
Abstract
Disease resistance genes in livestock provide health benefits to animals and opportunities for farmers to meet the growing demand for affordable, high-quality protein. Previously, researchers used gene editing to modify the porcine CD163 gene and demonstrated resistance to a harmful virus that causes porcine reproductive and respiratory syndrome (PRRS). To maximize potential benefits, this disease resistance trait needs to be present in commercially relevant breeding populations for multiplication and distribution of pigs. Toward this goal, a first-of-its-kind, scaled gene editing program was established to introduce a single modified CD163 allele into four genetically diverse, elite porcine lines. This effort produced healthy pigs that resisted PRRS virus infection as determined by macrophage and animal challenges. This founder population will be used for additional disease and trait testing, multiplication, and commercial distribution upon regulatory approval. Applying CRISPR-Cas to eliminate a viral disease represents a major step toward improving animal health.
Introduction
Animal agriculture plays an important role in the global food system, providing 18% of the world's calories and 34% of its protein through the production of meat, dairy, and eggs. 1 Recent estimates by the World Organization for Animal Health suggest that 20% of livestock production is lost to disease annually. 2 While disease in livestock has profound economic consequences, the environmental impact, suffering endured by animals, and the psychological toll on farmers cannot be overlooked. Agriculture, like human medicine, can now modernize the treatment of disease through the adoption of new technologies that modify genes necessary to improve health.
Porcine reproductive and respiratory syndrome (PRRS) is a viral disease affecting domestic pigs. First recognized in the United States in 1987, this disease is now found in pig production facilities worldwide. The disease can occur across the lifespan of an animal, with symptoms that include anorexia, fever, lethargy, and depression. Respiratory distress is particularly problematic for young pigs, where mortality can approach 100% in suckling animals. As the name suggests, the disease affects the reproductive success of pigs through symptoms including late-term abortions, stillborn or mummified piglets, and weak piglets at birth. A PRRS virus (PRRSV) infection compromises the respiratory immune system, leading to increased severity of secondary infections that are commonly treated with antibiotics.
With annual global losses estimated at $2.7B, PRRS is a disease of primary concern to the pig industry and farmers worldwide. 3 The causative agent of PRRS is one of two species of enveloped, positive-strand RNA viruses, PRRSV-1 or PRRSV-2. To date, the primary strategy to prevent PRRSV infection has been through the administration of either modified-live virus or killed virus vaccines. Unfortunately, vaccines have shown limited effectiveness, 4 as PRRSVs have a high rate of mutation due to an error-prone viral RNA-dependent RNA polymerase and a significant rate of genetic recombination.
Work by several labs identified CD163 as the host entry receptor for PRRSV.5,6 CD163 is a type I transmembrane protein composed of nine tandem cysteine-rich scavenger receptor superfamily (SRCR 1–9) domains, a C-terminal transmembrane domain, and an intracellular cytoplasmic tail. CD163 is expressed on the surface of mature macrophages and monocytes, and functions in mediating inflammation and the removal of hemoglobin-haptoglobin complexes from the blood.7,8
SRCR domain 5 of CD163 is an interaction site for PRRSV 9 and is encoded entirely within exon 7. Previous research demonstrated that a dual guide RNA (gRNA) approach, wherein two Cas9-gRNA complexes were targeted to the introns flanking exon 7, could result in deletion of SRCR domain 5 in pigs. 10 This editing strategy resulted in animals with PRRSV-resistant pulmonary alveolar macrophages (PAMs) and peripheral blood monocytes, and these pigs showed no signs of infection or viral replication in lung and lymph node tissue when challenged with PRRSV. 11 The ability to confer PRRSV resistance through modification of a single gene represents an opportunity to improve porcine health and reduce farm production costs globally.
Translation of proof-of-concept work to a commercial-scale gene editing program requires additional considerations beyond identification of a target sequence: the allele conferring disease resistance must exist in genetically advanced breeding lines with sufficient diversity to allow for continued genetic gain, and the modified allele must not negatively impact commercial performance. 12 Four breeding lines of domestic pigs (Sus scrofa domesticus) are typically utilized in commercial food production 12 : Landrace and Large White are elite dam lines with excellent maternal traits, such as age of puberty, fertility, and litter size; Duroc and a white composite line are elite sire lines with excellent production traits, such as growth rate, feed efficiency, and carcass composition. Any edit to CD163 conferring PRRSV resistance must be introduced into all four lines to maintain their high genetic merit and accelerate time to market. Introducing the same edit across all lines ensures consistent molecular composition and expression of the CD163 allele, while enabling a more straightforward regulatory review process.
In this report, a scaled gene editing program for the generation of a founder population of non-transgenic, PRRSV resistant pigs for commercial breeding is described. CD163 was modified using a dual gRNA-Cas9 ribonucleoprotein (RNP) strategy to remove exon 7, which has been previously shown to confer resistance to PRRSV infection while retaining known biological activities.11,13
The unique allele was generated in four different breeding lines, with sufficient genetic diversity captured to enable immediate population expansion and maximize future genetic potential. Extensive optimization and validation was employed to identify editing reagents with optimal on-target efficiency and low-off target activity. Further steps involved the molecular characterization of the modified CD163 allele across three generations and the identification and removal of off-targets during the breeding process
In agreement with earlier reports,11,13 these homozygous CD163 exon 7 deletion (CD163ΔE7/ΔE7) pigs were healthy and indistinguishable in appearance and behavior when compared to a cohort of unedited pigs. Further, animals homozygous for CD163ΔE7 and monocyte-derived macrophages (MoMØs) from these animals were completely resistant to both Type I and Type II PRRSV infection.
Materials and Methods
Cell culture, transfection, and indel detection
Porcine fetal fibroblasts (PFFs) were used for initial evaluation of editing reagents. PFFs were prepared as described 14 and cultured at 38.5°C with 5.0% O2 and 5.0% CO2 in Dulbecco's Modified Eagle Medium (high glucose, GlutaMAX Supplement, pyruvate; Thermo Fisher Scientific) supplemented with 12% fetal bovine serum (Cytiva), 1% Minimal Essential Medium (MEM) non-essential amino acids (Thermo Fisher Scientific), penicillin-streptomycin (100 U/mL and 100 μg/mL, respectively; Thermo Fisher Scientific), and 100 μM 2-Mercaptoethanol (Sigma-Aldrich).
Streptococcus pyogenes Cas9 RNPs (20 pmol) were nucleofected into 150,000 PFF cells using the P1 Primary Cell kit in the 4D-Nucleofector using program CM137 or using the P3 Primary Cell kit in the 4D-Nucleofector using program CM138 (Lonza). Transfected cells were grown for 48 h, and gDNA was harvested with QuickExtract Solution (LGC Biosearch Technologies) according to manufacturer protocols.
Target sites were amplified in a two-step PCR reaction. In brief, 3.75 μL (corresponding to ∼7500 cells) of lysate was used as a template for PCR amplification with Q5 Hot-Start High Fidelity DNA Polymerase (NEB) and unique primer pairs containing both an internal, locus-specific region and an outer Illumina-compatible adapter sequence. A second PCR targeting the outer adapter sequence was performed to append unique indices to each amplicon, and sites were sequenced in triplicate using the Illumina platform to an average depth of ∼10k reads/sample.
Generation of edited pigs
Edited pigs were generated by injection of S. pyogenes Cas9 RNPs into porcine zygotes. The estrous cycles of donor and surrogate gilts (∼40 and 20, respectively, per surgery round) were synchronized by a combination of MATRIX® (altrenogest) Solution 0.22% (Merck Animal Health), pregnant mare serum gonadotropin (PMSG; ProSpec), and human chorionic gonadotropin (hCG; ProSpec). Donor animals were artificially inseminated twice using elite, line-specific semen following standard practices.
Approximately 16 h post-insemination, presumptive zygotes were harvested and used for RNP injections. The approximate number of harvestable embryos ranged from 10 to 20 for Landrace, Large White, and the white composite line, and less than 10 embryos for Duroc gilts. On average, 50–70% of harvestable embryos were used for injection of editing reagents. Purified in vitro transcribed gRNAs and Cas9 protein were prepared as described. 15 For zygote injections, 4.56 μL of each gRNA (1.5 μg/μL) was denatured separately at 95°C for 2 min in a thermal cycler, cooled to room temperature, and added to H2O (1.41 μL) and Cas9 (1.0 μL) to form two separate RNP complexes.
RNP complexes were combined by mixing and stored on ice. Immediately before use, 7 μL of combined RNP complex was diluted with 193 μL of H2O to provide a working concentration of 50.2 ng/μL (0.31 pmol) of Cas9 and 17.2 ng/μL (0.468 pmol) of each single gRNA (sgRNA). The RNP solution was injected into the cytoplasm of presumptive zygotes with a single pulse from a FemtoJet™ 4i microinjector (Eppendorf).
Microinjection was performed in ABT TL Hepes (ABT 360; LLC) supplemented with 3 mg/mL BSA (Proliant) on the heated stage of an inverted microscope equipped with Narishige micromanipulators (Narishige International USA). Estimated volume injected per zygote was ∼20 pL (40 fg or 1.5 amol RNP complex). The injected zygotes were cultured in PZM5 medium 16 for 36–48 h in a humidified incubator at 38.5°C with 5% CO2 and 5% O2 to 4-cell stage embryos. On average, 30 developmentally competent, injected embryos were surgically transferred into synchronized gilts.
DNA isolation and sequencing of edited animals
Tail samples from edited piglets were collected in a conical tube and frozen for subsequent molecular analysis by Illumina sequencing. Genomic DNA was extracted from tissue samples using the QIAGEN DNeasy Blood and Tissue kit in 96-well format (QIAGEN) based on manufacturer instructions. The region surrounding exon 7 was amplified via a dual, three-primer strategy allowing Illumina sequencing.
Short amplicon sequencing primers are listed and contain standard Illumina adapters appended to the 5′ end for use in library construction and indexing. Intron 6-Exon 7 junction assay primers: P1-F: 5′-ATCGGCTAAGCCCACTGTAG-3′, P2-R: 5′-TTCACCAAGCGGATTTGTGT-3′, PC-3: 5′-CAACCAGCCTGGGTTTCCTG-3′. Exon 7-Intron 8 junction assay primers: P1-F: 5′-ATCGGCTAAGCCCACTGTAG-3′, P2-R: 5′-TTCACCAAGCGGATTTGTGT-3′, PC-4: 5′-GGACATGTAGCCACAGCAGG-3′.
Oxford Nanopore Technologies and DNA hybridization capture sequencing of CD163 alleles in animals
Oxford Nanopore Technologies (ONT) long-read sequencing was used to evaluate CD163 structural integrity in edited animals, whereas DNA hybridization capture sequencing was used to determine CD163 sequence at high resolution. Primers flanking the exon 7 deletion region were used to generate and sequence a ∼2.9 kb DNA region using the ONT GridION platform following manufacturer protocols. ONT primers: ONT-F: 5′-AGGATGCCAGTCTGTGTAGAG-3′ and ONT-R: 5′-ATGGGGGAATCCCTTTCACTTC-3′.
For DNA hybridization capture sequencing, a custom DNA oligo bait library (Arbor Biosciences) was generated to capture the CD163 gene, excluding regions of moderate to high repetition but allowing for saturation of the region to ensure uniform coverage within the non-repetitive regions of the gene. Whole-genome shotgun libraries were generated for sequencing on the Illumina platform from DNA of selected parents, E0, and E1 animals. The hybridized and sequenced fragments were compared to Sus scrofa genome assembly Sscrofa11.1 using standard alignment tools such as BWA-MEM 17 and Samtools. 18
Off-target sequence generation and detection
Potential off-target cut sites were identified using the SITE-Seq® assay 15 (Caribou Biosciences, Inc.). Briefly, purified genomic DNA derived from a composite line boar was incubated with S. pyogenes Cas9 RNP, followed by labeling, enrichment, and sequencing of DNA cleavage sites. Probes against 2 kb regions of DNA centered on sites identified by the SITE-Seq® assay were designed (Arbor Biosciences) and used in sequence capture experiments to identify off-target indels in selected E0 and E1 animals that contained the desired CD163 exon 7 deletion.
Whole genome sequencing and informatics
Reads were aligned to S. scrofa genome assembly Sscrofa11.1 using standard bioinformatics methods, including BWA-MEM, Samtools, and GATK MarkDups. 19 Variants were then joint called using GATK. De novo variants were identified using methods proposed by Kaplanis et al. 20 Briefly, variants were required to have coverage ≥15 × and ≤100 × , have only a single alternate allele, and have the variant not present in any other samples (wild-type [WT] founders or other E0 animals).
To remove additional sequencing artifacts, putative de novo variants were also compared against the remaining samples. De novo variants required ≥4 reads in E0s and ≥2 reads in all other samples. Putative de novo variants were removed as sequencing artifacts if the variant had supporting reads in any other sample. Copy number variants (CNVs) were detected using CNVpytor. 21
Virus propagation
Viruses were propagated on MARC-145 cells 22 in MEM (Thermo Fisher Scientific) supplemented with 7% fetal bovine serum and Penicillin-Streptomycin (80 U/mL and 80 μg/mL, respectively) for at least 4 days before titration. For titration, viruses were serially diluted 1:10 in media, added in quadruplicate to confluent MARC-145 cells in a 96-well plate to a final volume of 200 μL per well, and incubated for 4 days at 37°C in 5% CO2.
The titration endpoint was identified as the last well in each dilution series with cytopathic effect. The 50% tissue culture infectious dose (TCID50/mL) was calculated using the method of Reed and Muench. 23 Primary cultures of porcine alveolar macrophages were substituted for MARC-145 cells in the propagation and titration of the UIL21-0712 isolate.
Infection of macrophages
Macrophages derived from blood monocytes (MoMøs) were maintained in RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% FBS, and penicillin-streptomycin (100 U/mL and 100 μg/mL, respectively). Viruses were serially diluted 1:10 in media and added to macrophages on a 24-well plate. After incubation overnight, the cells were washed with phosphate buffered saline (PBS) and fixed for 10 min with 80% acetone.
To detect infected macrophages, cells were stained with PRRSV N-protein mAb SDOW-17 (RTI) diluted 1:1000 in PBS with 1% fish gelatin (PBS-FG; Sigma-Aldrich). After a 30 min incubation at 37°C, the cells were washed with PBS and stained with Alexa Fluor 488-labeled anti-mouse IgG (Thermo Fisher Scientific) diluted 1:200 in PBS-FG. Plates were incubated for 30 min in the dark at 37°C, washed with PBS, and viewed under a fluorescence microscope. The dilution of virus showing the maximum level of infection was used to report the result.
Immunoblot analysis of CD163 protein from porcine macrophages
To prepare MoMØs, whole blood was collected in 10 mL lithium heparin tubes and centrifuged at 1200 RCF for 10 min. Buffy coat was transferred into ACK Lysis Buffer (Thermo Fisher Scientific), incubated at room temperature for 8 min, and centrifuged at 1200 RCF for 10 min. The cell pellet was resuspended in PBS, and cell counts were determined using a Countess™ 3 Automated Cell Counter (Thermo Fisher Scientific).
Cells were added to six-well plates coated with 0.1% Type A gelatin from porcine skin (Sigma-Aldrich) in RPMI 1640 supplemented with 10% FBS, 1% MEM Non-Essential Amino Acids, Penicillin-Streptomycin (100 U/mL and 100 μg/mL, respectively), and 50 ng/mL recombinant porcine granulocyte-macrophage colony-stimulating factor (rpGM-CSF; R&D Systems). Cells were cultured for 7–10 days at 37°C in 5% CO2 and 20% O2.
CD163 protein expression in MoMØs or pulmonary alveolar macrophages (PAMs) was assessed by immunoblotting. Cells were lysed in RIPA Buffer (Thermo Fisher Scientific) containing 1 × Halt™ Protease Inhibitor Cocktail, EDTA-Free (Thermo Fisher Scientific) and protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Cell lysates were prepared in 4 × Bolt™ LDS Sample buffer (Thermo Fisher Scientific) and 10 × Bolt Sample Reducing agent (Thermo Fisher Scientific) and then heated at 70°C for 10 min.
Proteins were electrophoresed on an 8% Bolt Bis-Tris Plus Mini Gel (Thermo Fisher Scientific) in 1 × Bolt MES SDS Running Buffer (Thermo Fisher Scientific) and transferred to nitrocellulose membrane in 1 × Tris/Glycine Buffer (Bio-Rad) with addition of 10% methanol. Membranes were blocked in 5% non-fat dry milk in 1 × TBS with Tween™ (TBST; Thermo Scientific Chemicals) and incubated at 4°C overnight with anti-CD163 antibody (#ab8709; Abcam9) diluted 1:1000 or anti-Actin pan mAb MA5-11869 (Thermo Fisher Scientific) diluted 1:2000.
Membranes were washed three times with 1 × TBST, incubated 1 h at room temperature with goat anti-Rabbit IgG (H+L)-HRP (#1706515; Bio-Rad) diluted 1:10,000 or Goat Anti-Mouse IgG (H+L)-HRP (#1721011; Bio-Rad) diluted 1:10,000, washed three times with 1 × TBST, processed using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific), and imaged on an iBright Imaging System (Thermo Fisher Scientific).
Animal challenges inoculum and infection
A 3 mL inoculum containing between 4 and 5 log TCID50 PRRSV was administered to each pig. Half of the inoculum was administered intramuscularly using a 21-gauge needle, after which the needle was removed and the remainder of the inoculum was administered intranasally. Ten milliliters of blood was collected from each pig at 0, 3, 7, 10, 14 and days 17–21 post-infection and separated into serum. Serum samples were stored at −80°C.
PRRS assays
PRRSV nucleic acid in serum was measured using the EZ-PRRSV™ MPX 4.0 Master Mix (Tetracore). Results were reported as quantitative PCR (qPCR) cycle threshold (Ct), with a Ct value less than 37 considered positive for PRRSV nucleic acid. 24 PRRSV antibody was measured by enzyme-linked immunosorbent assay (ELISA) using the IDEXX PRRS X3 Ab Test (IDEXX), with a sample/positive (S/P) ratio greater than 0.39 considered positive for the presence of PRRSV antibody.
Statement on the ethical use of animals
Experiments involving the use of viruses and animals were performed in accordance with the Federation of Animal Science Societies Guide for the Care and Use of Agricultural Animals in Research and Teaching, the USDA Animal Welfare Act and Animal Welfare Regulations, and approved by the University of Illinois Institutional Animal Care and Use Committee and the Institutional Biosafety Committee. Pigs were humanely euthanized by intravenous pentobarbital injection according to American Veterinary Medical Association Guidelines on Euthanasia.
All research conducted in this report was subject to authorization and oversight by the U.S. Food and Drug Administration (INAD I-012878) for investigational development of pigs containing an intentional genomic alteration (IGA) of the CD163 gene. Claims of safety and efficacy of the IGA are under review by FDA.
Results
gRNA screening and selection
A dual gRNA strategy based on Cas9 RNP delivery into porcine zygotes eliminates the risk of foreign DNA integration, increases the frequency of porcine zygotes containing a CD163 exon 7 deletion, and minimizes the occurrence of Cas9-induced off-target insertion-deletions (indels). 25 To optimize this approach, RNP pairs would first be screened to find the combination of gRNAs that maximizes the desired on-target repair outcome. However, screening RNP pairs presents a challenge as there are multiple combinations, and accurately quantifying novel allele frequencies is complicated by the preferential PCR amplification of larger deletions when genotyping.
As such, on-target activity (indirectly determined by indel frequency) was initially determined for individual RNPs, based on the assumption that high-efficiency RNPs are promising candidates for subsequent dual gRNA screening. Specifically, 58 Cas9 RNPs targeting either upstream (i.e., within the 3′ 500 bp region of intron 6) or downstream (i.e., within intron 7) of exon 7 (Fig. 1A) were assayed for activity.
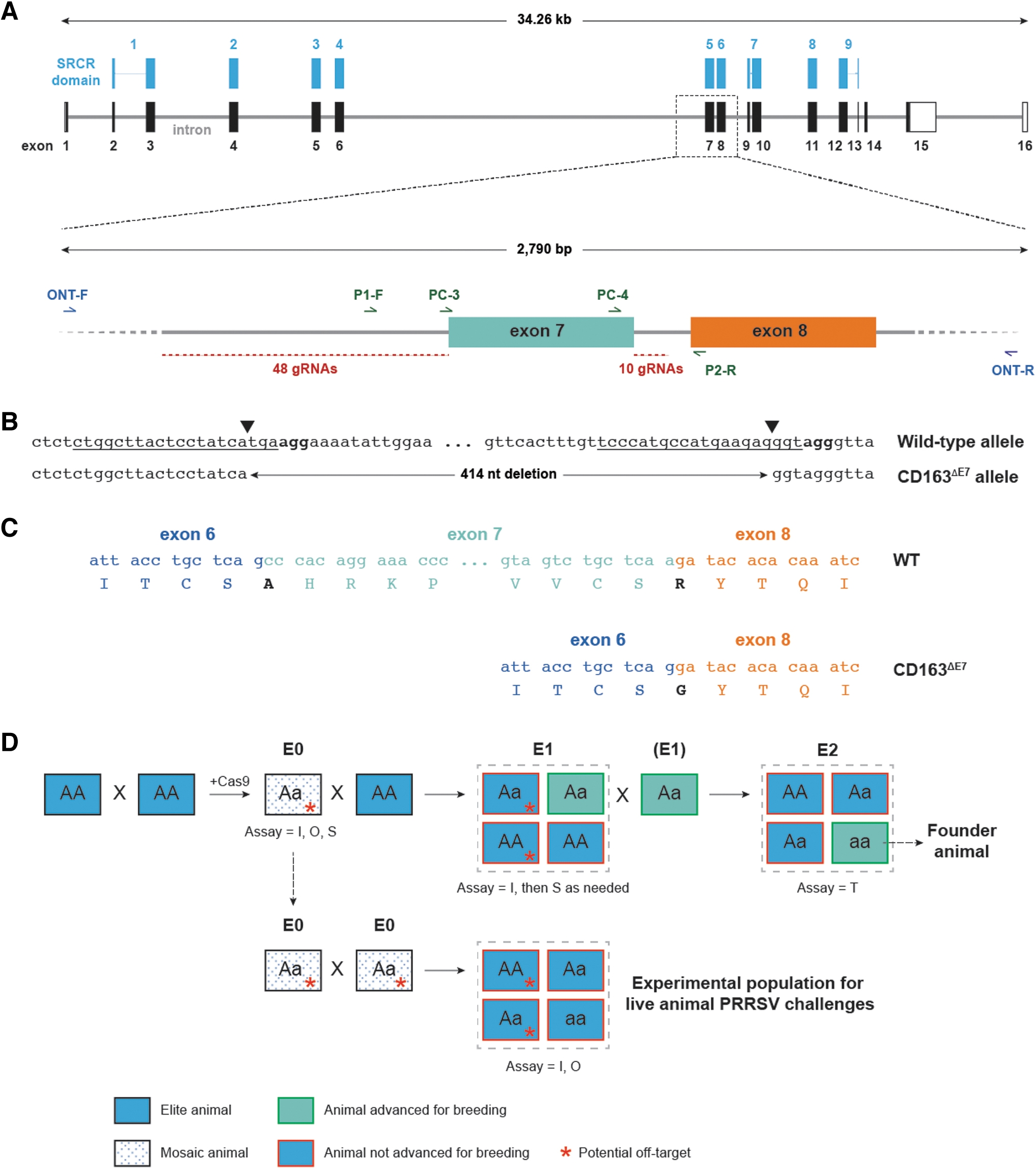
Generation of a CD163 allele lacking exon 7 using CRISPR-Cas and breeding strategy for establishing a founder population.
Cas9 was complexed with individual sgRNAs and the resulting RNPs were nucleofected into PFFs. PFFs were cultured for 48 h, lysed, and short DNA amplicons spanning this region of CD163 were sequenced using an Illumina MiSeq platform. Across RNPs, indel frequencies ranged from <1% to ∼60% (Fig. 2). Eleven 5′ sgRNAs and three 3′ sgRNAs were selected for further characterization.
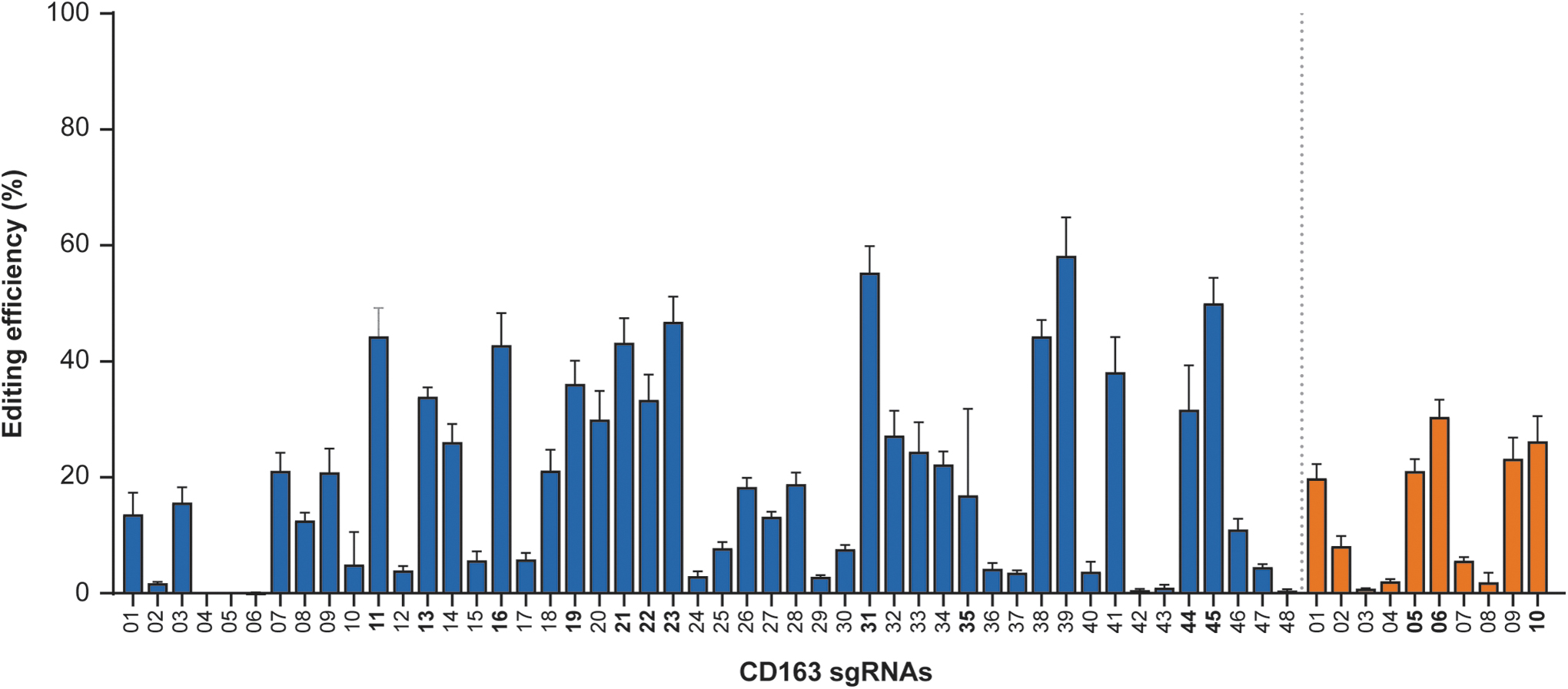
On-target editing activity of Cas9 RNPs flanking CD163 exon 7. gRNAs were combined with Cas9 to form RNPs and delivered to PFFs via nucleofection. Illumina short amplicon sequencing was used to query the regions around the Cas9 cleavage site. RNP editing efficiency is based on the fraction of indels to total reads, and accounts for both nucleotide and amplicon length differences compared to amplicons from unedited control cells. sgRNAs upstream of exon 7 (blue) and sgRNAs downstream of exon 7 (orange) are shown with errors bars indicating standard deviation. PFF, porcine fetal fibroblast; sgRNA, single gRNA.
Biochemical identification of Cas9-sgRNA off-targets
The SITE-Seq® assay was used to map potential Cas9-sgRNA off-target cleavage sites in vitro and assist in prioritization of sgRNAs for editing CD163 in zygotes. The SITE-Seq® assay was performed in triplicate on the 14 sgRNAs identified above across multiple RNP concentrations (4, 16, 64, and 256 nM), ranging from limiting to saturating amounts of RNP, to recover both high- and low-sensitivity cellular off-targets, respectively.
On-target recovery was observed at all RNP concentrations, and the number of recovered cleavage sites scaled with RNP concentration. The number of cleavage sites varied widely across RNPs, strongly suggesting that some RNPs had higher in vitro cleavage specificity than others (Fig. 3).

Off-target identification for selected guides. The SITE-Seq® assay was performed in triplicate at the RNP concentrations indicated. Numbers at each concentration refer to the average number of cleavage sites identified and include on-target site. The number of sites identified at a given concentration includes those identified at lower concentrations.
Dual guide characterization and selection
The intended deletion of CD163 exon 7 described in this report consists of end-to-end repair of Cas9 cleavage sites, with no extraneous insertions, deletions, or substitutions at the cut sites (Fig. 1B, C and Supplementary Fig. S1). Based on initial sgRNA screening and off-target analysis, 14 RNPs were assembled in pairwise combinations and introduced into PFFs via nucleofection to quantify their ability to generate end-to-end repairs. PCR amplicons targeting the putative Cas9 cleavage sites were again queried by Illumina short amplicon sequencing.
Across RNP combinations, the percentage of reads representing the desired repair outcome varied widely, from no detectable deletions up to 38% (Fig. 4A). While RNP screening in PFFs is useful for initial evaluation of activity, the editing frequency in zygotes that develop into blastocyst stage embryos more closely matches that of edited animals. Therefore, a subset of RNP pairs was tested for their ability to generate end-to-end repairs in porcine embryos.
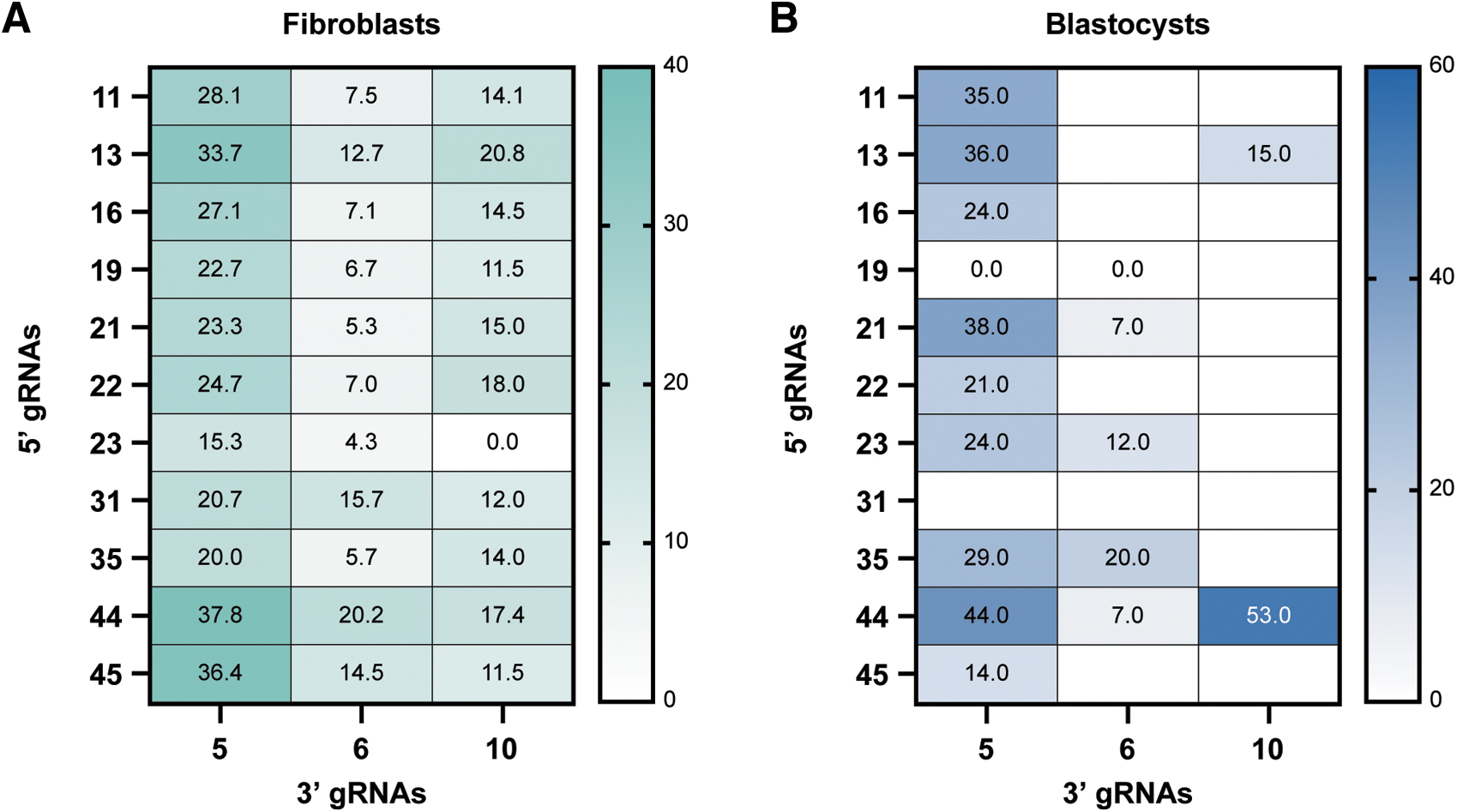
Paired guide analysis.
RNPs were injected into in vitro fertilized porcine zygotes that were cultured to blastocyst stage and queried by Illumina short amplicon sequencing. Figure 4B shows the percentage of blastocysts containing the desired edit. Based on the editing efficiencies in PFFs and blastocysts and proximity to the exon 7/intron 7 junction, 5′ sgRNA 44 and 3′ sgRNA 5 were chosen for generation of edited animals.
Generation and analysis of edited animals
Fertilized zygotes obtained from synchronized gilts artificially inseminated with line-specific semen were microinjected with RNP complexes to generate the first generation of edited animals (E0). The ex vivo injected embryos were cultured to 4-cell stage embryos and surgically transferred into oviducts of synchronized recipient gilts. Pregnant gilts farrowed the piglets naturally ∼112–116 days after embryo transfer.
Post-farrowing, tail tissue from E0 piglets was screened for the presence of the intended edit and possible off-target edits using three methodologies: (1) Illumina short amplicon sequencing to query the immediate area of the targeted locus, (2) ONT long-read sequencing to query a larger window around the targeted locus, and (3) hybridization-based sequence capture to query the entire coding region and adjacent regulatory regions of CD163, as well as potential off-targets identified by the SITE-Seq® assay.
Illumina short amplicon sequencing was employed to screen all E0 animals for the presence of the intended edit, as well as other unintended alterations contained within, or immediately adjacent to, the targeted CD163 locus. The size of the deletion generated by the dual RNPs (Fig. 1B) necessitated the design of a dual 3-primer PCR assay to produce similar-sized amplicons to address PCR amplification and sequencing bias.
Across four surgery rounds, which included all four lines, ∼21% of animals (90 of 435 piglets) contained the intended edit as determined by Illumina sequencing (Table 1). It is well established that microinjection of Cas9-sgRNA complexes into mammalian zygotes can generate mosaic E0 animals, with more than two unique alleles present at the target locus.26–28 Consistent with these studies, Illumina sequencing identified E0 animals with multiple alleles, consisting of various exon 7 deletions and/or indels at either sgRNA cut site. For surgery Round 1, Illumina sequencing revealed one or two alleles in ∼60% of the animals, with three or more alleles present in the remaining 40% of animals (Supplementary Table S1). In animals that contained the CD163ΔE7 allele, the Illumina read count frequency for this edit ranged from 14% to 99%.
Generation of E0 animals
L, Landrace; LW, Large White; C, white composite line; D, Duroc.
Larger structural events (e.g., insertions, deletions, inversions, and more complex arrangements of these types of alterations) are also known to occur as a byproduct of the editing process and can be either missed or misclassified by Illumina or Sanger sequencing due to loss or rearrangement of PCR primer binding sites. 29 Such structural events can impact interpretation of short amplicon sequencing results, where animals appear homozygous but harbor additional alleles.
To overcome this shortfall, 2.8 kb PCR amplicons were used to generate ONT long-read sequencing libraries to evaluate the structural integrity adjacent to, and encompassing, the intended edit (Fig. 1 and Supplementary Fig. S2). The ability of ONT to provide contiguous sequence of these PCR amplicons allowed for identification of larger structural events as well as determining whether those events are in cis or trans orientation with the intended edit.
The mosaicism observed by Illumina short amplicon sequencing was confirmed by ONT long-read sequencing, with more than two alleles observed in animals and at varying frequencies. ONT also revealed the presence and frequency of larger structural events in edited animals, with ∼20% of Round 1 animals having large (>450 bp) deletions or inversions (Supplementary Fig. S2). These structural events are likely the cause of the apparent homozygosity in some E0 animals as determined by short amplicon sequencing.
To identify edited animals which maintained sequence integrity outside of the intended exon 7 deletion, a DNA hybridization sequence capture strategy was employed. Due to the size of CD163 (35 kbp, including exons, introns, and flanking regulatory regions), a long PCR strategy to span the entire locus would not be feasible from a technical perspective.
The process for developing and analyzing the CD163 sequence capture library was adapted from the previously published experimental work evaluating the sequence variation within the exons of CD163. 30 A “trio” arrangement was utilized in this analysis pipeline, where the wild-type parents of E0 animals were evaluated along with their progeny. This approach allowed for discrimination between inherited parental sequence variation and that resulting from the editing process. Any unexpected variation present in the E0 animals and absent in the parents was flagged for more detailed evaluation.
In addition to using sequence capture to identify sequence variation at the CD163 locus, this methodology was also used to identify sequence variation at predicted off-target sites identified by the SITE-Seq® assay. Biochemical cleavage sensitivity in the SITE-Seq® assay was previously shown to predict off-target editing in vivo. 15 Accordingly, all cleavage sites identified by the SITE-Seq® assay at an RNP concentration of 16 nM, which includes the 4 nM sites, were queried in selected E0 animals, under the rationale that these sites represent the potential in vivo off-targets.
A combined total of 182 sites were identified for both guides at this concentration (Supplementary File S1). Given the number of loci to be queried and the number of E0 animals being screened, a hybridization-based sequence capture library consisting of tiled oligonucleotides spanning a 2 kb region centered on the SITE-Seq® cleavage sites was synthesized and validated. SITE-Seq® cleavage sites that showed sequence variation in E0 animals relative to the parents were prioritized for screening in the subsequent E1 generation progeny.
Generation and analysis of E1 and E2 edited animals
E0 animals containing the desired edit were crossed to line-specific wild-type animals with high genetic merit to produce a heterozygous E1 generation (Fig. 1D). To rapidly expand this E1 population, CD163ΔE7 E0 boars from each of the four lines were mated to 20–25 WT gilts, producing litters of 10–20 piglets. This breeding strategy served to simplify genetic analysis of edited animals due to mosaicism and allowed an opportunity to continue genetic improvement by introducing the most current high genetic merit mates to the multiplication process.
Illumina short amplicon sequencing of E1 progeny confirmed germline transmission of the desired CD163 edit. For the 24 E0 boars from surgery Rounds 1–4, transmission frequencies of the desired edit ranged from 5% and 100% (Supplementary Table S2). E1 progeny containing the desired edit were further screened by sequence capture to characterize the edited CD163 allele and identify transmitted off-target indels (now in a heterozygous state) at the 182 sites identified by the SITE-Seq® assay. Off-targets were not detected in the majority of E1 animals containing the desired edit. However, 9 of the 24 boars had detectable off-targets, resulting in a reduction in the number of animals brought forward for breeding to generate the E2 animals (Supplementary Table S2). Chromosome locations and sequences of in vivo off-targets identified by sequence capture are shown in Figure 5.
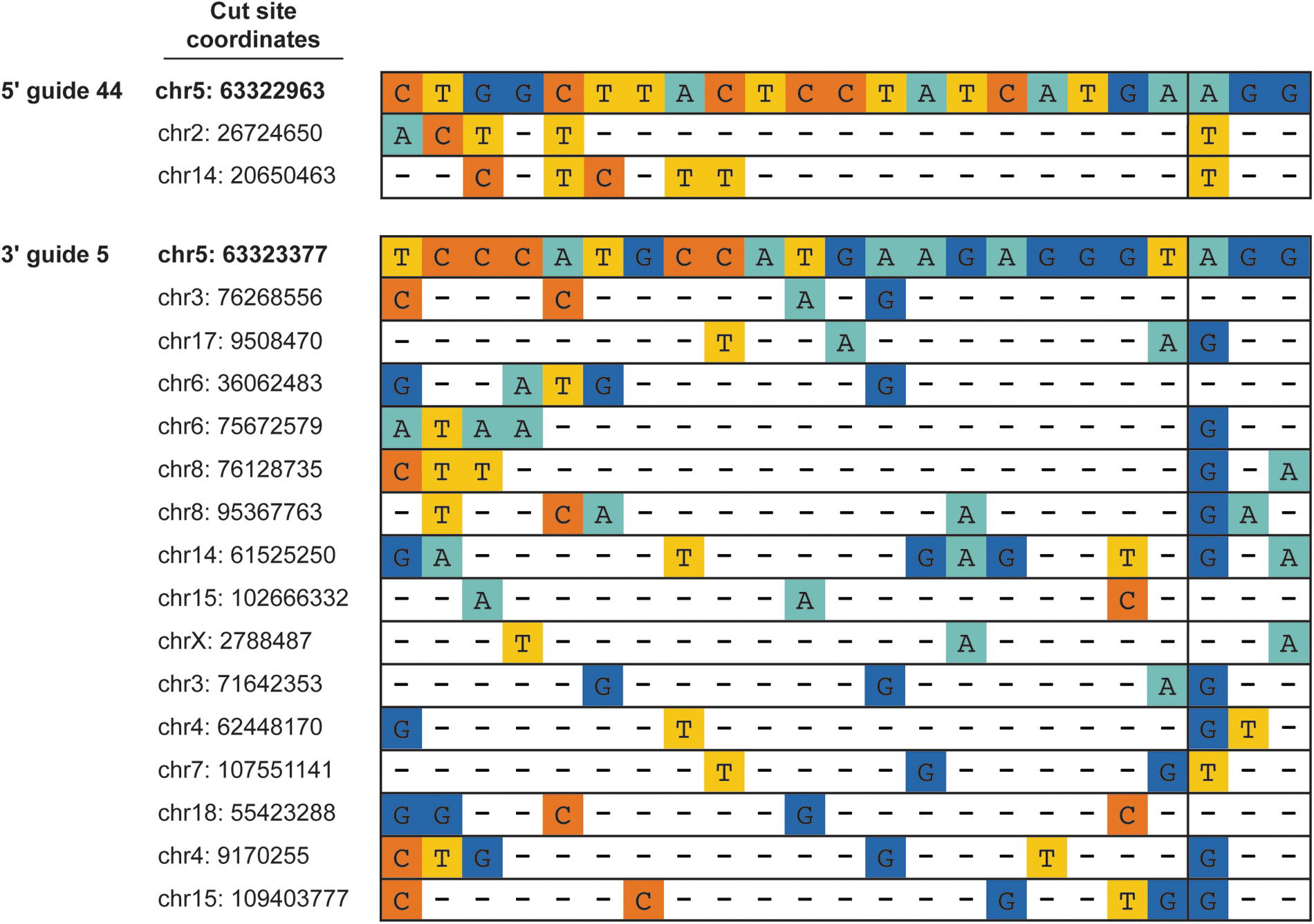
Off-targets identified in edited animals. Potential off-target cleavage sites identified by the SITE-Seq® assay were queried by sequence capture in E0 animals and in E1 animals that contained the desired CD163 edit. Sites above were found in one or more edited animals. Top of each panel shows Cas9 spacer sequence and PAM for 5′ guide 44 (top) and 3′ guide 5 (bottom). Conserved nucleotides are depicted with a dash (−).
To ensure high genetic merit animals served as parents in subsequent matings, the genetic index of CD163WT/ΔE7 E1 animals that did not contain off-target indels was estimated using Geneseek GGP Porcine 50K SNP Chip (Neogen, Lincoln, NE, USA) analysis. 31 Select heterozygous E1 animals were crossed in a line-specific manner to generate segregating E2 populations.
As the desired CD163 edit was confirmed in the E1 generation, and E1 animals containing off-targets were not used to generate E2 progeny, genotyping of the CD163 locus was simplified in the E2 generation. An end-point TaqMan assay was used to determine zygosity at the CD163 locus. Homozygous CD163ΔE7/ΔE7 pigs appeared healthy and indistinguishable in appearance and behavior when compared to unedited pigs of the same line (Fig. 6A). Immunoblot analysis verified that heterozygous and homozygous pigs expressed a CD163 protein that was smaller than the WT protein due to presence of the CD163ΔE7 allele (Fig. 6B).
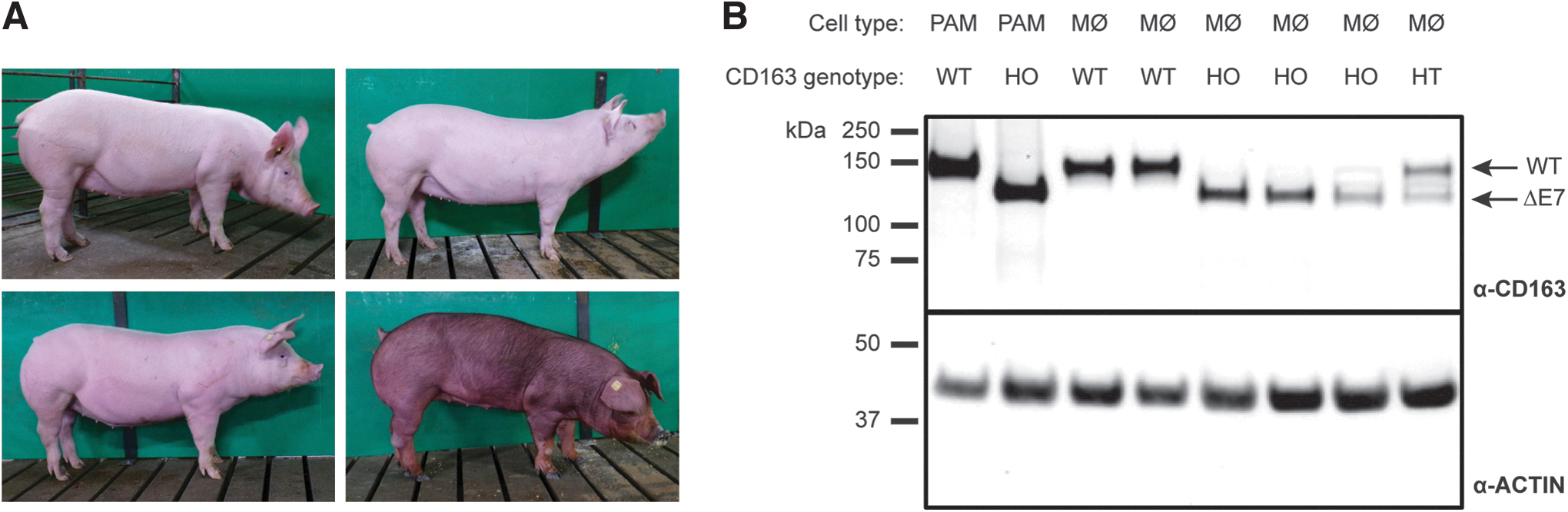
PPRSV-resistant pigs are healthy and lack CD163 exon 7.
Whole genome sequencing of edited animals
To validate identification of off-targets in edited pigs and verify their removal from the population through breeding, pedigree-matched parents of a small number of E0 edited pigs and their progeny were analyzed by whole genome sequencing (WGS). Ten WT parents, their 5 E0 progeny, 6 E1 animals, and 3 E2 animals (24 animals total) were selected for WGS (Fig. 7). WGS was also conducted on a litter of 13 WT animals and pedigree-matched parental dams and sires as controls. Due to mosaicism in E0 pigs, the E0 animals were sequenced to an average depth of 50 × while the remaining animals were sequenced to an average depth of 30 × . Based on sequence capture experiments, four of the five E0 pigs had evidence of indels at off-target sites identified by the SITE-Seq® assay, with indels at read frequencies of 4.5–14.5% (Fig. 7).
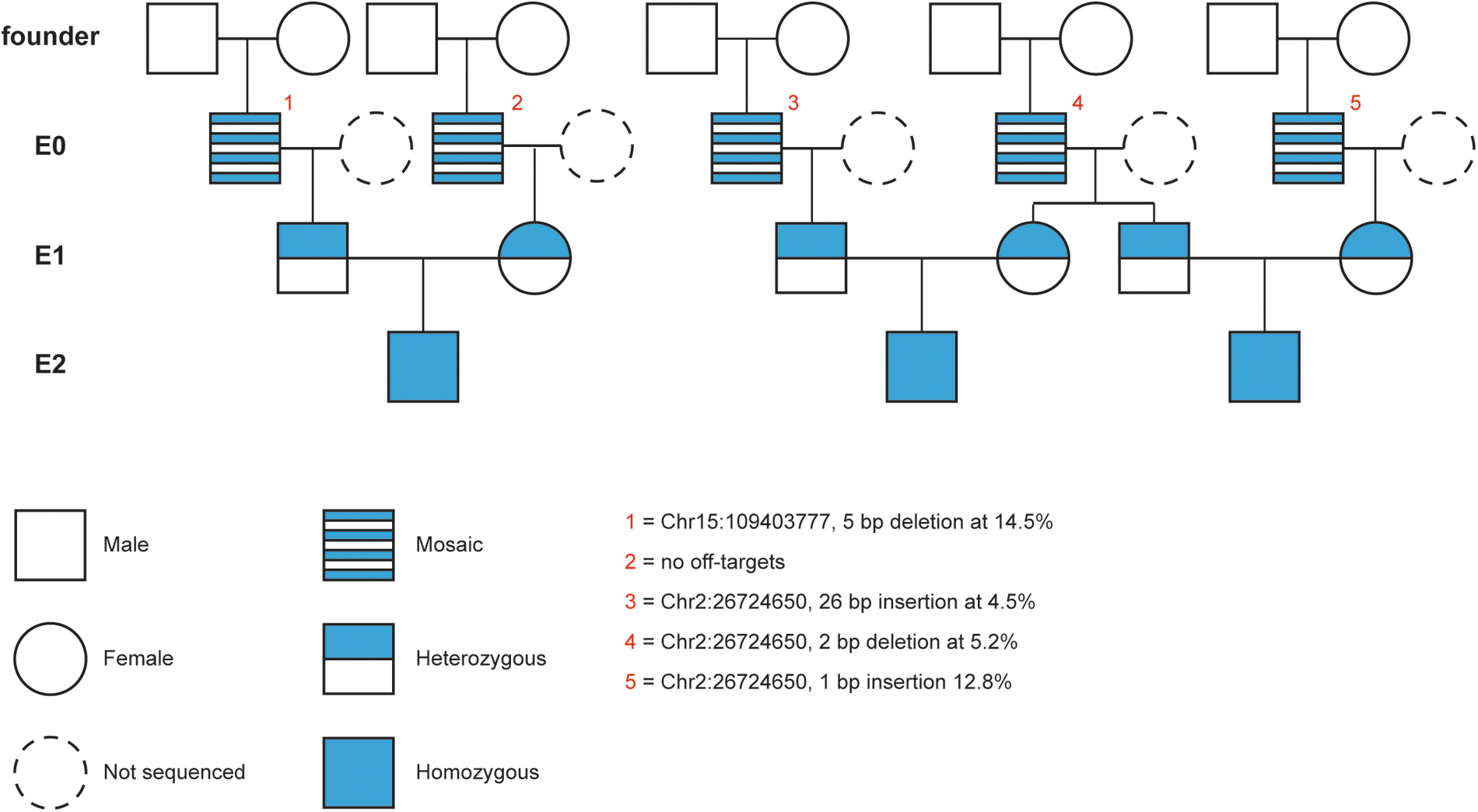
WGS analysis of edited animals. Pedigree of animals sequenced in this study. Zygote injection of editing reagents occurs in the founder generation to produce mosaic E0 animals. E0 boars were crossed to unedited, line-specific females to produce heterozygous E1 litters. E1 animals were crossed to produce segregating E2 litters. E0 animals are numbered in red. For each E0 animal, the location of any off-target, nature of edit, and frequency (as determined by sequence capture) are indicated. WGS, whole genome sequencing.
WGS identified an average of 63 (standard deviation [SD] = 8) de novo mutations in the E0 animals, compared to an average of 95 (SD = 26.6) and 80 (SD = 15) de novo mutations in the E2 trios and WT litter, respectively. Despite greater sequencing depth in the E0 animals, the number of de novo mutations in E0 animals was not greater than the number of de novo mutations identified in E2 or WT animals and was similar to that reported for mice and humans.32–34
These data support the findings that preimplantation embryo culture, embryo transfer, and Cas9 microinjection are not associated with an increase in de novo mutations.34–36 While the de novo mutations identified by WGS analysis of E0 animals included the off-targets identified by sequence capture, the remaining de novo mutations identified in E0, E1, or E2 animals did not correspond to the sites identified by the SITE-Seq® assay, even at the highest RNP concentration of 256 nM. This suggests that Cas9 RNP-mediated off-target indels at sites identified by the SITE-Seq® assay were effectively removed through a combination of screening by DNA sequence capture and selective sexual crosses within the E1 population.
PRRSV challenges
To verify that edited CD163ΔE7 pigs resisted PRRSV infection at the macrophage and animal level, an experimental set of segregating E1 animals was generated by crossing select, line-specific E0 boars and gilts (Fig. 1D). E0 animals possessing the desired CD163 edit and containing only one or two other CD163 alleles were used for crosses within lines, with resulting E1 progeny screened at the CD163 locus by Illumina short amplicon sequencing and ONT long-read sequencing.
For cell-based viral challenges, blood monocytes from edited Landrace and the white composite line pigs were isolated and cultured into MoMØs according to Patton et al. 37 Two cell-based challenges were conducted using MoMØs derived from unedited, heterozygous, and homozygous CD163ΔE7 pigs. Infection with PRRSV-1 or PRRSV-2 isolates was not detected in homozygous CD163ΔE7 MoMØs, in contrast to unedited CD163WT/WT or heterozygous CD163ΔE7/WT MoMØs (Fig. 8). A second trial using MoMØs derived from a different set of animals and challenged with additional PRRSV strains produced similar results (Table 2).
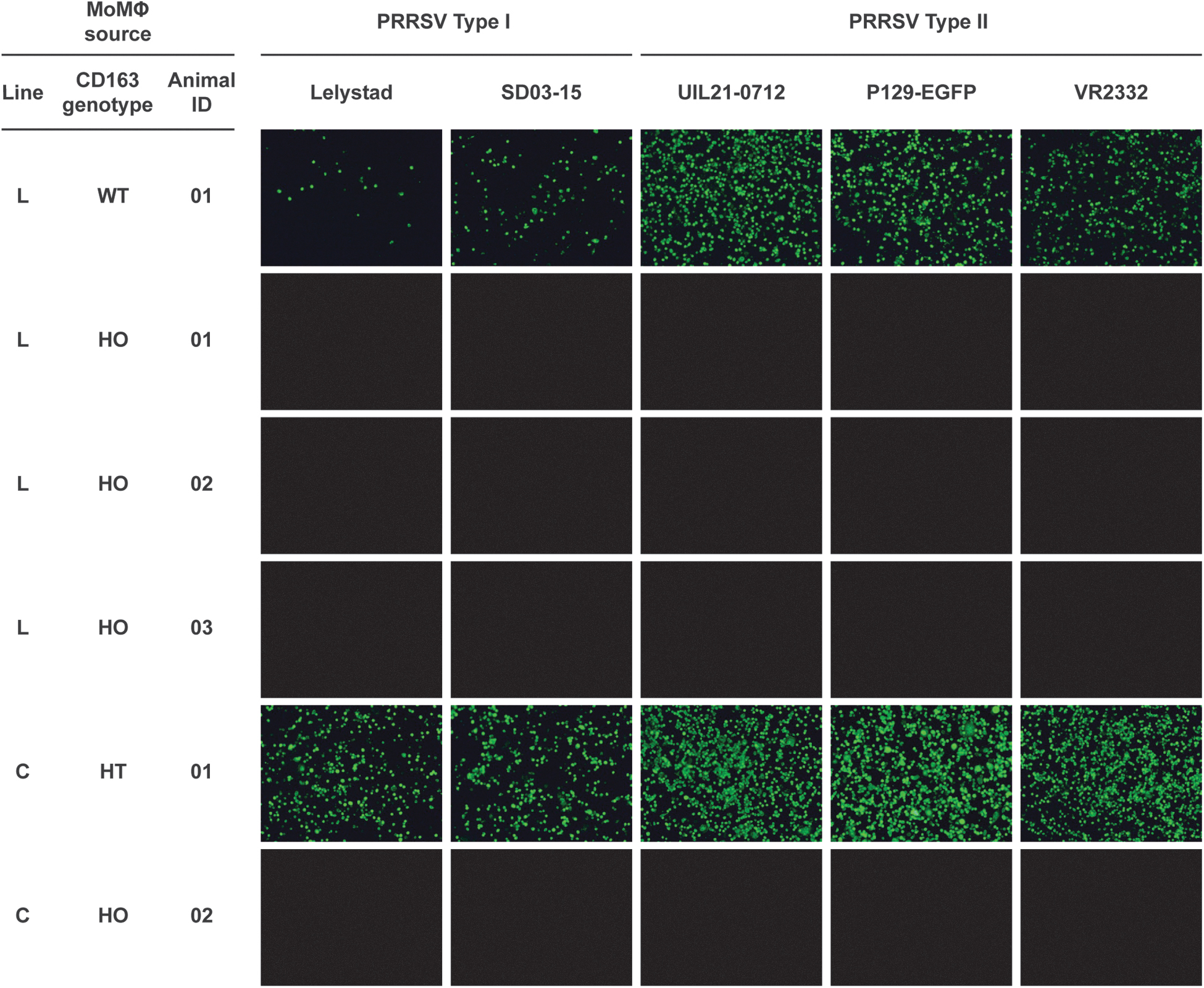
In vitro viral challenges. Blood monocytes from edited Landrace (LR) and white composite line (C) animals were cultured into MoMØs and challenged with the PRRSV strains indicated. Infection was detected using the PRRSV N-protein mAb SDOW-17. MoMØs, monocyte-derived macrophages.
Summary of in vitro porcine reproductive and respiratory syndrome virus challenges
MoMØ, monocyte-derived macrophage; PRRSV, porcine reproductive and respiratory syndrome virus; pos, PRRSV infection; neg, no PRRSV infection; n.d., not determined; L, Landrace; C, white composite line; UE, unedited; HO, homozygous (CD163ΔE7/ΔE7); HT, heterozygous (CD163ΔE7/+).
For animal studies, piglets from all four lines were inoculated intramuscularly and intranasally with either PRRSV-1 isolate SD03–15 (originally isolated from a herd in the United States experiencing PRRS) or PRRSV-2 isolate NVSL 97–7895 (originally isolated in 1997 from a herd in southeast Iowa, USA, experiencing a PRRS abortion storm). For these challenges, a mixture of 10–20 homozygous CD163ΔE7 and WT 3-week-old piglets, typically litter mates, were inoculated and then maintained in the same room to allow the continuous exposure of edited pigs to virus shed by infected WT pigs.
In agreement with the cell-based challenges, and in contrast to WT piglets, homozygous CD163ΔE7 pigs inoculated with either SD03–15 or NVSL 97–7895 were negative for virus and antibody over the course of the infection period (Table 3 and Supplementary Tables S3–S8).
Summary of live animal challenges
Viral replication as measured by qPCR.
Antibody response as measured by ELISA.
C, white composite line; L, Landrace; LW, Large White; D, Duroc; HO, homozygous (CD163ΔE7/ΔE7); UE, unedited; pos, PRRSV infection; neg, no PRRSV infection.
Discussion
Gene editing technologies, including those based on the CRISPR-Cas system, enable the introduction of alleles not readily attainable through classical breeding and selection and provide another tool that can be used to combat disease in livestock. While there are reports of using gene editing to generate fish and animals for food production,38–41 commercial-scale generation and integration of a CRISPR-Cas modified allele directly into a livestock genetic breeding program has not been reported.
Here, a founder population of non-transgenic, PRRSV-resistant pigs was generated by removing exon 7 of CD163 using a dual sgRNA-Cas9 RNP strategy. A requirement of any breeding program is to maintain sufficient genetic diversity both within and across the sire and dam lines, which allows for continued performance gain through heterosis. Accordingly, a PRRSV resistance allele (CD163ΔE7) was generated in four different breeding lines (two maternal and two paternal) to enable rapid population expansion and reduce time to market once approved for sale.
In addition, to reduce the potential of unintended trait performance consequences by breeding different CD163 exon 7 deletion variants, a single CD163 allele was introgressed into the four lines. Thus, as opposed to efforts where a single animal with defined genetics served as starting germplasm, the process outlined here fulfills the needs of breeders for continued genetic gain and rapid deployment of a trait directly into a commercial breeding program.
In agreement with other studies, the pigs described here were healthy and indistinguishable in appearance (Fig. 6A) and behavior when compared to unedited, line identical pigs. Importantly, CD163ΔE7/ΔE7 pigs were found to be completely resistant to infection by multiple Type I and Type II PRRSV strains as determined by both cell and live animal challenges (Fig. 8 and Tables 2 and 3). Having achieved these two important criteria, homozygous CD163ΔE7/ΔE7 animals were used for multiplication and further disease and trait testing across commercial lines. Importantly, these animals had no detectable difference in meat composition, mortality, birth defects, growth rate, body structure, or reproductive ability, relative to unedited pigs. 42
The process described here generated a founder population of breeding boars (10–15 per line) and gilts to serve as a gene edited nucleus herd for ultimate commercial pork production and sale using classical breeding. To achieve this goal, breeders were required to provide donor gilts with high genetic merit and line-specific semen from genetically diverse boars to establish the population for introduction of the PRRSV-resistance allele.
This was a critical step in the process, allowing for the generation of enough E1 boars to maintain genetic improvement outside of the traditional nucleus breeding population. Combining a new breeding tool like CRISPR-Cas with a strong breeding program allows for a step change in porcine genetic improvement.
Generation of this founder population was not without challenges. Despite optimization of sgRNAs to maximize on-target and minimize off-target cutting, only ∼20% of E0 animals carried the intended commercial CD163 allele and often contained multiple (>2) and different CD163 alleles, supporting the notion that piglets derived from injection of RNPs into porcine zygotes were mosaic.
While it was initially anticipated that sufficient numbers of E0 pigs could serve as a source for disease resistance testing, the utility of this generation for such studies was constrained by the low number of animals containing the CD163ΔE7 allele in a homozygous state. Therefore, to build the population and introduce the most current genetics to this generation, it was necessary to outcross selected E0 pigs containing the desired CD163ΔE7 allele to WT, line-identical, high genetic merit mates.
This crossing strategy eliminated mosaicism, enabling a more straightforward analysis of the CD163ΔE7 allele and simplifying identification and elimination of any off-target indels transmitted from the E0 animals. However, selected crosses were made between a few, line-identical E0 animals in parallel to enable early experimental disease testing in a small number of E1 progeny, using Illumina short-amplicon and ONT long-read sequencing to verify genotypes.
Given the investment and commitment toward meeting trait objectives in a commercial breeding program, removal of non-random CRISPR-mediated indels is prudent to avoid unintended consequences that may negatively impact genetic performance. Several studies have used WGS to examine off-target editing of CRISPR-Cas systems in plant and mammalian cells.34,43–45 However, such an approach may not be suitable in a commercial breeding program.
It would be challenging to deploy WGS on enough zygotes to unambiguously identify all potential off-targets for the numerous gRNAs under consideration during selection and prioritization of editing reagents. Further, given the complexity of E0 animals and the large number of E1 progeny generated in a commercial breeding program, WGS falls short as a cost-effective off-target screening tool. To overcome these shortcomings, the SITE-Seq® assay and DNA sequence hybridization were employed as the methods of choice to evaluate potential gRNAs for off-target editing and identify CRISPR-mediated off-target indels in edited animals, respectively.
Sequence capture screening, informed by SITE-Seq® data, was able to identify off-targets transmitted at varying frequencies to the E1 generation, enabling the advancement of pigs without off-target indels (Supplementary Table S2). This pairing of technologies was validated by WGS analysis, which found no off-target indels in pigs screened by DNA sequence capture. The agreement between the methodologies used in this report provides strong support for the use of WGS alternatives, like SITE-Seq®-informed sequence capture, for identification of off-target indels.
The tools and methods described here may serve as a model for others to introduce disease resistance alleles into a breeding program. However, approaches to optimize and streamline the generation of such a founder population should be mentioned. Improvements in Cas9 and gRNA technology, which include the generation of high-fidelity46–49 and short half-life Cas9 protein variants 50 and synthetic51,52 or RNA-DNA hybrid gRNAs, 53 could be adopted to improve specificity, reduce mosaicism, and maximize animal generation.
Carefully designed qPCR assays could replace Illumina short amplicon sequencing to identify primary animals containing the desired allele. E1 animals containing the desired allele could then be analyzed by DNA sequence capture to interrogate the structural integrity of the allele and eliminate off-target indels. The ability to generate E0 animals containing the edited allele in a homozygous state and absent off-target indels would be a major advancement.
In one scenario, porcine embryonic stem cells could be edited, screened for the on-target edit by qPCR or short-read sequencing, then analyzed by DNA sequence capture or other advanced methodologies to eliminate cells containing off-target edits. Edited cells, free of off-target indels, could be differentiated into blastocysts and surgically transplanted into surrogates, ultimately farrowing pigs containing the edited allele in a homozygous state with no off-target indels. Incorporation of improved reagents and screening technologies, together with advancements in cell biology, could lower the investment threshold and allow for increased adoption of gene editing to improve animal welfare through the introduction of novel disease resistance alleles.
Conclusions
Gene editing holds great promise for combating disease and improving both human and animal health. Thus far, the focus of gene editing applications has been on individualized medicine to treat human diseases such as leukemia, hemophilia, blindness, and cancer. Crop and livestock breeders can now incorporate CRISPR-Cas as a tool to address present and emerging diseases and ensure food security. CRISPR-Cas has been used in this report to enable a commercial-scale editing program with the intent of solving PRRS disease in swine.
This is a groundbreaking accomplishment in agriculture toward improving animal health, reducing waste, lowering production costs, and potentially reducing antibiotic use on the farm. Further, applying these learnings to eliminate other livestock diseases that are not only harmful to animals (African Swine Fever) but also to humans (swine influenza) would be a major step to benefit consumers, society, and the environment.
Footnotes
Acknowledgments
The authors gratefully acknowledge the PIC staff for animal coordination and reproductive husbandry: Tom Somrack, Brandy Eversole, Greg Ellis, Deanne Hemker, Perry Harms, Ricardo Ramos Cruz, Scott Norwood, Marney Jobin, Brian Melody, and Justin Holl. The authors would like to thank Stephen White and Michael McLachlan for critical reading of the manuscript, and Laura Gabrovsek for critical reading of the manuscript and assistance with figure preparation. The authors thank Jon Lightner and Bill Christianson for their support of this work. The authors gratefully acknowledge the thoughtful comments of the reviewers.
Authors' Contributions
B.T. Burger: Conceptualization; investigation; methodology; supervision; visualization; writing—original draft; and writing—review and editing. B.P.B.: Conceptualization; data curation; investigation; methodology; resources; supervision; and writing—review and editing. M.A.C.: Conceptualization; investigation; methodology; resources; supervision; writing—original draft; and writing—review and editing. B.T. Brett: Conceptualization; data curation; formal analysis; investigation; methodology; software; supervision; writing—original draft; and writing—review and editing. M.S.R.: Project administration; data curation. S.P., D.B., and J.A.B.: Investigation; methodology. K.J. and S.N.: Data curation; formal analysis; investigation; software; and validation. J.L. and A.O.: Data curation; formal analysis; investigation; and software. E.A.: Data curation; formal analysis; and software. G.R.: Supervision. S.R.: Conceptualization; supervision. J.B. and N.R.M.: Investigation; methodology; and validation.. J.H.: Investigation; methodology; and writing—review and editing. C.J.D.: Data curation; investigation; and methodology. P.C., C.K.F., and B.P.T.: Investigation; supervision. A.M.L.: Project administration. M.J.I., D.B.N., K.-E.P., J.W., S.S., and A.B.-N.: Investigation. S.G. and S.B.K.: Supervision. B.C.S.: Data curation; formal analysis; project administration; investigation; and resources. R.R.R.R.: Resources; supervision. M.C.: Resources. E.R.: Conceptualization; resources; and writing—review and editing. A.M.C.: Conceptualization; supervision; visualization; writing—original draft; and writing—review and editing.
Data Availability
Data described in this publication may be available for non-commercial research purposes on acceptance and signing of a materials transfer agreement. Cells, animals, or derivatives are derived from the PIC breeding program and will not be made available.
Author Disclosure Statement
B.T. Burger, B.P.B., M.A.C., B.T. Brett, M.S.R., S.P., D.B., K.J., S.N., J.L., A.O., E.A., G.R., S.R., J.B., N.R.M., J.A.B., J.H., C.J.D., N.R.M., J.B., J.A.B., M.L.R., and A.M.C. are current or former employees of Genus plc, a company that is working toward commercialization of PRRSV-resistant pigs. P.C., A.M.L., M.J.R., D.B.N., C.K.F., S.G., and S.B.K. are current or former employees of Caribou Biosciences from which Genus plc has licensed CRISPR-Cas9 gene editing technology. K.E.P., J.W., S.S., and B.P.T. are employees of RenOVAte Biosciences. B.C.S., A.B.N., and R.R.R.R. are employees of the University of Illinois at Urbana-Champaign. B.T.B., B.P.B., M.A.C., D.B., and A.M.C. have filed patent applications related to the manuscript.
The publication of this paper under the CC-BY license does not confer by their respective owners any patent rights, trade secret rights, or other knowhow that may be associated with the subject matter of this paper, and the authors do not waive or disclaim any such rights by this paper's publication.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.