
Abstract
Precision genome editing has become a reality with the discovery of base editors. Cytosine base editor (CBE) technologies are improving rapidly but are mostly optimized for T
Introduction
Scientists have worked for decades to develop genome engineering technologies safe enough for use in human gene therapy (reviewed by Refs.1,2). However, these technologies have been limited by concerns over insertion mutagenesis (integrating retroviruses) 3 and DNA breakage (zinc finger nucleases, transcription activator-like endonucleases) 4 and, in all instances, the possibility of off-target mutagenesis and cancer (reviewed by Refs.1,2). In addition, these technologies are not ideal for correcting single-nucleotide variants, which are the most common cause of genetic disease in humans.5,6 Examples include reversion of point mutations in the β-globin gene responsible for β-thalassemia and sickle cell anemia, reversion of an LMNA point mutation in Hutchinson–Gilford progeria, correction of splicing defects in spinal muscular atrophy, and introduction of new point mutations to modulate immune responses.7–12
Thus, for these and other diseases, there is a significant need for technologies that enable precise genome engineering, and particularly for correcting single-nucleotide mutations in a safe and efficient manner.
The feasibility of precision genome engineering improved dramatically with the landmark discovery of efficient cytosine-to-thymine (C-to-T) conversion in human cells using a complex consisting of rat apolipoprotein B mRNA-editing catalytic polypeptide 1 (rApobec1, rA1) linked to a Cas9 nickase (Cas9n) complex (Streptococcus pyogenes Cas9-D10A) and bound by an appropriate guide (g)RNA 13 (e.g., cytosine base editor [CBE] complex shown in Fig. 1A). The complementary base-pairing of the gRNA to a specific DNA target creates a short single-stranded (ss)DNA region (an R-loop), which becomes a substrate for deamination by the covalently linked rA1 enzyme. Cytosine-to-uracil (C-to-U) editing events within the exposed ssDNA region can then become immortalized as C-to-T mutations due to the Cas9n-catalyzed ssDNA break, which directs repair enzymes to the opposing DNA strand (the G-containing strand).13,14
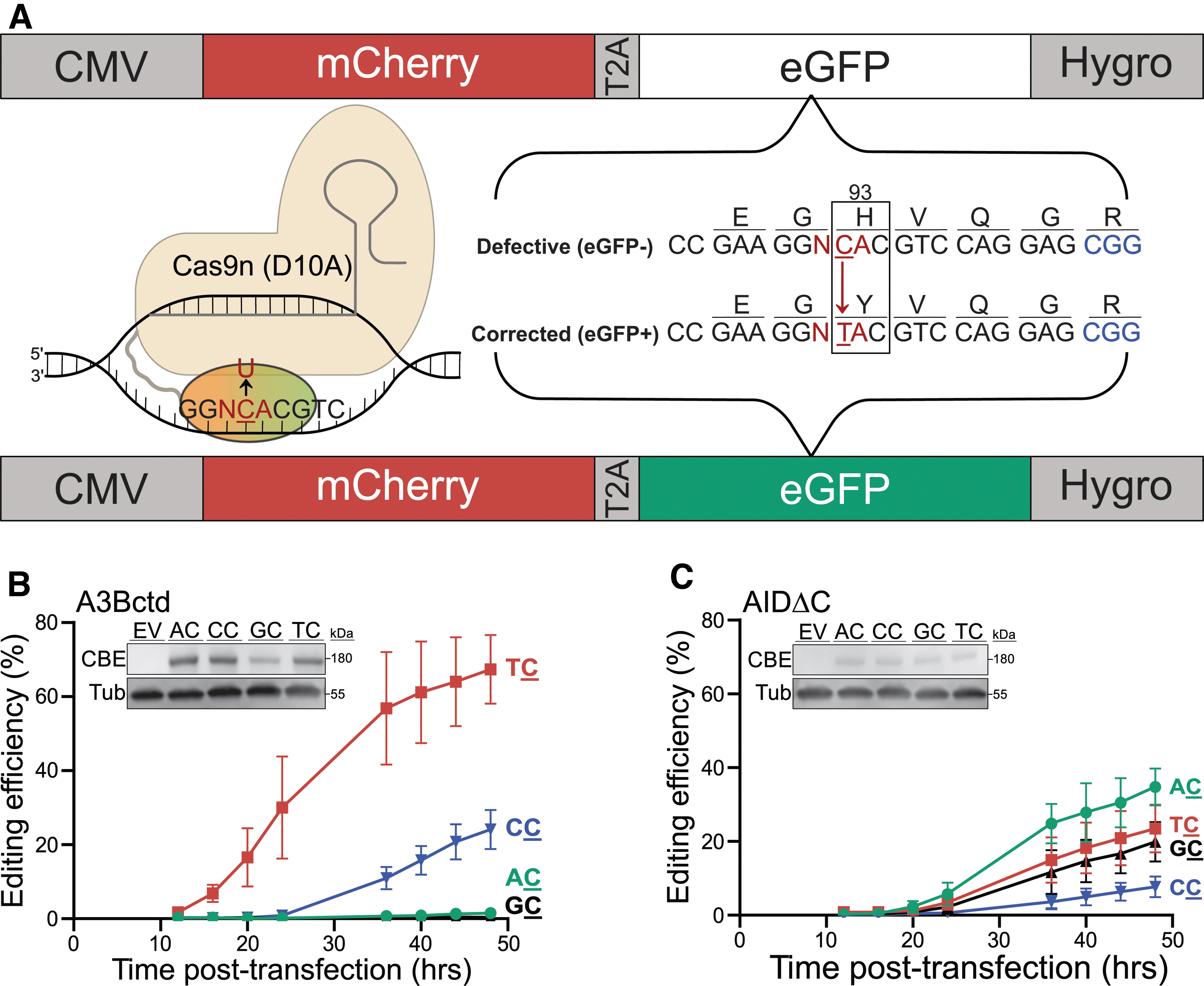
ARSENEL episomally.
Uracil persistence and overall editing efficiencies are further improved by linking the rA1-Cas9n complex to a phage-derived uracil DNA glycosylase inhibitor (Ugi) to prevent uracil excision and error-free repair. This design—rA1-Cas9n/gRNA-Ugi—is known popularly as base editor 3 (BE3). 13 Since this discovery, a multitude of CBEs have been created by fusing hundreds of different APOBEC family enzymes (wild-type proteins and mutants) to different Cas9 nickase complexes (also wild-type and mutants), as well as varying copy numbers of Ugi.13,15–19 Several of these published constructs are used here including BE3, 13 BE4max (codon optimized rA1-Cas9n-Ugi-Ugi), 19 APOBEC3B (A3B) C-terminal domain (ctd)-Cas9n-Ugi,17,18 full-length A3B-Cas9n-Ugi,16–18 activation-induced cytidine deaminase (AID)ΔC-Cas9n-Ugi,20–22 and APOBEC3A-N57G-Cas9n-Ugi, 8 as well as new constructs such as derivatives of A3Bctd-BE3/BE4max.
Despite thousands of constructs, hundreds of thousands of editing reactions, and millions of quantified edits, CBEs still have significant room for improvement.1,2 The most challenging issue is achieving the “right balance” between on-target specificity (the ability of a CBE to specifically deaminate a single-target cytosine and not bystander or off-target cytosines) and editing efficiency (the rate at which the on-target cytosine is edited). Efficient base editors such as BE3, BE4max, and A3A invariably catalyze off-target editing events proximal to the target cytosine (here called bystander edits) and gRNA-dependent and -independent events throughout the genome of cells.23–25 Conversely, a few editors have been engineered to exhibit higher degrees of on-target specificity, such as YEE-BE4max and eA3A, but an increase in specificity often comes at the cost of a decrease in editing efficiency.8,24
Two decades of research have shown that the −1 nucleobase directly upstream of the target cytosine has the largest effect on cytosine deaminase substrate specificity because it makes direct intermolecular contacts with the editing enzyme.26–35
Thus, most CBEs show a strong bias for T
Here, we describe a set of four reporters that can be used episomally or chromosomally to compare CBE dinucleotide specificities and editing efficiencies in real time. These APOBEC Reporter Systems for Evaluating di
Last, the ARSENEL system is comprised of four different plasmid constructs that can be separately transfected into cells for direct episomal editing reactions or used to produce VSV-pseudotyped lentiviral supernatants for transduction into almost any cell line and chromosomal DNA editing reactions, thus mimicking many different human genomic DNA disease alleles that can be corrected by precise single C-to-T mutations. The ARSENEL system is used here to evaluate the dinucleotide preferences of a variety of CBEs. In addition, the ARSENEL system can provide a quantitative readout of regulators of CBE activity including deaminase inhibitors.
Materials and Methods
Reporter constructs
All plasmids used in this study are available through Addgene (MA), and oligonucleotide sequences are listed in Supplementary Table S1. The T
CBE constructs
Human A3A (No. 109425; Addgene), 17 human eA3A, 8 human full-length A3B (No. 198889; Addgene), 17 human A3Bctd (No. 109426; Addgene), 17 human AIDΔC CBE (No. 198890; Addgene), 36 and rat A1 (No. 73021; Addgene, deposited by D. Liu lab) 13 CBEs have been reported. A3Bctd-L7G was PCR subcloned from an existing construct 38 and used to replace rA1 in BE3 by restriction digestion and ligation (No. 198886; Addgene). A3Bctd-D314E (No. 198887; Addgene) and eA3A (No. 207165; Addgene) were created using site-directed mutagenesis. The BE4max plasmid was a gift from the B. Moriarity laboratory (No. 112093; Addgene, deposited by D. Liu lab). 19 The BE4max plasmid was reconstructed by removing the deaminase and inserting Esp3I sites 5′ from the Cas9 gene (No. 198891; Addgene). Deaminase open reading frames were generated by PCR and cloned into the reconstructed BE4max backbone via Esp3I Golden Gate cloning 37 (A3Bctd-max, No. 198888; Addgene, A3Bmax, No. 207167; Addgene, eA3Amax, No. 207166; Addgene).
BORF2 constructs have been reported.39,40 All CBE constructs were confirmed by Sanger sequencing (Azenta/Genewiz) or whole-plasmid sequencing (Plasmidsaurus). WT constructs match the following GenBank accessions: Homo sapiens A3B (NM_004900), H. sapiens A3A (NM_145699), H. sapiens AID (NM_020661.4), Rattus norvegicus APOBEC1 (NC_051339.1), S. pyogenes Cas9 (WP_038431314.1), and Epstein-Barr virus (EBV) BORF2 (V01555.2).
Episomal base editing experiments
Semiconfluent 293T cells in three 24-well plates were transfected (in parallel) with 50 ng gRNA, 100 ng reporter, and 150 ng of each base editor (25 min at RT with 0.9 μL of TransIT LT1 [Mirus] and 50 μL of serum-free RPMI). One plate was imaged using a Cytation1 Multi-Mode Reader (Agilent, CA) as described 36 over a 48-h period (configured with Texas Red [586/647] and GFP [469/525]). After 48 h of incubation, the second and third plates were harvested for immunoblots and flow cytometry. For flow cytometry analysis, trypsinized and PBS-washed cells were placed in a 96-well round-bottom plate. A minimum of 20,000 events were acquired for each condition using a BD LSRFortessa or a BD FACSCanto flow cytometer (BD, OR). Here, percent editing was calculated by dividing the number of eGFP and mCherry double-positive cells by the total number of mCherry-positive cells and multiplying by 100 (FlowJo software; BD).
Chromosomal base editing experiments
Semiconfluent six-well plates of 293T cells were transfected with 500 ng of an HIV-1 Gag-Pol packaging plasmid and of a VSV-G expression plasmid cocktail (a 2:1 ng ratio), and 500 ng of each base editing reporter. Viruses were harvested 48 h post-transfection and used to transduce target cells (multiplicity of infection, MOI = 0.3). Forty-eight hours post-transduction cells were subjected to hygromycin selection at a 400 μM concentration. After selection, >95% of cells were mCherry positive by flow cytometry. Transduced, mCherry-positive cells were transfected with 425 ng of CBE and 75 ng of targeting gRNA into four parallel semiconfluent 24-well plates. As with the episomal experiments above, one plate was imaged using a Cytation or Incucyte instrument over a 72-h period.
The other three plates prepared in parallel were harvested after 72 h of incubation for editing quantification by flow cytometry as explained in the previous section, genomic DNA isolation for reporter target analysis, and for expression confirmation immunoblotting.
Live-cell imaging analyses
The Cytation1 Multi-Mode Reader (Agilent) configured with Texas Red (586/647) and GFP (469/525) light cubes was used to image and analyze data for Figures 1, 2, and 4. Live-cell images were captured using a 4 × objective at several time points after the initial transfection (e.g., Supplementary Fig. S1A). Temperature was maintained at 37°C for the duration of imaging. Nine images were taken per well in a fixed 3 × 3 grid. After background subtraction, a dual-mask analysis was used to quantify edited versus nonedited cells. All mCherry-positive cells were first identified as the primary mask using the fluorescence threshold as well as size exclusion. Next, a secondary mask was created within the primary mask to identify eGFP-positive cells using a GFP fluorescence threshold. The ratio of double-positive cells to mCherry-positive cells was then used to calculate relative editing frequencies over time.
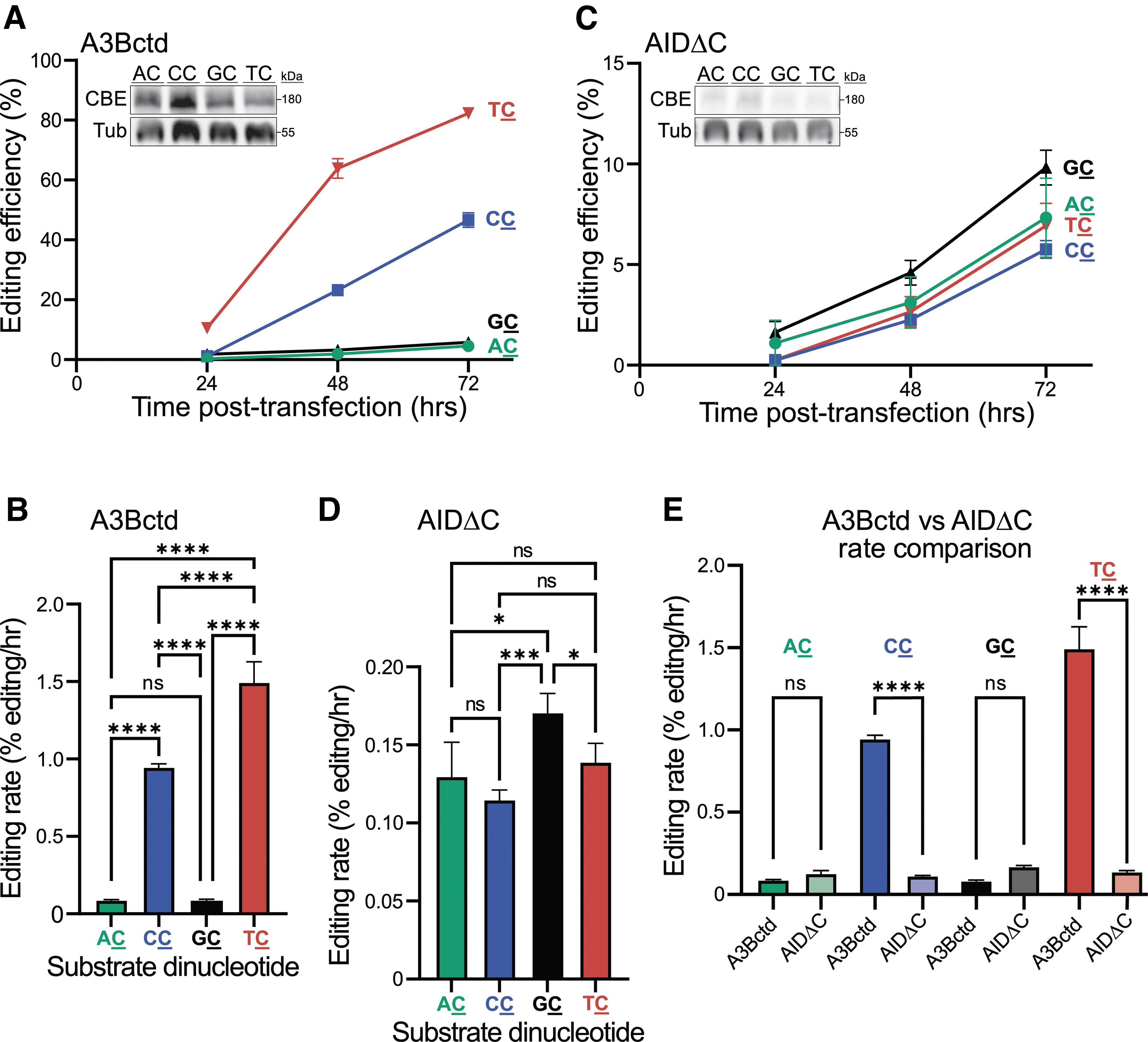
ARSENEL chromosomally.
Data for Figure 5 were collected and analyzed using an Incucyte (Sartorius, Germany). Live-cell images of orange, green, and phase image channels were captured with a 4 × objective every 4 h after the initial transfection with five images per well in a fixed grid. mCherry- and GFP-positive cells were identified with internal cellular analysis software.
Editing quantification by Sanger sequencing
After sample collection, genomic DNA was isolated (Qiagen Gentra Puregene). A 712 bp segment of the eGFP target region was amplified by high-fidelity PCR using Phusion polymerase. PCR products were gel purified and submitted for Sanger sequencing by Azenta/Genewiz, ACGT (MD), or Eurofins (TX). Chromatograms received from Sanger sequencing were uploaded to EditR for peak quantification. 41 For individual clone analysis, genomic DNA was prepared and the target editing region PCR amplified as described above, and then inserted into pJET1.2/blunt Cloning Vector using the CloneJET PCR Cloning Kit (Thermo Fisher Scientific, MA). Individual colonies were picked after transformation into Escherichia coli, miniprepped, and then submitted for Sanger sequencing.
Immunoblotting
Cells were collected and lysed in 2 × reducing sample buffer, and then incubated at 98°C for 15 min. All samples collected from an individual experiment were run on the same gel to compare protein expression levels as accurately as possible. Samples were separated by a Criterion 4–20% gel (No. 5671095; Bio-Rad), and then transferred to PVDF membranes. Antibody incubation was conducted as previously reported. 42 Primary antibodies used include rabbit anti-Cas9 (No. ab189380, 1:5000; Abcam), mouse anti-tubulin (No. T5168, 1:10,000; Sigma-Aldrich), and rabbit anti-FLAG (No. F7425, 1:5000; Sigma-Aldrich). Secondary antibodies used were goat anti-rabbit IRdye800 (No. 925-32211, 1:10,000; LI-COR) and goat anti-mouse IRdye680 (No. 926-69020, 1:10,000; LI-COR).
Statistical analyses
GraphPad Prism 9.0 was used for statistical analyses of quantitative data. Sample size, biologically independent experiments, and statistical analyses are given in the figure legends. Unless otherwise stated in the legends, error bars represent one standard deviation from the mean, and two-way ANOVA tests were used to assess significance (ns = not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Results
Episomal ARSENEL
We previously developed eGFP-based reporters for quantification of APOBEC-catalyzed base editing in living cells.17,36 One of these constructs relies on editing of a mutant
Here, we report the creation of an isogenic set of four constructs in which the wobble position of the Gly92 codon (GG
To test whether these constructs accurately report known intrinsic deaminase preferences, we first compared the dinucleotide editing preferences of an A3Bctd CBE and an AIDΔC CBE in transient transfection experiments using 293T cells. The intrinsic preference of A3Bctd is T
As anticipated, A
In comparison, the AIDΔC CBE exhibits lower overall editing efficiencies in transient reactions and prefers A
Nevertheless, taken together, the results with the A3Bctd and AIDΔC CBE constructs combine to show that the ARSENEL system enables an assessment of dinucleotide editing efficiencies of CBEs in real time in living cells.
Chromosomal ARSENEL reduces experimental variability and reflects intrinsic CBE dinucleotide preferences
Next, retroviral transduction was used to introduce each reporter construct stably into the chromosomal DNA of 293T cells (MOI = 0.3). This pool-based approach ensures that most cells have only one integrated reporter and simultaneously minimizes integration position effects common for single clones. Thus, results with chromosomal ARSENEL are anticipated to provide a less biased population-based readout of the efficiency and specificity of CBE editing of single-genomic cytosine nucleobases in each of the four dinucleotide contexts. Using this system, the A3Bctd CBE exhibits a strong dinucleotide specificity for T
Flow cytometry data confirm high frequencies of correction of the T
A different picture emerges from AIDΔC CBE chromosomal ARSENEL experiments. First, in comparison with the episomal editing data described above, a significant drop in editing efficiency is observed even for the best substrate, G
The use of editing rates (percent editing per hour) enables cross-comparisons of the dinucleotide preferences of different CBE constructs within the same chromosomal editing experiment. For example, the A3Bctd CBE significantly outperforms the AIDΔC CBE on the T
Sanger sequencing of chromosomal reporter pools enables quantification of on-target and bystander editing efficiencies
An additional property of the ARSENEL system is that the gRNA binding region in chromosomally transduced pools with one reporter per cell can be recovered easily in bulk by high-fidelity PCR and then Sanger-sequenced to assess frequencies of on-target specificity for the target cytosine and bystander editing (in contrast, editing rates of a transiently transfected reporter cannot be quantified in this manner because wild-type reporter copy numbers can far exceed one per cell and the vast majority of targets are not edited). The gRNA binding window has a total of six cytosines in three different dinucleotide contexts that can be edited by a CBE (numbered 1, 2, 9, 11, 14, and 15 in Fig. 3A).
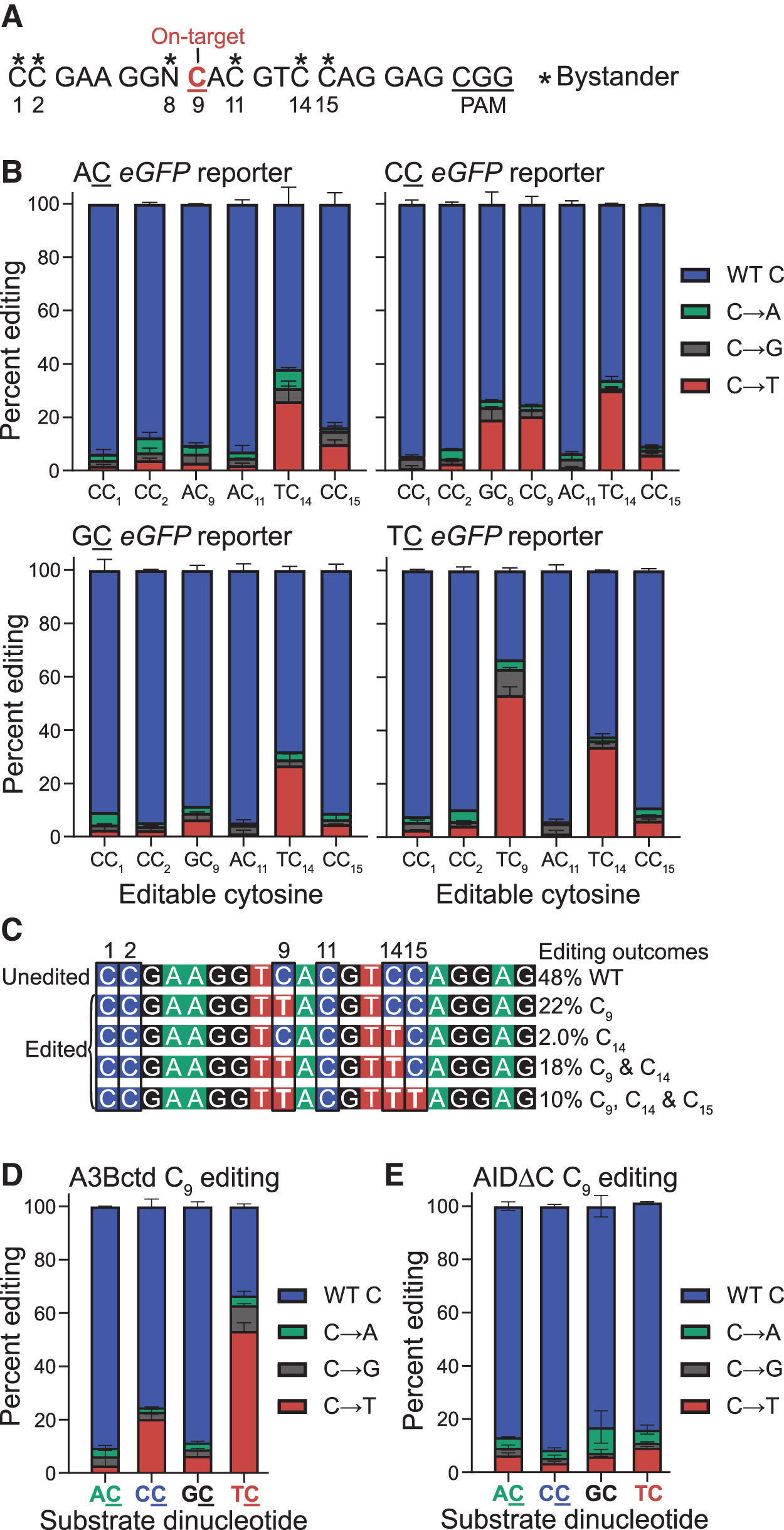
On-target and bystander editing events in chromosomal editing reactions with ARSENEL system.
C-to-T editing at C9 restores eGFP function, whereas editing at the majority of the other cytosines is expected to be aphenotypic, with the exception of a TAG stop codon produced by C15-to-T editing events (Supplementary Fig. S1B). Thus, Sanger sequencing data, analyzed using the chromatogram peak quantification program EditR, 41 can provide a simultaneous assessment of both on-target C9 and bystander cytosine editing frequencies.
As anticipated from eGFP fluorescence data above, an on-target edit of T
To investigate coincident editing events at C9 and C14 and other cytosine positions within the gRNA region, genomic DNA collected from the A3Bctd-edited T
Interestingly, this individual clone analysis also reveals that 10% of the sequences have an identical combination of multiple edited sites (C9, C14, and C15) and likely represent hyper-edited targets incapable of encoding a functional eGFP protein (due to C15-to-T creating a stop codon). No insertion–deletion (indel) mutations were recovered using this approach suggesting that such outcomes may only comprise a small minority of all A3Bctd CBE editing events.
Notably, the A3Bctd CBE exhibits >25% editing at T
However, such noncanonical outcomes are not detected in 50 individual clone Sanger sequence reads (Fig. 3C). The Sanger sequencing results also suggest that the duplex DNA adjacent to the gRNA-binding region may be protected from editing as no mutations in flanking regions are detected.
Bar graphs afford another way to summarize and compare PCR product sequencing data for ARSENEL editing outcomes at the C9 position (Fig. 3D, E). Editing by A3Bctd CBE leads preferentially to C-to-T mutations at T
Loop 7 changes alter the dinucleotide editing preferences of A3Bctd CBE
To further investigate the utility of the ARSENEL system, we asked whether structure-guided changes to the loop 7 region of A3Bctd can alter dinucleotide editing preferences in the same manner as reported for recombinant proteins in vitro.
26
Crystal structures of A3Bctd and ssDNA-A3Bctd have demonstrated that the target deoxy-cytosine nucleotide is deep within the active site pocket and the upstream deoxy-thymine nucleotide (−1 T) is engaged by three distinct hydrogen (H)-bonds (two directly with Asp314, one water-mediated; Fig. 4A).26,28,35,51 Structural and biochemical studies have shown that swapping the entire loop 7 region of A3Bctd with the corresponding region from A3Gctd changes the intrinsic dinucleotide preference from T
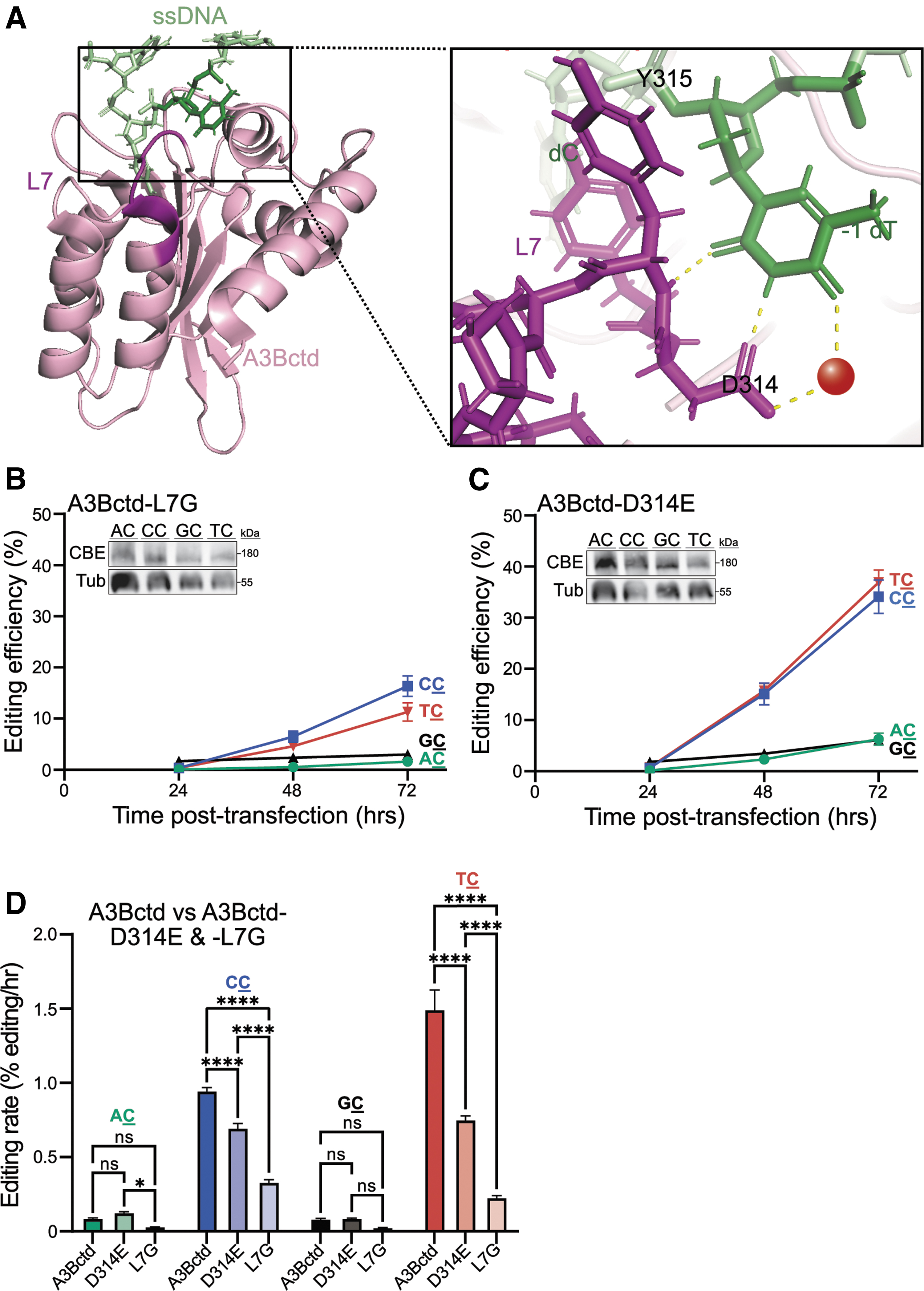
A3Bctd loop 7 dictates substrate specificity and editing efficiency.
Moreover, a single amino acid substitution in A3Bctd loop 7, Asp314-to-Glu, extends the amino acid side chain by one carbon and is hypothesized to break the H-bond network with the −1 T and create a more favorable H-bond environment for accommodation of a −1 C.
35
Thus, these two changes to loop 7 (full loop swap and Asp314-to-Glu) are expected to change the dinucleotide editing preference of the A3Bctd CBE from T
This mechanistic connection between loop 7 and dinucleotide preference was tested using chromosomal ARSENEL. In comparison with the parental A3Bctd CBE construct, which has a clear preference for T
Taken together, these results combine to indicate that loop 7 changes (full loop swap or Asp314-to-Glu) are able to diminish the intrinsic preference of A3Bctd for T
High-efficiency CBEs exhibit elevated bystander editing events
Next, we used the chromosomal ARSENEL system and editing rates to compare the A3Bctd CBE with established and novel CBEs to identify editing complexes with the highest on-target T
As described above, A3Bctd CBE also edits C
These results are likely due to A3A having a larger active site than A3Bctd and the highest documented rates of ssDNA C-to-U editing,16,17,35,50,51 as well as the A3A CBE having the same Cas9n-Ugi editing platform as the A3Bctd CBE.
Because A3Bctd-max has a lower C
Next, we used Sanger sequencing chromatograms from high-fidelity PCR products to compare the repertoire of on-target versus bystander editing events in the gRNA binding region of eGFP. Bar graphs enable visual comparisons of C9 C-to-T editing for each CBE with data for each of the four reporters above the x-axis line, and cumulative bystander C-to-T editing events below the line, again, for each of the four reporters. Interestingly, of all T
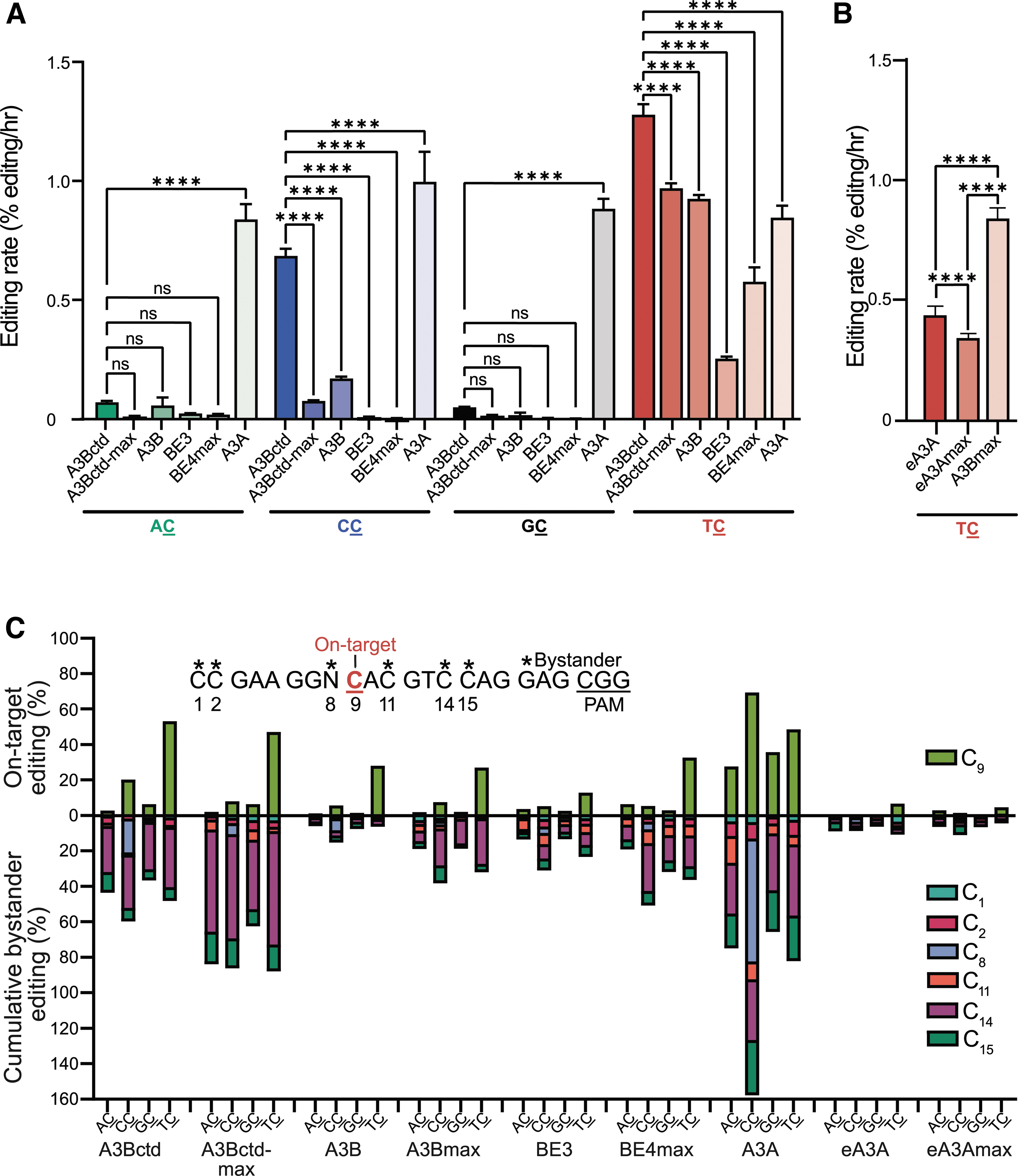
Comparisons of CBE editing rates, dinucleotide specificities, and bystander events.
Interestingly, inserting full-length A3B into the BE4max backbone increased bystander editing at C14 compared with full-length A3B in the BE3 backbone. A3A CBE shows almost no specificity with similarly high rates of editing of C9 in all four dinucleotide contexts, and this liability is compounded by the highest levels of bystander editing events. On the opposite end of the spectrum, both eA3A and eA3Amax exhibit low rates of T
Characterization of CBE inhibitors using ARSENEL
Methods to systematically inhibit CBE activity may be desirable to prevent bystander and distil off-target events after the primary target has been edited successfully. The biological activity of A3 family members is virus restriction and, accordingly, viruses have evolved mechanisms to fight back by inhibiting A3 activity (reviewed by Ref. 57 ). We recently discovered a mechanism of A3B inhibition in which the Epstein–Barr virus viral ribonucleotide reductase large subunit, BORF2, binds directly to the A3B active site and blocks catalytic activity.38–40,58
We therefore asked whether BORF2 is sufficiently potent to inhibit A3Bctd in the context of a powerful, gRNA-directed, nuclear-localizing CBE complex. Using the chromosomal T
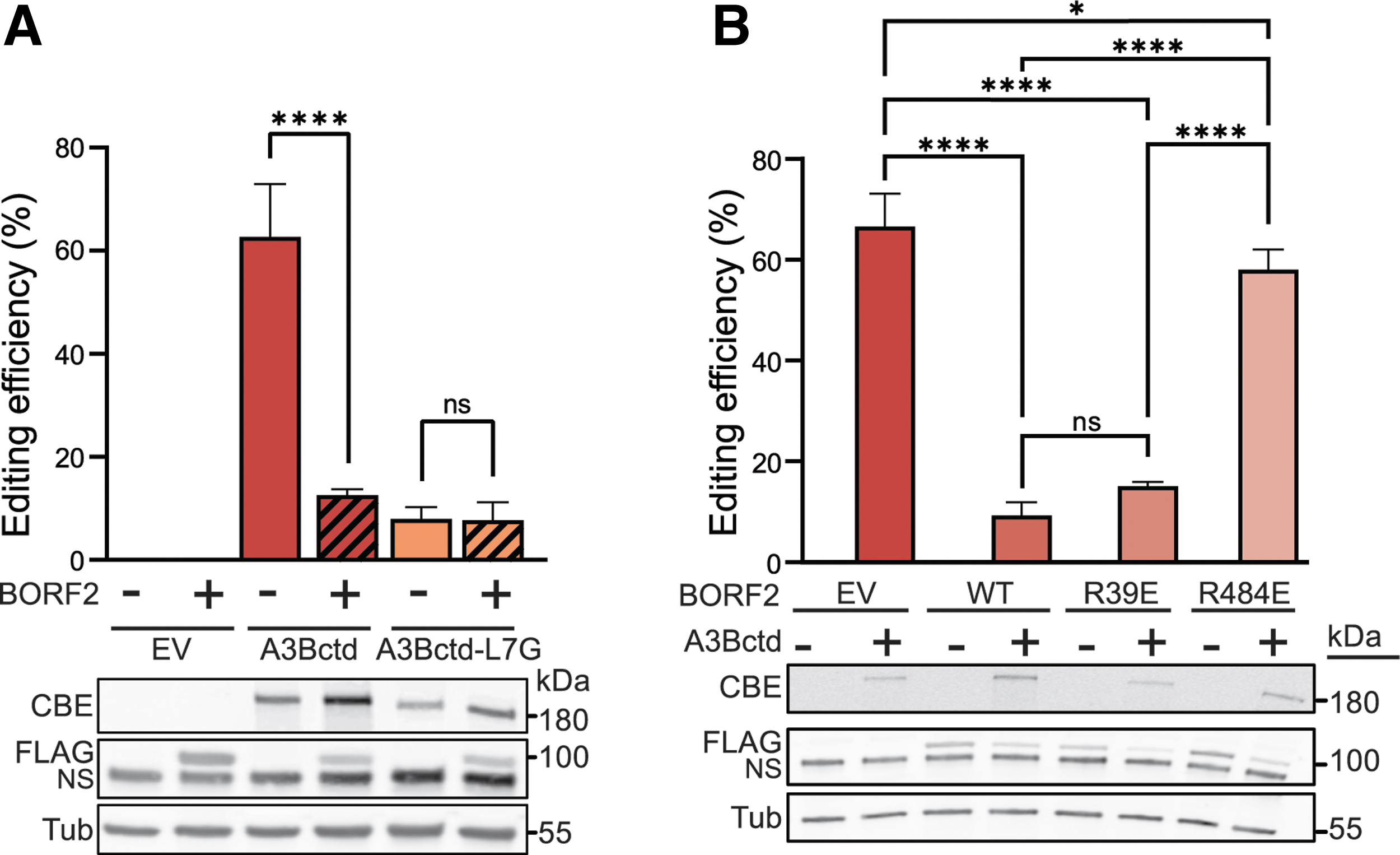
Demonstration of a virus-encoded inhibitor of A3Bctd CBE editing.
In support of a direct inhibition mechanism, the single Arg484-to-Glu substitution prevents BORF2 from inhibiting chromosomal T
Discussion
Here we describe ARSENEL, which are live-cell fluorescence-based assays for quantifying the dinucleotide editing specificities and efficiencies of CBEs. ARSENEL can be used for assessing individual editing complexes and for testing candidate inhibitors, as exemplified here for the EBV protein BORF2. ARSENEL may also be useful for future studies using medium- to high-throughput screens to identify new CBEs with enhanced properties. ARSENEL is the first system to our knowledge to enable direct isogenic comparisons of CBE dinucleotide editing efficiencies, although other fluorescent base editing reporter systems such as BE-FLARE and TREE may be adapted for this purpose (as described in the last paragraph of Discussion). ARSENEL also has added value by enabling the simultaneous assessment of on-target and bystander editing events within the gRNA binding region by Sanger sequencing, as in studies here, or deep sequencing of high-fidelity PCR products.
An important observation from experiments with ARSENEL is that the wild-type full-length A3B CBE exhibits the highest overall specificity for T
These results illustrate the tremendous challenge of identifying a CBE with a high on-target efficiency and no liabilities. A precise molecular explanation of the desirable attributes of the full-length A3B CBE will require high-resolution structural studies but is likely to involve a role for the noncatalytic N-terminal domain (A3Bntd), which is unique to this construct. In comparison, the A3A CBE lacks specificity as evidenced by high-frequency editing of all four ARSENEL dinucleotide substrates (Fig. 5A, C, and Supplementary Fig. S3A). The A3A CBE also exhibits the highest levels of bystander editing events (Fig. 5C). This editing construct is therefore the most active and promiscuous of all CBEs tested here, which could be useful in mutagenesis studies where many different mutations are desirable and phenotypic screens can be leveraged to help identify useful mutants.
A notable curiosity from studies here with ARSENEL is that all CBEs are not equal in producing transversion mutation outcomes. If both uracil excision by UNG2 (despite all constructs including at least one Ugi) and translesion synthesis by REV1 are stochastic events, then all CBEs should yield transversion mutation outcomes at rates proportional to their overall editing and C-to-T transition mutation capabilities. This is not likely to be the case because, for example, the AIDΔC CBE appears to produce a C-to-A transversion bias at its preferred dinucleotide target in the G
A provocative interpretation of this result is that AID (or an AID-associated factor) may somehow have the capacity to influence downstream mutagenic DNA repair outcomes (reviewed by Refs.59,60). This property may be intrinsic to AID and advantageous for its physiological function in somatic hypermutation and class switch recombination, and it may be harnessed for use as a C-to-A transversion base editor. 61
A limitation of the studies here is that we focus on a single gRNA binding site in eGFP to isogenically compare editing of the four different dinucleotide contexts by a panel of CBEs. It is possible that other reporters and/or chromosomal sites will yield different results. In addition, at least for T
This could explain why here, for instance, the AIDΔC and A3B-L7G CBE constructs exhibit lower than expected editing rates (ARSENEL editing motifs are subideal for AID and A3G, which prefer WR
Previous studies including our own have used fluorescence to measure the editing activity of CBEs.17,36,51,68–72 BE-FLARE
69
and TREE
72
both utilize the same target site that through a single C-to-T mutation converts blue fluorescent protein (BFP) fluorescence to GFP via a His66 to Tyr66 amino acid change. Because the upstream Thr65 codon in BFP can be wobbled, either of these systems could be expanded in future studies to examine dinucleotide editing preferences as we have done here for ARSENEL (and enable the examination of a different repertoire of bystander cytosine editing events relative to the PAM site; e.g., C2, C4, C5, C13, and C18 for BE-FLARE). In comparison, GO
71
is a gain-of-signal system that restores GFP function, but it is restricted to editing of a phenotypically constrained A
Conclusions
The ARSENEL system described here is unique in that it is the first to enable a systematic comparison of dinucleotide editing preferences of virtually any CBE. In addition, we found that a full-length A3B CBE has the best overall on-target T
Footnotes
Acknowledgments
We thank the members of the Harris laboratory, the Moriarity laboratory, and the UMN Center for Genomic Engineering for support and constructive feedback. We thank David Liu for the BE3 plasmid and Brandon Moriarity for the BE4max plasmid. R.S.H. is an investigator of the Howard Hughes Medical Institute, a CPRIT scholar, and the Ewing Halsell President's Council Distinguished Chair at the University of Texas Health San Antonio.
Authors' Contributions
A.E.R.: Conceptualization, methodology, validation, formal analysis, investigation, resources, writing, and visualization. Y.C.: Validation, formal analysis, investigation, and resources. D.J.S.: Conceptualization, methodology, resources, and supervision. S.N.M.: Resources. R.S.H.: Conceptualization, writing, supervision, project administration, and funding acquisition.
Data Availability
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by NCI P01 CA234228 (to R.S.H.), NIAID R37 AI064046 (to R.S.H.), and a Recruitment of Established Investigators Award from the Cancer Prevention and Research Institute of Texas (CPRIT RR220053 to R.S.H.). Salary support for S.N.M. was provided by NIAID F31-AI161910 and subsequently a HHMI Gilliam Fellowship. Salary support for D.J.S. was provided by NIAID K99/R00-AI147811.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.