
For analysis of retinal differentiation, qPCR measurements were normalized to GAPDH then to undifferentiated iPSC using the ΔΔCT method. qPCR products were run on 1.5% agarose gel by gel electrophoresis and purified using the Wizard SV Gel and PCR Clean-Up System (A9281; Promega, Madson, WI), then analyzed by Sanger Sequencing (Australian Genome Research Facility, Nedlands, Australia). Primers used in this study are listed in Supplementary Table S1.
CRB1 fragments were amplified by polymerase chain reaction (PCR) and gene specific primers used to identify splicing abnormalities. Purified PCR products were sent to AGRF for Sanger Sequencing analysis to confirm the identity of the amplicons. Primers used in this study are listed in Supplementary Table S1.
Results
CRISPRa of CRB1 gene expression in fibroblasts
Dermal fibroblasts from two healthy control subjects and three RP12 patients (Table 1) were cultured and cotransfected with Edit-R dCas9-VPR-EGFP mRNA and synthetic guide RNAs (gRNAs) targeting promoter regions of the CRB1 gene. Although the mRNA construct was designed to express enhanced green fluorescent protein (EGFP) from an internal ribosomal entry site (IRES), no EGFP fluorescence was detected in transfected fibroblasts by microscopy (data not shown). Therefore, we used EGFP-specific qPCR to assess transfection efficiencies (Fig. 1B). Untransfected control fibroblasts showed very low or undetectable EGFP and CRB1 levels.
Fibroblasts cotransfected with dCas9-VPR-EGFP mRNA and CRB1-targeting crRNA guides showed upregulation of CRB1 expression by qPCR analysis (Fig. 1C). Transfection efficiency varied between the two control fibroblast lines, with the 1271 line showing a ninefold increase in EGFP mRNA levels compared with the 1938 line. Concomitant with this increase in transfection efficiency, CRB1 expression was increased ninefold in 1271 fibroblasts compared with 1938 fibroblasts after CRISPRa, suggesting gene activation was correlated with transfection efficiency in control fibroblasts.
In contrast, despite equal or higher transfection efficiencies than the 1271 control, lower CRB1 levels were detected after CRISPRa in RP12 patient fibroblasts (Fig. 1B, C), suggesting that mutant CRB1 transcripts may be degraded after transcription. Sanger sequencing of gene-activated control fibroblast qPCR products confirmed the identity of the CRB1 amplicons (data not shown). Together, these results indicate successful transcriptional activation of the CRB1 gene in human fibroblasts by CRISPRa.
All three retinal CRB1 transcripts were induced by CRISPRa
Three main isoforms of CRB1 (CRB1-A, CRB1-B, and CRB1-C) have been reported to be expressed in the retina, with CRB1-B representing the most abundant isoform. 22 Since our qPCR assay targets an amplicon spanning exons 11–12 of the CRB1-A transcript, we designed additional primer sets to target the CRB1-B and CRB1-C isoforms (Fig. 1D). PCR analysis using primer sets specifically targeting each of the three retinal CRB1 isoforms demonstrated expression of all isoforms in CRB1 gene-activated fibroblasts (Fig. 1D). No bands were detected in untransfected fibroblast cDNA samples. The same bands were amplified from adult human retinal cDNA samples. PCR products were purified and Sanger sequencing confirmed the identity of the amplified products (data not shown).
The CRB1 c.4005 + 1G>A variant causes skipping of exon 11 in CRISPR-activated fibroblasts
PCR targeting exons 9 to 12 of CRB1-A amplified a 921 bp fragment from both CRB1 gene-activated fibroblast and adult human retinal cDNA samples. In addition to the full-length CRB1-A band, a smaller band was detected in CRB1 gene-activated fibroblasts derived from patient 1166, who carries a splice site mutation in the first base of intron 11 (Fig. 2A). Sanger sequencing of this band demonstrated skipping of exon 11 (Fig. 2B), providing functional evidence for the pathogenic mechanism of the c.4005 + 1G>A variant.
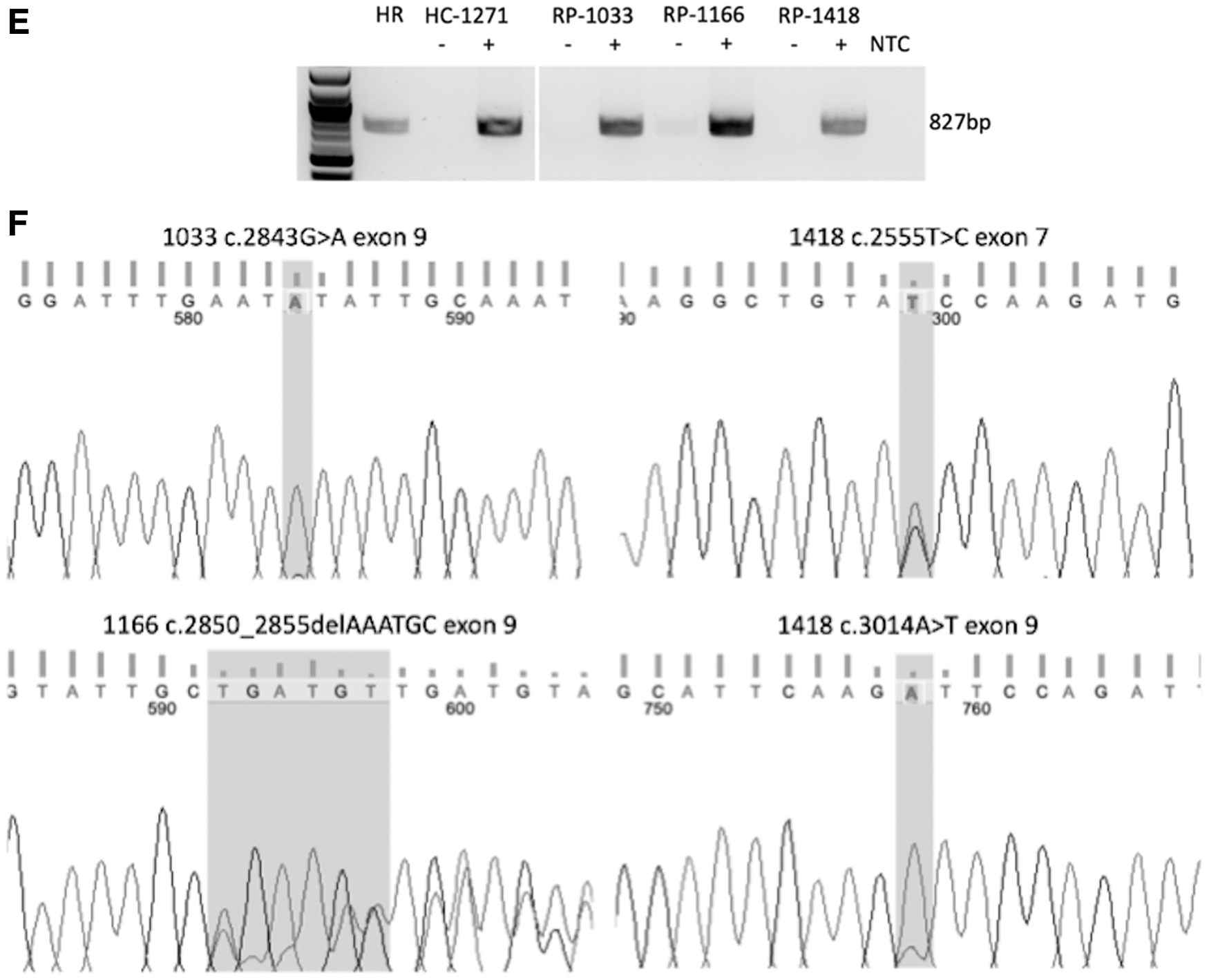
Skipping of exon 11 is predicted to cause a frameshift mutation, introducing a premature termination codon (PTC) 14 nucleotides into exon 12. Since this PTC affects the terminal exon of the CRB1-A transcript, it is predicted to escape nonsense-mediated decay (NMD) and be translated into a mutant CRB1-A protein with a 109 amino acid C-terminal truncation. These results demonstrate the utility of CRISPRa for identification of splice-altering genetic variants.
The CRB1 c.652 + 5G>C variant enhances exon 2 skipping in CRISPR-activated fibroblasts
The c.652 + 5G>C variant carried by patient 1033 was predicted to disrupt the donor splice site of intron 2. To test this prediction, we performed PCR targeting CRB1 exons 1–4 on cDNA prepared from CRISPR-activated patient fibroblasts. A strong 921 bp band, derived from full-length CRB1-A and CRB1-C transcripts, was amplified from CRB1 CRISPR-activated fibroblasts derived from a control subject. In contrast, the 921 bp band was only weakly amplified from CRISPR-activated fibroblasts derived from patient 1033, which expressed a smaller 339 bp band (Fig. 2C). Sequencing of this band demonstrated skipping of exon 2 (Fig. 2D).
Interestingly, exon 2 skipping was also detected in cadaveric human retina, suggesting the intact intron 2 donor splice site may be inefficiently utilized. However, since the 339 bp band was not expressed by CRISPR-activated control fibroblasts, this result suggests the c.652 + 5G>C variant further weakens the donor splice site. Skipping of exon 2 is predicted to cause an in-frame deletion of 194 codons in the coding sequence.
Expression of mutant CRB1 alleles in CRISPR-activated fibroblasts
Three of the six variants carried by the three RP12 patients were located in exon 9, with another located in exon 7. To assess expression of mutant alleles in gene-activated fibroblasts carrying these variants, we performed PCR targeting CRB1 exons 7–9, which are present in both CRB1-A and CRB1-B transcripts. A single band was amplified from control and patient-derived gene-activated fibroblasts, as well as adult human retinal cDNA (Fig. 2E). Sanger sequencing of this band demonstrated differences in the detection of specific variants.
Interestingly, patient 1033 fibroblasts carrying the c.2843G>A variant in exon 9 showed a pure A signal at the mutation site, with no contribution from the wild-type G, which is expected to be present in cis with the c.652 + 5G>C splice site variant in the other CRB1 allele in this patient (Fig. 2F). The lack of the wild-type G signal at the c.2843 position indicates that the c.652 + 5G>C variant results in complete loss of both CRB1-A and CRB1-B transcripts containing exons 7–9 in CRB1 gene-activated fibroblasts.
The c.2850_2855delAAATGC introduces an in-frame deletion into exon 9 of the CRB1 coding sequence and is carried in trans with the intron 11 c.4005 + 1G>A variant in patient 1166. Consistent with PCR results targeting CRB1 exons 9–12, Sanger sequencing demonstrated the expression of CRB1-A transcripts from both alleles in gene-activated 1166 fibroblasts, with frameshifted sequence evident downstream of the deletion site (Fig. 2F).
Patient 1418 fibroblasts carrying the c.2555T>C missense variant in exon 7 and the c.3014A>T missense variant in exon 9 showed heterozygous nucleotide signals at both mutation sites (Fig. 2F).
Detection of CRB1 transcripts in ROs
To validate our CRB1 CRISPRa results in patient-derived retinal cells, we differentiated ROs from iPSCs derived from the three RP12 patients and one control subject (1271). After 2 months of retinal differentiation, organoids formed neuroretinal tissues and upregulated expression of retinal genes, including PAX6, CRX, RCVRN, and RHO (Supplementary Fig. S1 and DeRoach et al. 23 ). In addition, expression of all three CRB1 isoforms (CRB1-A, CRB1-B, and CRB1-C) was detected in ROs (Supplementary Fig. S1).
Screening of CRB1 transcripts showed skipping of exon 11 in ROs derived from patient 1166, demonstrating that the splicing defect associated with the c.4005 + 1G>A variant was conserved between CRISPRa fibroblasts and ROs (Fig. 3A, B). PCR and sequencing of exons 7–9 showed that mRNA was expressed from both mutant alleles in ROs from all three RP12 patients. In contrast with data from CRISPRa fibroblasts, ROs derived from patient 1033 showed a heterozygous signal at position c.2843, indicating mRNA transcribed from the allele containing the c.652 + 5G>C splice site variant was expressed in retinal cells (Fig. 3C, D).
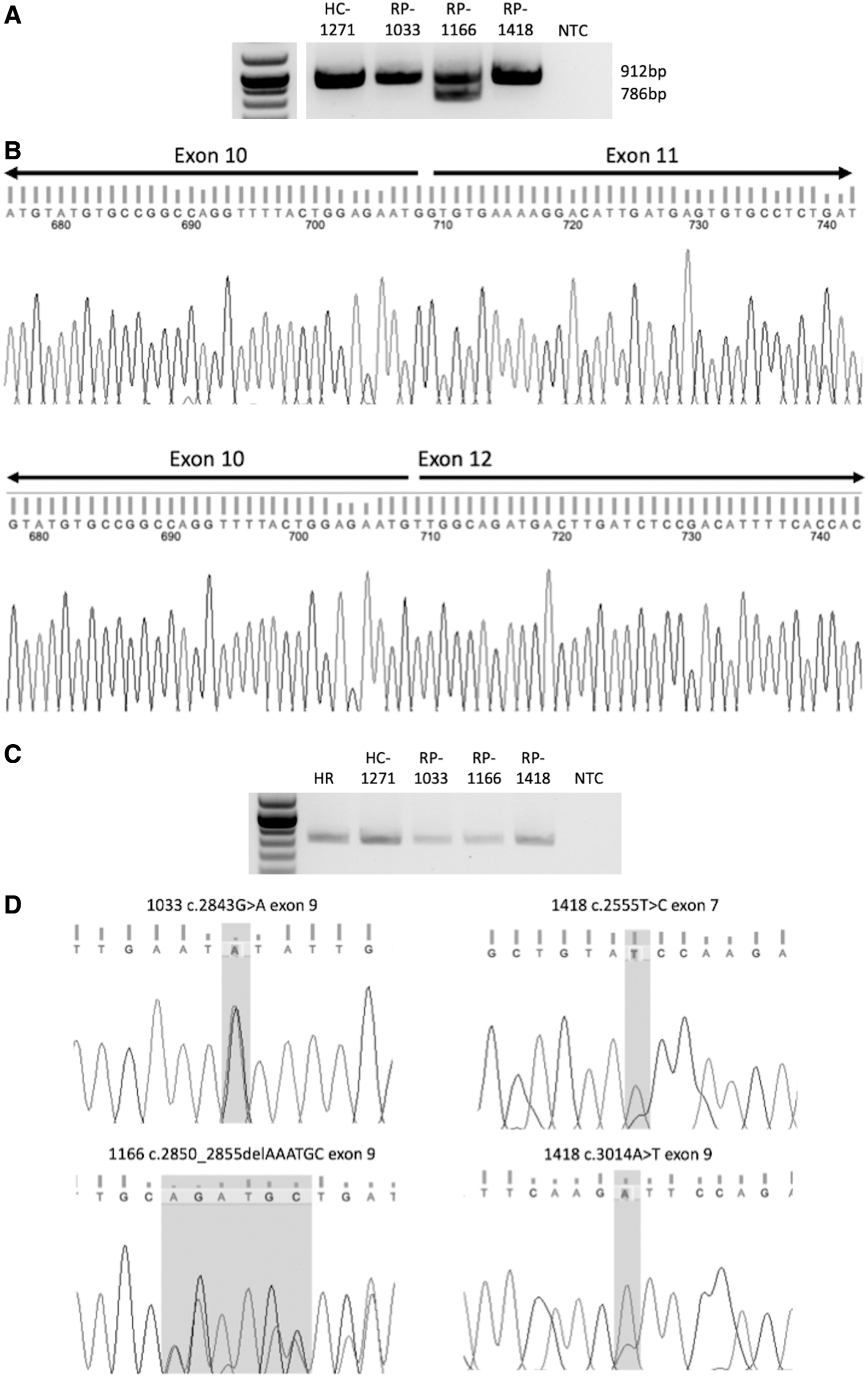
(
CRISPRa selectively upregulated the targeted gene
To confirm whether transcriptional activation was specific for the targeted gene, we measured RHO mRNA levels in CRB1-CRISPRa-treated fibroblasts. CRB1-CRISPRa treatment induced CRB1 expression in fibroblasts derived from healthy controls, but did not induce RHO expression, indicating gene activation was specific for targeted promoter. Transfection of Cas9-VPR-EGFP mRNA without gRNAs did not induce expression of CRB1 or RHO in fibroblasts (Fig. 4).
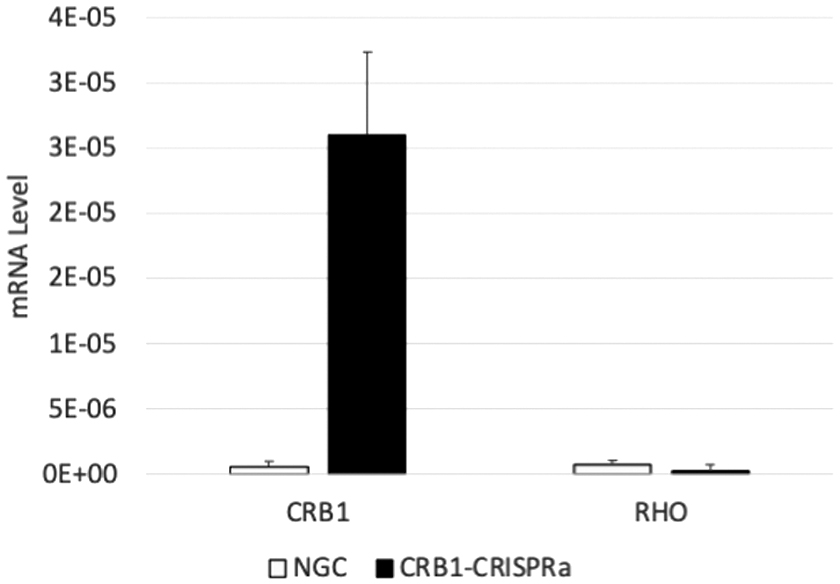
Fibroblasts derived from HC subject 1271 were cotransfected with dCas9-VPR-EGFP mRNA and crRNA/tracrRNA complexes (CRB1-CRISPRa) or with dCas9-VPR-EGFP mRNA alone (no guide control). Total RNA was harvested after 48 h for qPCR analysis. CRB1-CRISPRa selectively upregulated CRB1 expression, but not RHO expression, whereas transfection of dCas9-VPR-EGFP mRNA alone did not upregulate either gene. The graph shows mean expression levels normalized to GAPDH expression. Error bars indicate standard deviation from triplicate qPCRs.
Discussion
In this study, we demonstrate the use of CRISPRa for transcript screening of human retinal genes in cultured dermal fibroblasts. Transfection of dCas9-VPR-EGFP mRNA and predesigned guide oligonucleotides provided a simple and effective method of activating CRB1 expression in control and patient fibroblasts. Since the EGFP IRES was not active in fibroblasts, the Cas9-VPR mRNA could potentially be used instead, which would increase the number of gene copies delivered per microgram of mRNA.
Gene activation was generally proportional with transfection efficiency, however, RP12 patient fibroblasts showed lower CRB1 activation relative to controls, suggesting the mutant CRB1 transcripts were less stable than wild-type transcripts in fibroblasts. Using transcript-specific primers, we detected all three retinal CRB1 transcripts after CRISPRa. CRB1-A represents the longest isoform and contains the sequence used to evaluate genetic variants for pathogenicity. In contrast, the CRB1-B and CRB1-C transcript variants were more recently discovered by long read sequencing, with CRB1-B reported to represent the most abundant retinal isoform.
CRB1-B transcription is thought to be initiated from an alternative transcription start site located in intron 6, 22 which is ∼150 kb from the targeted promotor sequences that lie within 2 kb of the CRB1-A transcriptional start site. This suggests that recruitment of transcriptional activators to the 5′ promotor of CRB1 can open distal intragenic chromatin for transcription.
CRISPRa enabled detection of CRB1 splicing defects in patient fibroblasts. For example, the CRB1 c.4005 + 1G>A variant is located in the first base of intron 11 and was predicted to cause exon 11 skipping due to disruption of the donor splice site. In this study, we demonstrated that CRB1-A exon 11 skipping occurred in both CRB1 CRISPR-activated patient fibroblasts and ROs derived from the same patient. Interestingly, this variant is not predicted to affect CRB1-B transcripts, which utilize an alternative exon 11 donor splice site located in intron 11, or CRB1-C, which lacks exons 7–12. 22
The recapitulation of the retinal CRB1 transcript variant splicing and detection of pathogenic splicing defects after CRISPRa demonstrates a robust assay for identifying splice-altering CRB1 mutations in patient fibroblasts. We further applied this assay to assess the CRB1 c.652 + 5G>C variant, which was previously classified as likely pathogenic. 24 Using PCR primers targeting exons 1–4 of the CRB1-A and CRB1-C transcripts, we demonstrated the CRB1 c.652 + 5G>C variant enhanced skipping of exon 2, leading to an in-frame deletion in the N-terminal coding sequence.
Interestingly, PCR amplification of exons 7–9 showed that CRB1-A and CRB1-B mRNA transcripts transcribed from c.652 + 5G>C were undetectable in CRISPR-activated fibroblasts, suggesting differential effects of this variant on different isoforms. Together, the results suggest that c.652 + 5G>C-driven exon 2 skipping occurs during CRB1-C splicing, whereas CRB1-A and CRB1-B mRNA transcripts are completely destabilized in CRISPRa fibroblasts. Based on these results, we reclassified the c.652 + 5G>C variant according to ACMG standards from likely pathogenic 24 to pathogenic (Table 1).
Protein-coding CRB1 transcripts bearing the c.2850_2855delAAATGC in-frame deletion or the c.2555T>C, c.2843G>A, or c.3014A>T missense variants were expressed in both CRISPR-activated fibroblasts and RO, suggesting these variants exert pathogenic effects at the protein level. It remains unclear whether CRISPRa can produce sufficient protein for functional analyses and further studies in ROs may be necessary to determine pathogenic mechanisms at the protein level (such as mislocalization, misfolding, or loss of domain functions).
One limitation of this study is that not all splicing defects can be detected using the methods employed here. Large intronic inclusions may not be amplified due to large product size, whereas splicing alterations affecting the exons targeted by PCR could also prevent detection of aberrant splice products. Combination of our CRISPRa methods with long read sequencing techniques would help to improve detection of splicing alterations. In addition, the addition of NMD inhibitors during CRISPRa may also be useful to detect highly unstable transcripts.
Together, our results show that CRISPRa is a useful tool for inducing retinal gene expression in patient-derived fibroblasts, providing a rapid assay for molecular interrogation of transcripts containing pathogenic variants.
Footnotes
Acknowledgments
The authors thank all patients and families associated with the Western Australian Retinal Degeneration study for their participation in this project and sample contributions to our biobank.
Authors' Contributions
S.Y.M. contributed to conceptualization, methodology, formal analysis, investigation, writing—original draft, writing—review and editing, and visualization. D.Z. was involved in methodology, writing—review and editing, and supervision. S.-C.C. carried out methodology and writing—review and editing. T.M.L. carried out investigation and writing—review and editing. J.A.T. carried out investigation, formal analysis, validation, and writing—review and editing.
T.L.M. took charge of investigation and writing—review and editing. F.K.C. was involved in resources, writing—review and editing, supervision, project administration, and funding acquisition. S.M. was in charge of conceptualization, methodology, validation, resources, writing—original draft, writing—review and editing, visualization, supervision, project administration, and funding acquisition.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by the National Health and Medical Research Council of Australia (GNT1188694, GNT1054712, MRF1142962), the Government of Western Australia (Department of Health, WANM Fellowship Award), the McCusker Charitable Foundation, Telethon, Retina Australia, and donations from the Constantine family.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.