
Abstract
Knockout mice for human disease-causing genes provide valuable models in which new therapeutic approaches can be tested. Electroporation of genome editing tools into zygotes, in vitro or within oviducts, allows for the generation of targeted mutations in a shorter time. We have generated mouse models deficient in genes involved in metabolic rare diseases (Primary Hyperoxaluria Type 1 Pyruvate Kinase Deficiency) or in a tumor suppressor gene (Rasa1). Pairs of guide RNAs were designed to generate controlled deletions that led to the absence of protein. In vitro or in vivo ribonucleoprotein (RNP) electroporation rendered more than 90% and 30% edited newborn animals, respectively. Mice lines with edited alleles were established and disease hallmarks have been verified in the three models that showed a high consistency of results and validating RNP electroporation into zygotes as an efficient technique for disease modeling without the need to outsource to external facilities.
Introduction
Animal models for human diseases constitute a valuable tool for the study of disease pathology and for the testing of new treatments in the preclinical stage. This is especially crucial for those metabolic inherited rare diseases in which the causal defective gene has been identified, elusive mechanisms of pathophysiology could be unraveled, and potential therapies tested. 1 In other cases, the implication of a particular gene in the developmental processes is challenging because of the lethality of existing knockout models. This is of great interest for the evaluation of new alternative genetically modified animal models that could uncover additional functions for the targeted gene. Among all animals, the mouse is the one most commonly used for disease modeling and research in human diseases. 2
Previously established methods for the generation of genetically modified mouse models, such as genome modification in embryonic stem cells followed by injection into morulae or blastocysts, entail a substantial amount of time, notable technical expertise, and the use of a large number of animals to obtain organisms with the desired modifications. 3
In recent years, the combination of two innovative areas, genome modification driven by the CRISPR-Cas9 system 4 and new procedures for delivery of exogenous material into zygotes, has resulted in efficient methods for generating modified mice in a controlled manner and in record time.5–7 The use of programmable nucleases that specifically recognize target sequences in the genome constitutes a remarkable advance for the generation of genome-modified organisms, including the efficient editing of several loci at the same time or the generation of mice carrying reporter or conditional alleles.8–10 In addition, the delivery of Cas9/guide RNA (gRNA) into pronuclear-stage embryos as ribonucleoproteins (RNPs) aims to narrow the time window in which editing tools are active to minimize mosaicism. 11
Furthermore, in an attempt to control the result of genome editing, pairs of gRNAs, instead of only one guide, have been used, which has resulted in a more precise genetic outcome in terms of controlled deletions.12–14 This could be extremely useful to simplify progeny characterization when generating genetically modified mouse models.
Traditional methods for the delivery of modification tools include mating of superovulated females, isolation of zygotes, microinjection of genome editing components into the zygotes, and transfer of microinjected zygotes into the oviducts of pseudopregnant females. 15 In particular, microinjection of this material into the pronuclei or the cytoplasm of mouse zygotes is the main impediment for an accessible and rapid generation of genetically modified mouse models. Zygote electroporation of nucleases appeared as a relevant improvement in the field6,11,16 because of its high efficacy generating mutations by nonhomologous end joining (NHEJ) and also homology-directed repair-mediated modifications. Furthermore, based on the live birth rate, electroporation is a less invasive method than microinjection. 6 Moreover, when delivering gene editing tools directly by in vivo electroporation of oviducts, the number of required animals is greatly reduced, which circumvents this additional limiting step of classical animal transgenesis.3,17,18
Available models for human diseases do not always reproduce all the hallmarks of the conditions to be modeled. More importantly, patients usually display a wide range of phenotype severities that in many cases is not fully represented by current models. Consequently, an efficient and robust method for generating different models of genetic diseases is of great interest. In the present work, we have tested in vitro or in vivo electroporation of gene editing tools to target three different loci independently, to generate new alternative mouse models of two different inherited metabolic rare diseases, Primary Hyperoxaluria Type 1 Pyruvate Kinase Deficiency (PKD), and of the lack of the cancer-related gene Rasa1 (Fig. 1 and Supplementary Fig. S1).
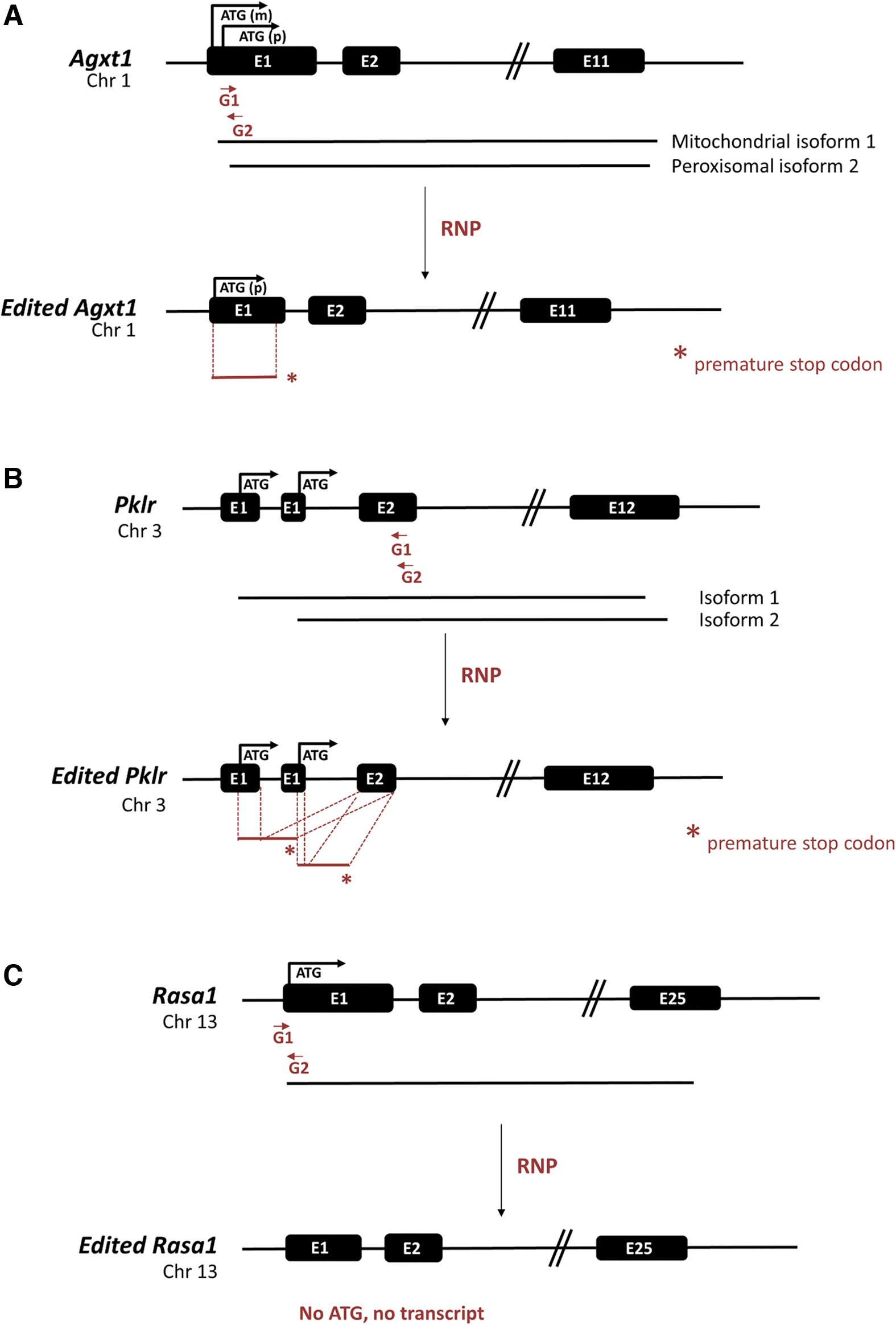
Scheme of gene editing strategy for different genes. Pairs of specific gRNAs were designed targeting each locus to assist precise and controlled deletion. Wild-type and edited versions are represented. For the generation of KO mice models of Prymary Hyperoxaluria Type 1
The outstanding efficiencies, reproducibility, and robustness that we have obtained in the generation of these three models validate the use of RNP electroporation into zygotes for these purposes and underscore the relative facility of the method, which can be performed in standard laboratories without the need to outsource to specialized core facilities.
Materials and Methods
Animals and procedures: ethical statement
Mice were maintained at the animal facility of CIEMAT (user code ES280790000183) with free access to standard chow and water. For the generation of edited mice by in vitro or in vivo electroporation, 10–12-week-old females from three immunocompetent strains [C57BL6J x DBA2J F1 (B6D2F1), Crl:CD1(ICR) (CD1), and B6.SJL-Ptprca/bPep3b/BoyJx-DBA/2 F1 (P3D2F1)] were used. In addition, 8–10-week-old females from an immunodeficient background [NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG)] were used for in vivo electroporation. All the animal experimentation procedures were approved by the Animal Protection Area of the Community of Madrid with reference numbers PROEX 165-18, PROEX 166-18, PROEX-192.0/21, and PROEX 066-19. All experiments complied with all the ethical regulations.
Gene editing tools and RNP assembly
gRNAs from Integrated DNA Technologies (IDT, Coralville, IA) (Supplementary Table S1) consisted of a duplex system: Alt-R CRISPR-Cas9 crRNA + Alt-R CRISPR-Cas9 tracrRNA. crRNA and tracrRNA were resuspended in nuclease-free duplex buffer from IDT at 200 μM and mixed in an equimolar concentration, followed by incubation at 95°C for 5 min to form the gRNA. For RNP complex formation, 6 μM crRNA:tracrRNA duplex and 1.2 μM of Cas9 nuclease were mixed and incubated for 20 min at room temperature. gRNA-Cas9 RNP complexes were mixed in equimolar quantities. For in vitro electroporation, 5 μL of RNP complexes was used for electroporation of 50 one-cell embryos. For in vivo electroporation (i-GONAD), RNP complexes were made by combining 3 μL of crRNA:tracrRNA complexes, 1 μL of Cas9 nuclease (at 61 μM), and 6 μL of Opti-MEM; 1.5 μL of RNP complexes was used per oviduct.
In vitro electroporation of zygotes
Pronuclear-stage embryos were mixed with preassembled Cas9/gRNA RNPs and subjected to electrical pulses to permeabilize the zona pellucida and the cell membrane in a transient manner to allow access of RNP to the nucleus (Fig. 2A). The NEPA21 electroporator system and a CUY501P1-1.5 electrode were used (NepaGene, Japan) following the provider's instructions and with a final impedance range of 0.18–0.22 kΩ.
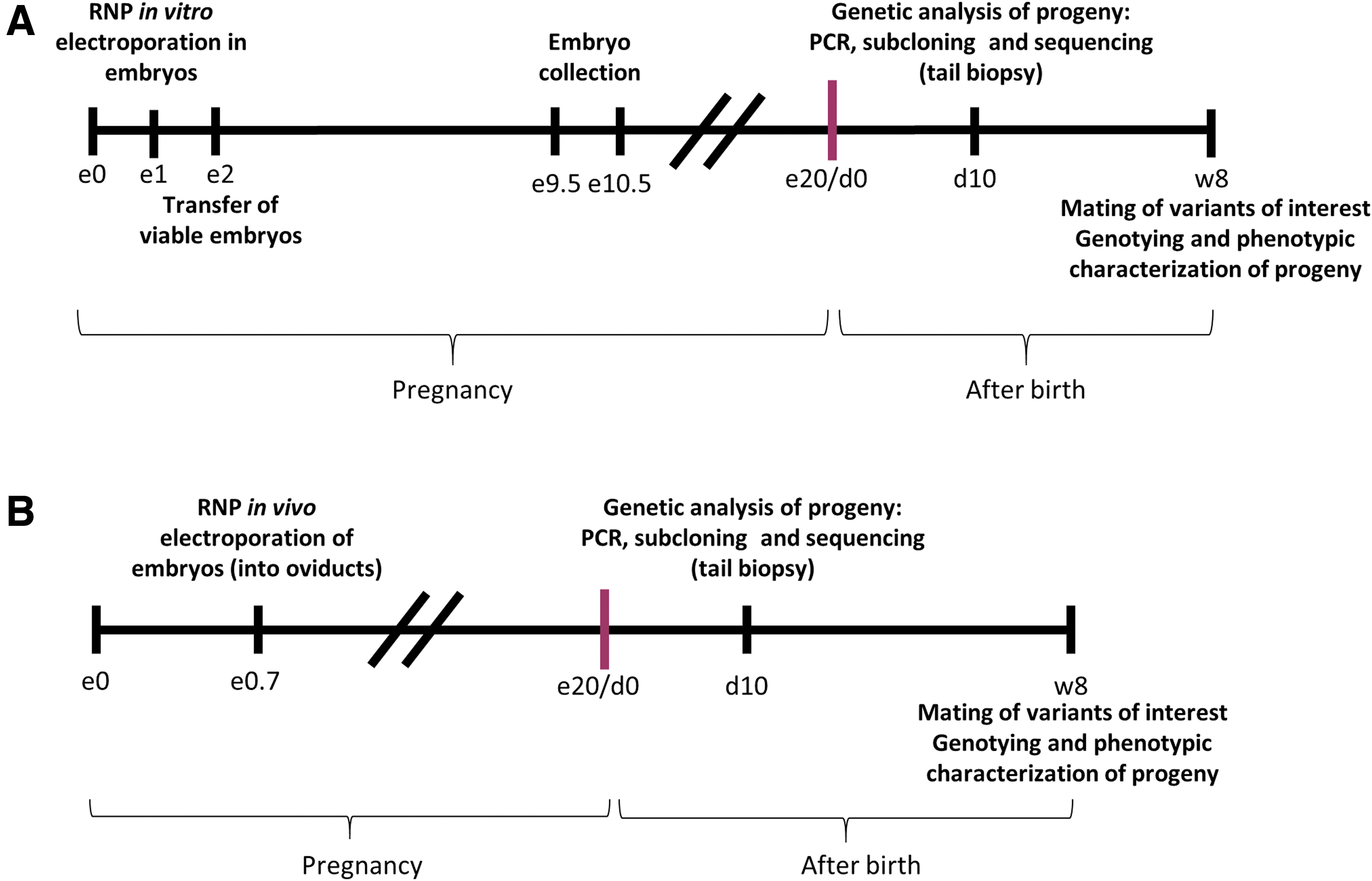
In vitro and in vivo genome editing in embryos.
Embryo culture, oviduct transfer
In vitro electroporated one-cell embryos were cultured under light paraffin oil (Nidoil) in EmbryoMax KSOM Mouse Embryo Media (Sigma-Aldrich) at 37°C in air containing 5% CO2 and 95% relative humidity. Two-cell embryos were surgically transferred to the oviduct of pseudopregnant B6D2F1 females following standard techniques. 19
i-GONAD procedure and microinjection
Microinjection of gene editing tools into oviducts was performed as described. 20 Briefly, matings were set up in the afternoon at 16:00–17:00 h and plugs were confirmed by visual inspection the next morning. Females with plugs were considered at day 0.7 of gestation at 16:00 h, when they were anesthetized. Two different RNPs were instilled into the oviducts close to the location of the 0.7-day-old embryos (i.e., the ampulla) and electroporated using the NEPA21 electroporator system and a CUY652P2.5X4 electrode (specially designed for oviduct electroporation) or a CUY650P3 electrode (Fig. 2B). The impedance range was 0.09–0.20 kΩ.
Characterization of edited genome variants
For variant characterization in born animals, tail or finger biopsies were lysed and purified genomic DNA was amplified. Primers listed in Supplementary Table S2 were used to amplify sequences surrounding the recognition sites of different gRNAs. Sanger sequencing of polymerase chain reaction (PCR) amplicons was performed to fully characterize the different variants caused by genome editing after subcloning using the TOPO cloning kit (Invitrogen). At least 10 subclones were sequenced per mouse.
Primers designed to identify large deletions as a consequence of the editing process in Agxt1 and Rasa1 loci are listed in Supplementary Table S2.
Genotype analysis
For subsequent genotyping of the progenies from founder mice and identification of homozygous mice, the primers described above for the characterization of mice were used, and DNA amplicons were analyzed by agarose gel electrophoresis.
Phenotypic characterization of edited mice
AGXT detection and nephrocalcinosis development in Agxt1-edited mice
Agxt1-edited mice were subjected to an ethylene glycol challenge, as an overloading of oxalate is needed to develop renal damage and nephrocalcinosis.21,22 The precursor of glyoxylate metabolism ethylene glycol (0.5% Ethylene glycol Reagent Plus; Sigma-Aldrich) was administered in drinking water for 7 days. Hematoxylin and eosin staining was conducted in kidney sections, and analysis was performed using an Olympus BX41 microscope and an analyzer (U-ant) and a polarizer (U-pot) set to detect crystal deposition (Olympus). AGXT protein reduction was verified by Western blot analysis. Liver homogenates were used, 23 and protein was detected using an anti-AGXT antibody (1/5000 dilution; ab178699; Abcam). Vinculin (ab129002; Abcam) was used as a load control. Nonedited littermates were used as controls for Western blot analysis and crystal deposition characterization.
Reticulocytosis and splenomegaly study in Pklr-KO mice
To define the anemic phenotype in Pklr modified mice, the reticulocyte percentage in blood was analyzed by hematological counter (Sysmex), and spleens were extracted and measured after sacrificing the animals. Nonedited littermates were used as healthy controls for both measures.
Rasa1 phenotype testing
Male and female mice carrying one copy of the mutated allele were mated and embryos were analyzed at e9.5D and e10.5D. At these ages, Rasa1 knockout embryos presented evident morphological defects that could be visually detected, such as distention of the pericardium and smaller size. Nonedited littermates were used as controls for morphological characterization. Genotype and phenotype analysis had 100% coincidence for homozygous animals.
Statistical analysis
Significant differences between groups were determined by the Mann–Whitney test. Analyses were performed with GraphPad Prism software. Significant values were considered when p < 0.05.
Results
In vitro electroporation of RNPs is efficient in three different loci and induces a high number of genomic variants
Pronuclear-stage embryos were mixed with preassembled Cas9/gRNA RNPs and subjected to electrical pulses. Electroporated one-cell embryos were maintained at 37°C O/N and then transferred as two-cell embryos into pseudopregnant females to allow embryo development. More than 90% of the mice obtained after in vitro electroporation and analyzed at 10 days of age carried modifications in the target sequence (13/14 in Agxt1, 11/12 in Pklr, and 6/8 in Rasa1) (Fig. 3A). Some mice carried more than two variants in all the models (Fig. 3B).
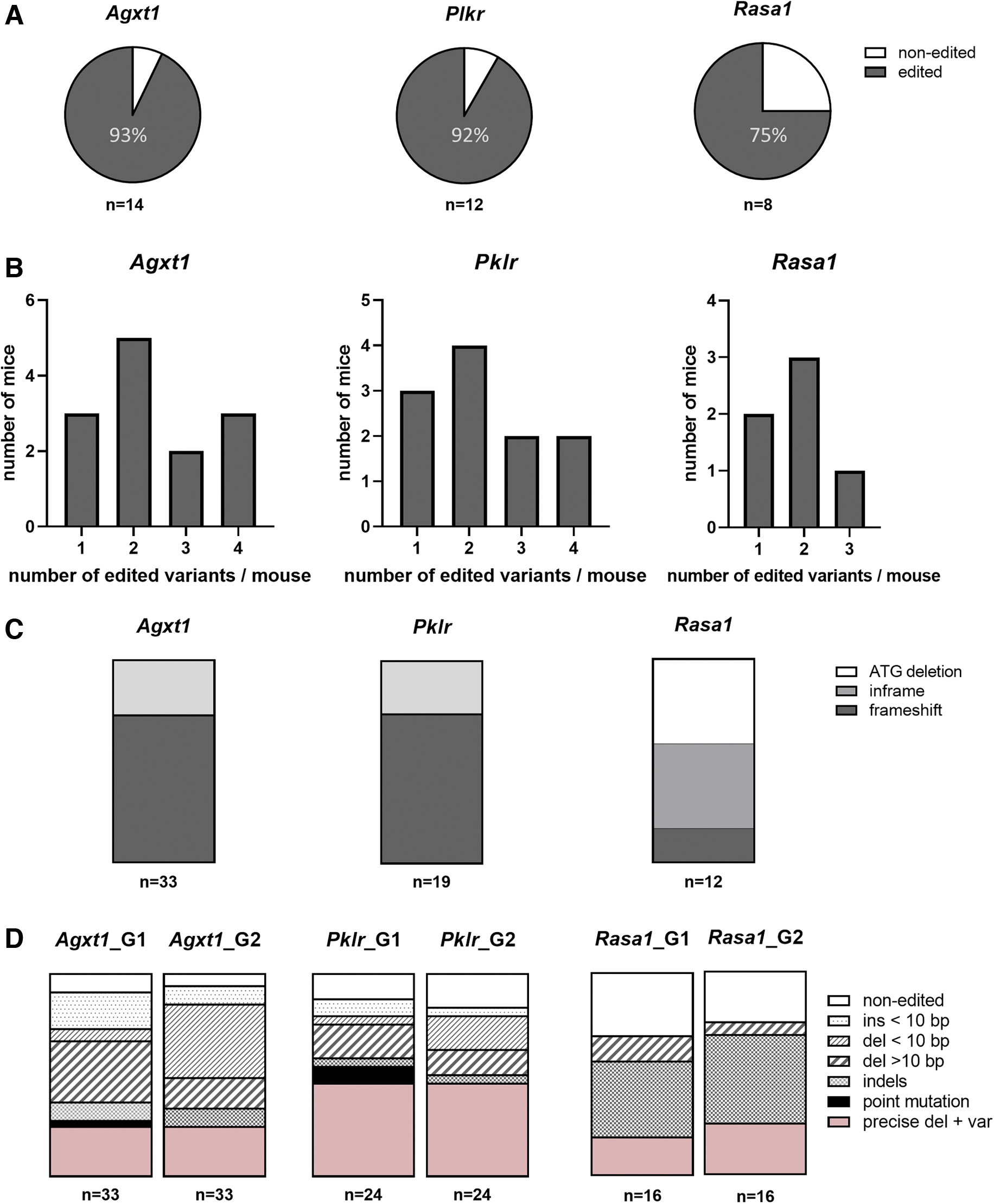
Generation of genetic variants by in vitro zygote electroporation of gene editing tools.
The characterization of CRISPR-Cas9-mediated gene editing outcome revealed different patterns of modifications depending on the different gRNAs. The most frequent changes were deletions for Agxt1-specific gRNAs, the precise deletion for Pklr-specific gRNAs, and small indels and deletions for Rasa1-specific gRNAs, respectively. Frameshift mutations were the most abundant result (more than 70% of characterized variants) in Agxt1 and Pklr editing, which led to a premature stop codon or a completely different protein and thus to the absence of wild-type (WT) protein (Fig. 3C, D). In addition, editing in Rasa1 produced the expected ATG deletion, and also a diverse spectrum of results consisting mainly of independent deletions of different lengths, pointing to two consecutive rounds of cutting and repairing.
This effect was also detected in the other two loci (Fig. 4). Precise deletion or its variants (precise deletion plus small indels in one or both cleavage sites) were more frequent in the Pklr locus, where they accounted for almost half of the edits (Fig. 3D). In the case of Agxt1 locus, specific small indels seemed to occur in homozygosity (data not shown), which was surprising taking into account the high number of different genetic variants identified. The finding of the same edit in the two alleles would seem improbable, for example, we found large deletions in one allele in an Agxt1-edited mouse in which a 21 bp deletion in the recognition site of G1 and a small insertion in the recognition site of G2 seemed to be in homozygosity.
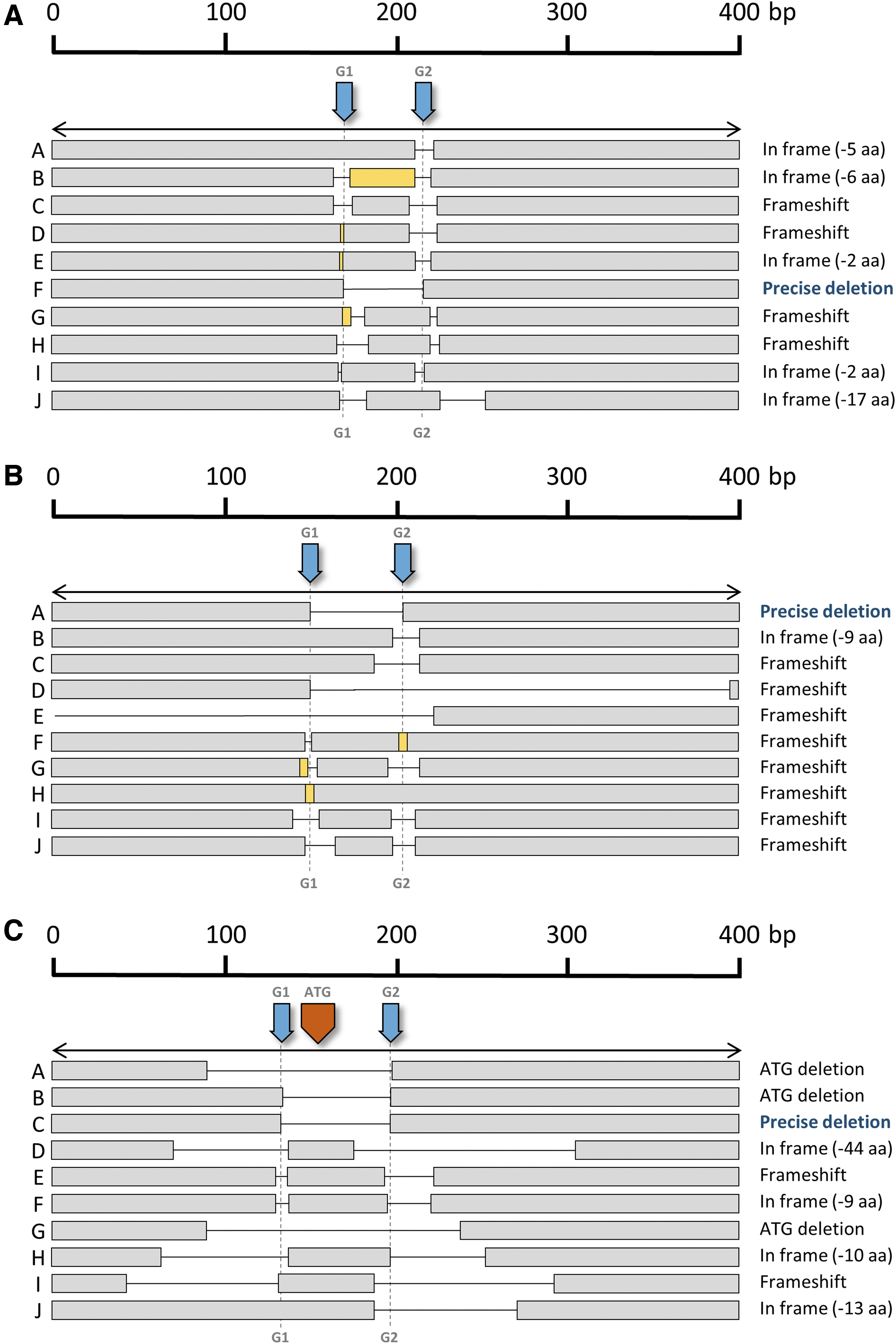
Generation of edited alleles after in vitro zygote electroporation. Diagram showing some of the different alleles found after editing of each locus.
A larger PCR revealed different large deletions in one of the two alleles (Supplementary Fig. S2), pointing to the possibility that this type of deletion could be underestimated in conventional PCR analysis of genomic DNA. In the case of Rasa1 edit, amplification of a 3 kbp fragment did not show evidence of larger deletions (data not shown).
In vivo electroporation of RNP rendered different editing outcomes depending on the target locus
In vivo electroporation of RNP in oviducts of females was performed to target each of these three loci. Editing efficiencies were significantly lower for each locus in comparison with those obtained by in vitro electroporation. Editing of Rasa1 was not detected in any of the analyzed mice (n = 22) and Agxt1 and Pklr loci were edited in 23% (n = 35) and 38% (n = 16) of the analyzed mice, respectively (Fig. 5A). Up to five different variants were identified per mouse after PCR cloning of the target sequence following Sanger sequencing (Fig. 5B). Differences were also observed in the frequency of frameshift mutations obtained in the different target loci, with the Pklr locus again being more favorably edited, with nearly 60% of the modifications leading to a shift in the reading frame, and representing less than 35% of the total detected variants in the Agxt1 locus (Fig. 5C).
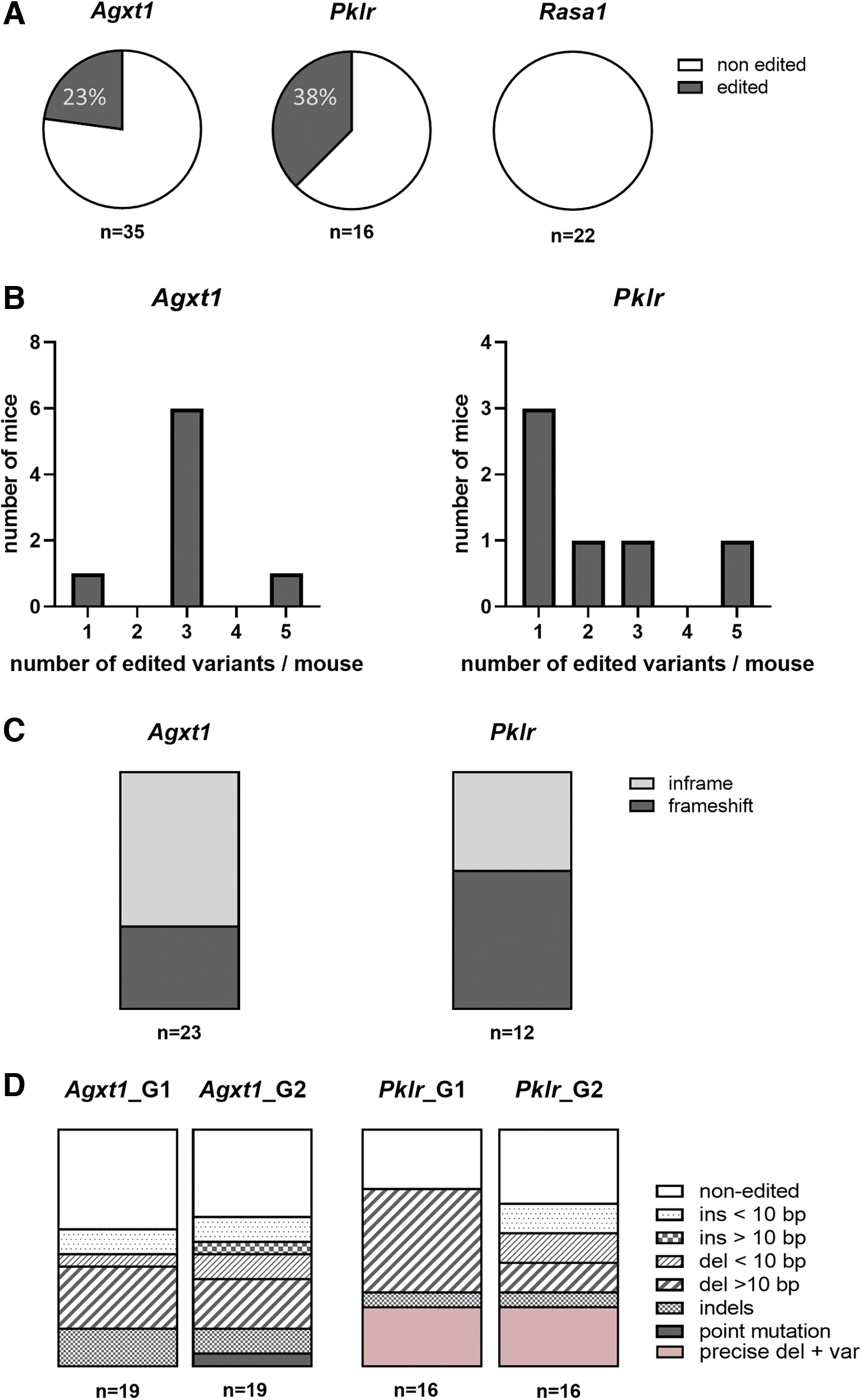
Generation of genetic variants by in vivo electroporation of gene editing tools in oviducts.
Precise deletion, as it occurred after in vitro electroporation of gene editing tools, was not as frequent as expected, being absent in Agxt1-modified mice (24 analyzed variants) and representing only 25% of the characterized variants in Pklr. Deletions were the most frequent modifications generated for both gRNAs in both target genes (Fig. 5D) and, as occurred after in vitro electroporation of zygotes, most variants seemed to arise from independent editing events of the two gRNA target sites (Fig. 6).
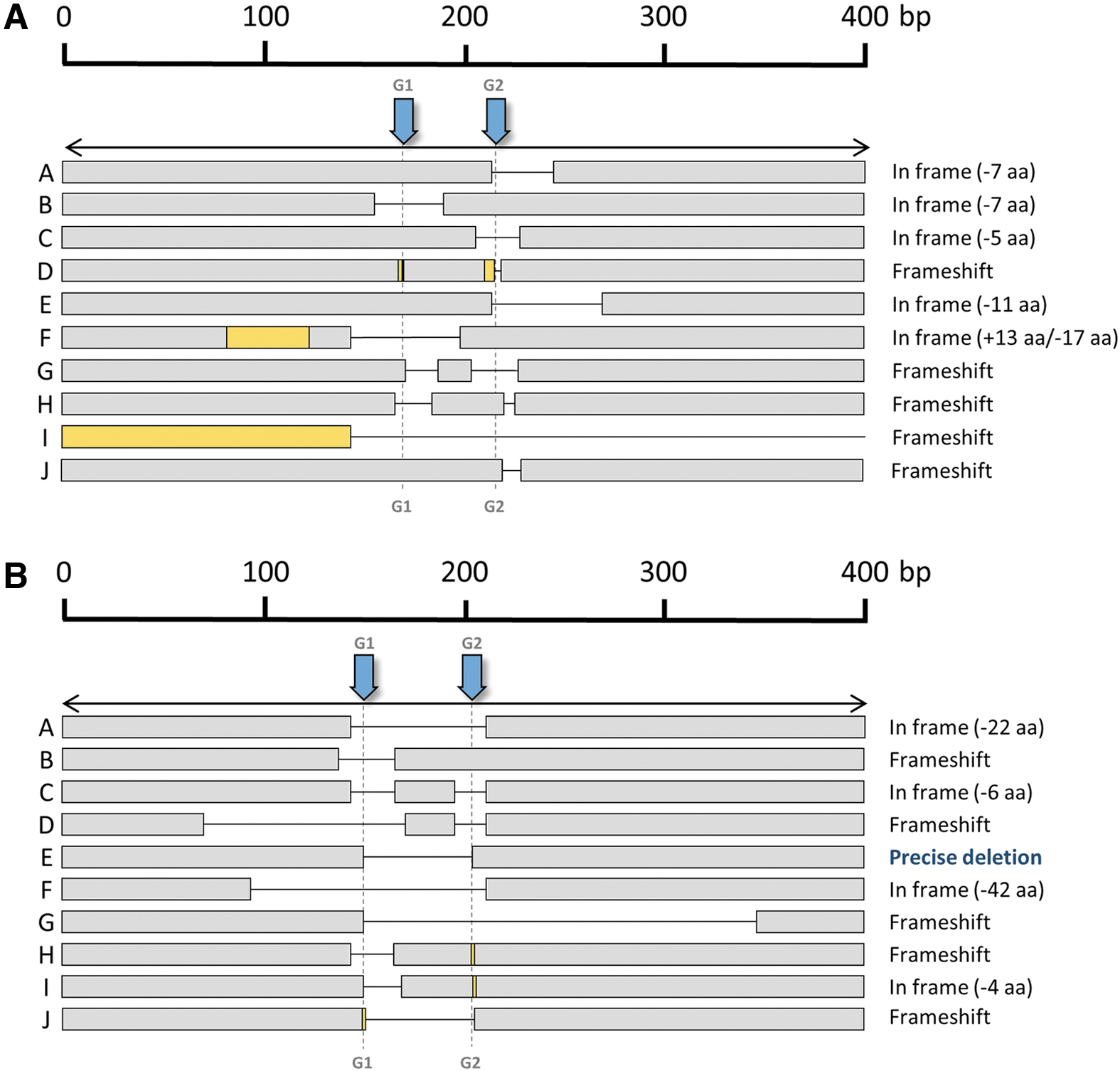
Generation of edited alleles after in vivo electroporation. Diagram showing some of the different alleles found after editing of each locus:
Agxt1-edited mice lack AGXT protein and accumulate oxalate crystals in kidneys
AGXT-deficient patients present an overproduction of glyoxylate in the liver that is converted into oxalate and excreted by kidneys. Accumulated oxalate, together with calcium, form calcium oxalate crystals, which are deposited in the kidney lumen and cause renal inflammation and subsequent renal disease.23–25 Metabolism in mice is known to be more active than in humans. Thus, mice need to be challenged with a precursor of glyoxylate metabolism to force oxalate accumulation and kidney damage.21,22 To test whether knock out of Agxt1 locus by genome editing in mice led to its loss of function, homozygous animals carrying the precise 46 bp deletion (named del46/del46) were obtained and subjected to an ethylene glycol challenge for a week. After that, animals were sacrificed and Western blot analysis showed a complete absence of AGXT in homozygous mice (Fig. 7A).
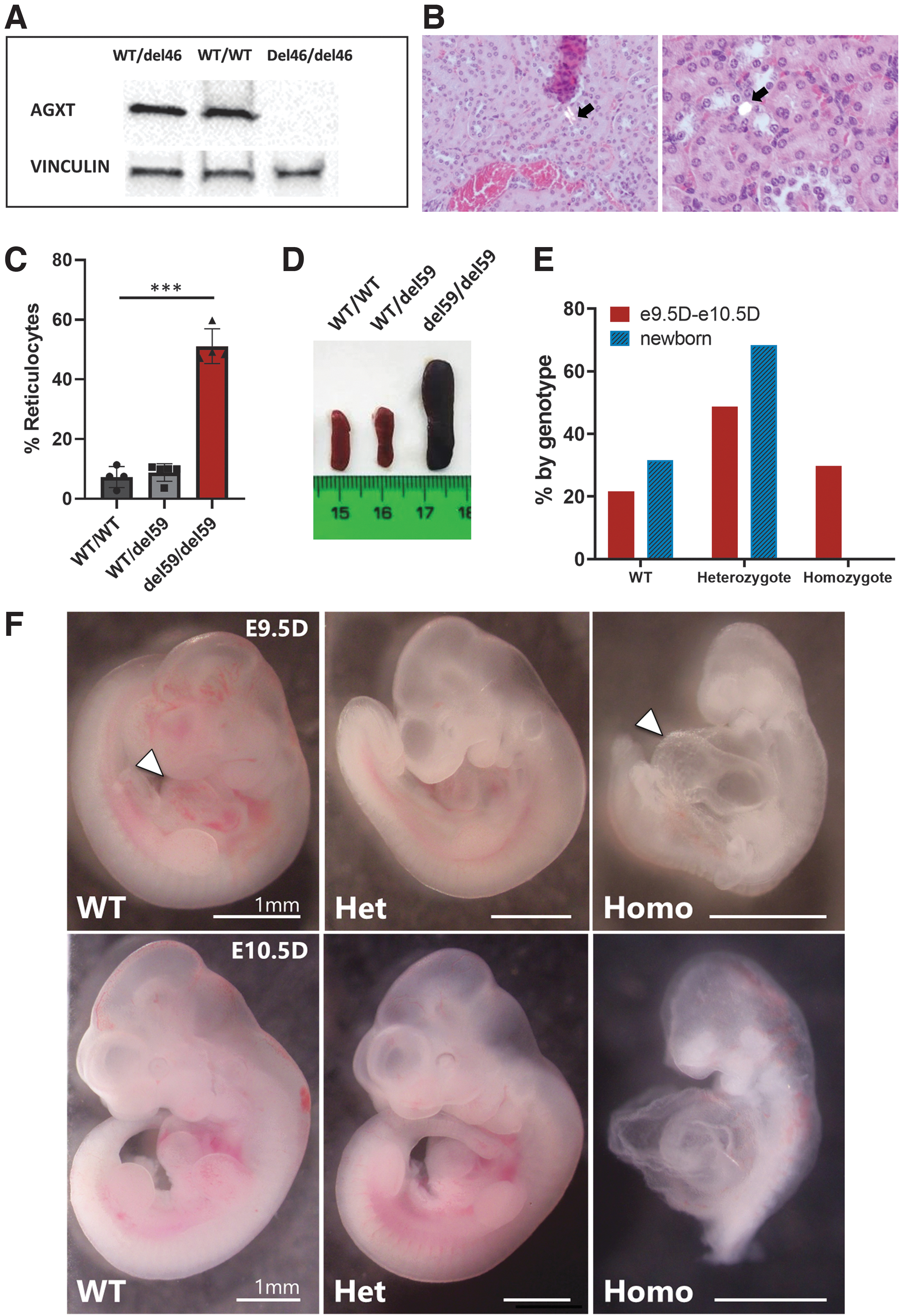
Phenotypes of edited mice.
Histological analysis of kidney sections revealed deposition of crystals in the urinary tract of edited mice (Fig. 7B). No crystals were identified when analyzing kidney sections from nonedited mice subjected to the ethylene glycol challenge.
Pklr-edited mice recapitulate hematological hallmarks of PKD patients
The animals with the precise deletion of 59 bp in both alleles (del59/del59; Fig. 7C) generated by the gene editing tools presented an increased percentage of reticulocytes, a primary feature of PKD profile. As expected, heterozygous individuals (WT/del59) did not show this reticulocyte increase and showed similar values to WT nonedited mice (WT/WT) (Fig. 7C). Homozygous edited mice also showed splenomegaly (Fig. 7D), another classic PKD feature, thus corroborating the modeling of the disease.26,27
Rasa1-edited mice undergoing frameshift recapitulate the phenotype of Rasa1 null animals
Half (5/10) of the Rasa1-edited alleles obtained underwent independent edits for each guide and resulted in the loss of two small fragments of the gene (Fig. 4C). In most cases, editing resulted in proteins carrying a short deletion that conserved the reading frame. However, two edits resulted in Rasa1 frameshift mutations that gave rise to a nonfunctional RASA1 protein. Crosses of these mice completely failed to yield newborn animals homozygous for the frameshift alleles. Knockout of Rasa1 is embryonic lethal at day e10.5D, 28 due to massive apoptosis. Therefore, to better characterize this effect, we crossed heterozygous animals bearing the frameshift alleles and analyzed embryos at day e9.5D and e10.5D.
PCR genomic analysis of DNA from amnion and chorion confirmed that at these early embryonic stages, all three genotypes appeared in proportions close to the Mendelian inheritance (Fig. 7E). e9.5D homozygous embryos were smaller than heterozygous and WT embryos, and presented an evident distension of the pericardium (Fig. 7F). Most of the e10.5D homozygous mice were dead and starting to be reabsorbed by the mother's uterus. These results mimic the phenotype observed in Rasa1 null animals28–30 and confirm that CRISPR-Cas9 in vitro editing of embryos can be used to easily obtain null animals.
Discussion
The combination of CRISPR-Cas9 system to increase editing by NHEJ in specific genome locations and its electroporation in zygotes is nowadays facilitating the generation of a relevant mouse model for biomedical research. 31 In this study, pairs of gRNAs complexed with Cas9 as RNPs were electroporated, in vitro or in vivo, into zygotes to delete specific genome sequences in a controlled manner. The expression of three different genes was abolished, with the aim of testing the effectiveness of these methods to generate three independent models of human diseases as a first step, with the objective of using the generated models in the respective research areas of the authors in a second phase. Both methods, in vitro electroporation of one-cell embryos or in vivo electroporation of 0.7-day-old embryos in oviducts, have led to the efficient modification of the target loci and therefore to the generation of disease mouse models.
In vitro electroporation rendered a clearly more efficient output in the three targeted loci, resulting in more than 75% of mice edited, with most edits leading to frameshift mutations or significant modifications in protein sequence (Fig. 3A, C). On the contrary, after in vivo electroporation of the RNP into the oviducts, less than 40% of mice harbored edited alleles in the case of Agxt1 or Pklr targeting and no edited mouse was identified in the case of Rasa1 targeting (Fig. 5A).
One main hurdle for using RNA-guided Cas9 nuclease is the diversity of allelic variants generated that causes heritage of multiple variants in progeny.10,31 In this study, two different guides were used simultaneously with the aim of deleting the intermediate sequence and restricting the range of genome modifications, as has been previously indicated.12–14 However, it did not appear as a solution in zygotes, which is contrary to what has been described in adult cells.12,32,33 The preferential pathway for DNA repair differs between embryonic and adult cells and, in addition, DNA repair is accelerated in embryonic cells compared with adult cells. 34 These facts would explain the differences observed in controlling the generated variants after gene editing of target loci.
We observed that two separate editing events occurred in many cases, which resulted in independent modifications of the two gRNA-hybridizing sites added at both sides of the targeted sequence. We observed that the central DNA tract between guide recognition sites was conserved in many variants, with insertions and deletions around both of these sites (Figs. 4 and 6). This can be best explained as a result of consecutive cleavage by the Cas9 protein, first using a guide, and, after accelerated embryo cell DNA repair, using the other guide. When using two guides, the distance between the recognition sites also affects the success of precise deletions.
In this study, despite similar distances between the two recognition sites for the gRNAs in the three loci, clear differences in the editing efficacy have been obtained. Other factors should be taken into account, such as the different orientations of the PAM in Agxt1 and Rasa1 editing and thus positioning of the Cas9 nuclease on the DNA. 12 The expression level of the to-be-edited locus could also play a role in the efficacy of editing. 35 However, in this particular case, neither Agxt1, Pklr, nor Rasa1 genes are expressed, to our knowledge, at the embryonic stages in which editing tools were electroporated and should act.
Another significant pitfall in genome modification assisted by the CRISPR-Cas9 system is the potential nonexpected changes at on-target or off-target genome locations. Large deletions (longer than 1000 bp) were detected at the on-target site for at least one of the three targeted loci. This points to a potential underestimation of editing frequency and highlights the importance of characterizing these events. Regarding off-target effects, the analysis in our edited mice is pending, although selection of gRNA was conducted based on in silico analysis and only those gRNAs with the best scores (high for on-target site and low or null for off-targets sites) were chosen. Unexpected editing events, both at on-target and off-target sites, should be addressed as they could be modifying the observed phenotypes.
Nevertheless, taking into account the reproducibility of the deficient phenotype in all the knockout (KO) animals generated in the three different models, a very low impact of the potential off-target events would be expected.
Conclusions
Despite the difference in efficacy, the fact that both methods worked as expected suggests that the generation of the three aimed mouse models of human diseases is feasible. The main phenotypic features of each disease have been confirmed and the models generated are currently used for disease-specific research. Based on these results, in vitro and in vivo electroporation of gene editing tools constitutes suitable methods for the fast generation of genetically modified mice. Furthermore, these techniques can be performed by research groups with basic transgenesis skills without the need for specialized transgenic mice facilities.
Footnotes
Acknowledgments
The authors thank Ian Blackstock (Sonidel Limited, Dublin, Ireland) for lending the Nepa 21 equipment and for advice in the generation of CRISPR-modified mice. The authors also thank Jesús Martínez, Edilia de Almeida, and Miguel A. Martin for the careful maintenance of animals; Federico Sánchez-Sierra for the histological processing and staining; and Aurora de la Cal, María del Carmen Sánchez, Soledad Moreno, Nadia Abu-sabha, Montserrat Aldea, and Sergio Losada for dedicated administrative help.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from “Ministerio de Economía, Comercio y Competitividad” (PID2020-119637RB-I00), Fondo de Investigaciones Sanitarias, “Instituto de Salud Carlos III” (RD21/0017/0027 and PI20/01173), and cofunded by the European Regional Development Fund (FEDER), “Comunidad de Madrid” (AvanCell, B2017/BMD-3692), and Oxalosis and Hyperoxaluria Foundation. The authors also received funds from Instituto de Investigación Sanitaria “Fundación Jiménez Díaz” and CIBERER (CB06/07/0014) and CIBERONC (CB16/12/00228), initiatives of the “Instituto de Salud Carlos III” and “Fondo Europeo de Desarrollo Regional (FEDER).” R.S.-B. is funded by the Consejería de Ciencia, Universidades e Innovación (Comunidad de Madrid), and the European Regional Development Fund (FEDER). “Ministerio de Ciencia, Innovación y Universidades” awarded grants to V.N.-R. (FPU16/02228), A.M.-V. (FPU17/02179), and I.O.-P. (SAF2017-84248-P).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.