
Abstract
Mutations in the GPR143 gene cause X-linked ocular albinism type 1 (OA1), a disease that severely impairs vision. We recently generated induced pluripotent stem cells (iPSCs) from skin fibroblasts of an OA1 patient carrying a point mutation in intron 7 of GPR143. This mutation activates a new splice site causing the incorporation of a pseudoexon. In this study, we present a high-performance CRISPR-Cas ribonucleoprotein strategy to permanently correct the GPR143 mutation in these patient-derived iPSCs. Interestingly, the two single-guide RNAs available for SpCas9 did not allow the cleavage of the target region. In contrast, the cleavage achieved with the CRISPR-AsCas12a system promoted homology-directed repair at a high rate. The CRISPR-AsCas12a-mediated correction did not alter iPSC pluripotency or genetic stability, nor did it result in off-target events. Moreover, we highlight that the disruption of the pathological splice site caused by CRISPR-AsCas12a-mediated insertions/deletions also rescued the normal splicing of GPR143 and its expression level.
Introduction
Ocular albinism type 1 (OA1) is a rare X-linked inherited disorder that impairs vision in affected individuals. It has a prevalence of approximately one male in 50,000 live born children.1,2 OA1 patients are affected with nystagmus, foveal hypoplasia, reduced visual acuity, hypopigmentation of the iris and the retinal pigment epithelium (RPE), photophobia, and aberrant optic pathway projections that result in stereoscopic vision loss. 3 A large number of OA1 patients have mutations in the GPR143 gene,2–4 also known as OA1, which encodes the intracellular G-protein-coupled receptor GPR143. GPR143 is a glycoprotein 5 with seven transmembrane domains6,7 that is highly expressed in the melanosomes of pigmented cells. Previous studies have suggested that GPR143 is involved in melanosome biogenesis and trafficking.8–14 Consistently, melanosomes are abnormally enlarged (macromelanosomes) in OA1 patients.15,16
We recently generated a line of induced pluripotent stem cells (iPSCs), named SEIi001-A or SEIOA1#1, from fibroblasts of an OA1 male patient 17 who carries the previously described GPR143 point mutation NC_0000023.8:g.25288G>A, 18 currently identified as NC_0000023.11:g.9740590G>A. This mutation, located in intron 7, creates a consensus binding motif for the splicing enhancer factor ASF/SF2 and activates a new 5′ acceptor site causing the predominant incorporation of a 165-bp pseudoexon.17,18 The resulting unstable mRNA codes for a premature stop codon that leads to loss of function of the GPR143 protein.
Although Vetrini et al. 18 were able to mask the pathological acceptor site with an antisense oligonucleotide (AON) and partially rescue the splicing defect in the patient's melanocytes, we wanted to use the SEIOA1#1 line to develop a gene editing strategy that would allow the permanent correction of the intronic GPR143 mutation. The CRISPR-Cas system, originally an adaptive immune response of bacteria to exogenous DNA material,19,20 has been extensively studied and engineered to become a valuable tool for genome editing experiments.21–23 It is composed of a guide RNA (gRNA), targeting a complementary DNA region, and a Cas endonuclease. A protospacer adjacent motif (PAM) has to be present next to the gRNA target for Cas' recognition. The gRNA drives the nuclease to the target region, where it creates a double-strand brake (DSB). The default non-homologous end-joining (NHEJ) pathway randomly repairs the DSB, and frequent insertions and deletions (indels) are incorporated at the cleavage site during this process. However, if a donor DNA is provided as a template, the homology-directed repair (HDR) can occur at a higher rate. 24
The most commonly used nuclease is SpCas9, 25 isolated from Streptococcus pyogenes. Nonetheless, other nucleases have been recently identified in different bacterial strains, 26 such as Cas12a, also known as Cpf1.27–29 Particularly interesting for mammalian applications 30 are the Acidaminococcus sp. BV3L6-derived AsCas12a31,32 and the Lachnospiraceae bacterium ND2006-derived LbCas12a. 33 Cas12a has a few differences compared with SpCas9 First, both AsCas12a (1,228 aa) and LbCas12a (1,307 aa) are smaller than SpCas9 (1,368 aa) and they work in combination with a shorter gRNA. The CRISPR gRNA (crRNA) for Cas12a is only 42–44 nt long compared with the ∼100 nt long single-gRNA (sgRNA, which is formed by crRNA + tracrRNA) for SpCas9. Second, the PAM sequence for SpCas9 is 3′ NGG, whereas that of Cas12a is 5′ TTTV. Cas12a is therefore more suitable for targeting T/A-rich sequences. Lastly, in contrast with the blunt ends created by SpCas9 cleavage close to the PAM sequence, Cas12a-mediated cleavage produces ∼5 nt overhanging ends farther from PAM, which may favor HDR events.
Another important aspect for genome editing experiments is the delivery of the CRISPR-Cas system into cells. Although plasmid-based delivery has been extensively used, the use of ribonucleoproteins (RNPs) is currently an attractive alternative, because they can rapidly start the editing process, are less toxic, and are more efficient.34–37 Moreover, since the CRISPR-Cas system is not continuously produced by the encoding plasmid, the RNP-delivered components are present for a limited time in the cell nuclei, reducing potential off-target events.34,38
In the past few years, CRISPR-Cas has become a particularly useful tool for editing mutations causing different hereditary blindness conditions, such as retinitis pigmentosa,23,39,40 enhanced S-cone syndrome, 41 Leber congenital amaurosis,42,43 and Usher syndrome type 2, 44 in patient-derived iPSCs or in mouse models of retinal degeneration. Thus, gene editing holds a promising potential for translational progress in the vision field.
With the aim of developing a novel therapeutic option for ocular albinism patients, we investigated gene editing tools to target the GPR143 gene. We exploited the RNP CRISPR-Cas technology to correct the intron 7 mutation in OA1 patient-derived iPSCs through HDR. Unexpectedly, we observed that the sgRNAs for SpCas9 were unable to satisfactorily mediate the cleavage of the patient's DNA close to the mutation site, hindering any chance of HDR. Conversely, the crRNAs for AsCas12a mediated a highly efficient cleavage (up to ∼86%) and, when coupled to a donor template, a high correction rate (up to ∼50%). We successfully generated OA1 patient-derived corrected iPSC clones, and showed that the CRISPR-AsCas12a-mediated correction did not alter cell pluripotency or genetic stability, nor did it result in off-target events at the top 10 predicted loci.
Interestingly, in this study, we highlight that the disruption of the pathological acceptor site caused by indels, and not only the HDR correction, can restore the normal splicing and increase GPR143 expression to normal levels.
Materials and Methods
Patient
A 27-year-old male patient provided written informed consent for this study, which received UCLA IRB #16-001277 approval. He carries the previously described NC_0000023.8:g.25288 G > A mutation, 18 currently identified as NC_0000023.11:g.9740590 G>A, in intron 7 of the GPR143 gene.
Design of sgRNAs for SpCas9, crRNAs for Lb-AsCas12a, and single-stranded oligodeoxynucleotide
The sgRNAs were designed using the online tool CRISPOR (http://crispor.tefor.net/), 45 whereas for the crRNAs, we used CRISPOR, DeepCas12a (http://deepcrispr.info/), Breaking-Cas (https://bioinfogp.cnb.csic.es/tools/breakingcas/), CHOPCHOP (https://chopchop.cbu.uib.no/), 46 and CRISPR-DT (http://bioinfolab.miamioh.edu/CRISPR-DT/). 47 The predicted efficiency score and the vicinity to the patient's mutation determined the final selection. sgRNA and crRNA sequences are listed in Supplementary Table S1. The sgRNAs, SpCas9 protein, and the positive control crRNA for LbCas12a were purchased from Synthego (Redwood City, CA), the LbCas12a protein from New England Biolabs (Ipswich, MA), and the crRNAs for AsCas12a, the AsCas12a Ultra protein, and the Electroporator Enhancer from Integrated DNA Technologies (IDT, Coralville, IA). The designed single-stranded oligodeoxynucleotide (ssODN) was purchased from IDT, with phosphorothioate and IDT-patented modifications at both ends to increase stability.
In vitro digestion assay
To obtain the RNP complexes, 1 μM gRNAs were incubated for 10 min at room temperature with the corresponding 1 μM CRISPR-nuclease. The genomic target regions were polymerase chain reaction (PCR)-amplified (primer sequences in Supplementary Table S2) with the AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific, Carlsbad, CA) and purified. Two hundred nanograms of each one was then incubated at 37°C for 1 h with the 0.05 μM RNP complexes. SpCas9 was inactivated at 65°C for 15 min, whereas Cas12a was inactivated by incubation with proteinase K at 56°C for 10 min. The samples were then electrophoresed on a 2% agarose gel and stained with the SYBR Safe DNA Gel Stain (Thermo Fisher).
iPSC culture, transfection, and sorting
Our previously generated SEIOA1#1 and the control FA0000010 48 iPSC lines were cultured on Corning hESC-qualified Matrigel-coated supports in an mTeSR Plus medium (STEMCELL Technologies, Vancouver, BC). Following single-cell dissociation with Accutase, ∼250,000 cells per condition were nucleofected (4D-Amaxa Nucleofector; Lonza, Koeln, Germany) using the P3 solution. The conditions tested were 0.5 μg of pMAX green fluorescent protein (GFP) plasmid (Lonza), 1 μg of GFP mRNA (System Biosciences, Palo Alto, CA), and the following RNP combinations: 160 pmol sgRNA + 100 pmol SpCas9, 160 pmol crRNA + 100 pmol LbCas12a, and 160 pmol crRNA + 100 pmol AsCas12a ± 78 pmol Electroporator Enhancer ± 4 μM ssODN. Before nucleofection (2 h) and for 24 h after it, the culture medium was supplemented with 10 μM of Rho-associated kinase (ROCK) inhibitor, Y-27632 (Millipore, Temecula, CA).
For the HDR experiment, 48 h postnucleofection, single cells were sorted with an FACSAria II (BD, BSCRC Flow Cytometry Core Facility, UCLA) into each well of a 96-well plate. The culture medium was supplemented with 10 μM of ROCK inhibitor and 10 × Clone R (STEMCELL Technologies) until cells formed big colonies, which were observed daily under a Lynx stereomicroscope (Vision Engineering, New Milford, CT). Surviving colonies were dissociated starting from day 12 post-sorting with a 0.5 mM ethylenediaminetetraacetic acid (EDTA) solution and expanded for screening. Due to the relatively high number of arising clonal cell lines, we performed short-term cryopreservation in 96-well plates 49 using CryoStor CS10 (STEMCELL Technologies).
Surveyor assay
Forty-eight hours post-nucleofection, genomic DNA was isolated using the PureLink Genomic DNA Kit (Life Technologies, Carlsbad, CA). The target regions were PCR-amplified from genomic DNA (50 ng, primer sequences in Supplementary Table S2) with the AmpliTaq Gold 360 Master Mix and used as a substrate in the Surveyor Mutation Detection Kit for standard gel electrophoresis (IDT), according to the manufacturer's instructions. The digested products were visualized on a 2% agarose gel with the SYBR safe DNA gel stain.
Indels and HDR screening
Forty-eight hours post-nucleofection, the genomic DNA target regions were PCR-amplified (50–100 ng, primer sequences in Supplementary Table S2) with the Phusion High-Fidelity PCR Master Mix (Thermo Fisher) and Sanger sequenced by Laragen, Inc. (Culver City, CA). The sequencing readings were used to run Inference of CRISPR Edits (ICE; https://ice.synthego.com/#/) and Tracking of Indels by DEcomposition 50 (TIDE; https://tide.nki.nl/) analyses for indel estimation, or Tracking of Insertion, Deletions, and Recombination events 51 (TIDER; https://tide.nki.nl/) analysis for detecting HDR events. The GPR143 intron 7 sequencing results of the isolated clones after single-cell sorting were analyzed for evaluation of the correction.
Reverse Transcription (RT)-PCR analysis
Total RNA was extracted using the QIAshredder and RNeasy Plus kit (Qiagen, Germantown, MD) and 1 μg was retro-transcribed with the 5 × All in One First Strand cDNA Synthesis Mix (Bioland Scientific LLC, Paramount, CA). The cDNA regions of interest were amplified with the AmpliTaq Gold 360 Master Mix (primer sequences in Supplementary Table S2) and visualized on a 2% agarose gel with the SYBR Safe DNA Gel Stain.
Quantitative PCR analysis
Ten nanograms of cDNA were used per amplification reaction, in combination with the TaqMan Gene Expression assay (specific for GPR143 and GAPDH) and the TaqMan Fast Advanced Master Mix (Thermo Fisher) according to the manufacturer's instructions. Reactions were run on a QuantStudio 3 and results were analyzed using the Quantum Design and Analysis Software (Thermo Fisher) and the Microsoft Excel program. Results were normalized to GAPDH expression, and quantification was performed using the ΔΔCt method, using healthy control cells as reference.
Immunofluorescence staining
Cells were treated as previously described. 17 The primary antibodies and used dilutions were as follows: anti-NANOG 1:400 (Cell Signaling, Danvers, MA), anti-OCT4 1:200 (Abcam, Cambridge, MA), anti-SOX2 1:1000 (Abcam), and anti-TRA1–81 1:200 (Thermo Fisher). The 488 Alexa Fluor secondary antibody was used at a 1:1000 dilution (Thermo Fisher). Cells were observed using the FluoView FV1000 confocal laser-scanning microscope (Olympus, JSEI Core facility).
iPSC in vitro differentiation assay
The corrected patient-derived iPSC clones were differentiated using the Human Pluripotent Stem Cell Functional Identification Kit (R&D Systems, Minneapolis, MN) following the manufacturer's protocol with minor changes. The 488 Alexa Fluor secondary antibody (Thermo Fisher) was used against the kit's primary antibodies and actin was labeled with 80 × 546 phalloidin (Thermo Fisher). Cells mounted using Fluoromount-G (Southern Biotech, Birmingham, AL) were imaged with the FluoView FV1000 confocal laser-scanning microscope.
KaryoStat and cell ID analyses
Non-treated (NT) and corrected patient-derived iPSC clones A8 and B10 were dissociated in a single-cell suspension with Accutase, stored as a dry pellet at −80°C, and analyzed by Life Technologies. Briefly, gDNA was extracted using the Genomic DNA Purification Kit (Thermo Fisher) and quantified using the Qubit™ dsDNA BR Assay Kit (Thermo Fisher). Two hundred fifty nanograms of total gDNA was used to prepare the GeneChip® array for KaryoStat™ according to the manual, in which 150k single-nucleotide polymorphism (SNP) probes across the human genome were considered for analysis. The same 150k SNPs were compared between NT and corrected clones in the Cell ID assay for DNA fingerprint matching to evaluate the genetic background.
Statistical analysis
Quantitative PCR (qPCR) statistical analysis was performed using a one-way analysis of variance followed by multiple comparisons against healthy control cells and NT patient cells using a Dunnett test. p < 0.5 values were considered statistically significant.
Results
The sgRNAs available for SpCas9 do not allow the correction of the GPR143 intronic G > A mutation in the OA1 patient-derived iPSCs
To design the sgRNAs for targeting the patient's GPR143 mutation, we used the online CRISPOR tool. Because the sequence surrounding the mutation is particularly T/A-rich, only 2 sgRNAs (named sgRNA-1 and sgRNA-2) were identified for the SpCas9 nuclease (Fig. 1A). The G > A patient's mutation corresponds to the N position of the NGG PAM sequence for sgRNA-1, and to the first nucleotide of sgRNA-2. Hence, due to the low tolerance to mismatches of the SpCas9 nuclease, it was impossible to test sgRNA-2 in non-mutated cells, such as immortalized human cell lines.
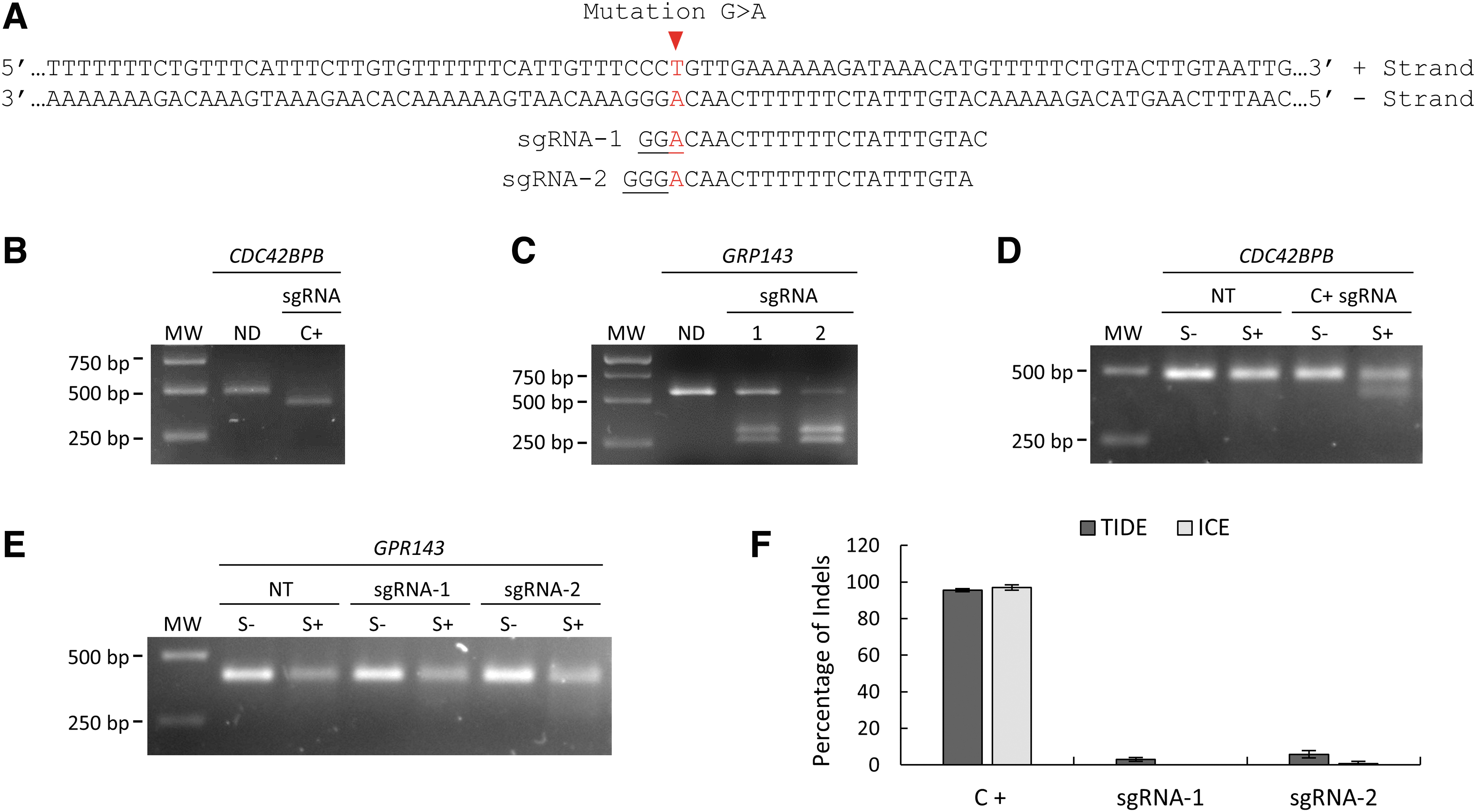
CRISPR-SpCas9 targets and sgRNA screening in OA1 patient-derived iPSCs.
Thus, we performed a first screening of the sgRNAs using an in vitro digestion assay. The DNA region of interest of the patient's iPSCs was PCR-amplified and then incubated for 1 h with the different RNP complexes of SpCas9 and sgRNAs, before migration on an agarose gel. The positive control (C+) sgRNA, a highly efficient sgRNA targeting the CDC42BPB gene, mediated the complete digestion of the PCR product. This resulted in two bands (one visible in Fig. 1B and the other too small to be seen in the same gel), whereas the sample incubated in the absence of the C+ sgRNA gave a non-digested (ND) full-length product (Fig. 1B). The GPR143 mutation-specific sgRNAs only mediated a partial digestion of the PCR product (Fig. 1C), with the sgRNA-2 showing a higher digestion compared with sgRNA-1, suggesting an overall lower cleavage efficiency than that of the C+ sgRNA.
To determine the RNP efficiency in cells, we first optimized the nucleofection conditions for the transfection of the patient-derived iPSCs with a GFP marker and observed the highest efficiency, >90% of GFP+ cells, with the DN-100 program, compared with the CA-137 and CB-150 programs that gave ∼65% and ∼40% of GFP+ cells, respectively (Supplementary Fig. S1). Thus, we further optimized the DN-100 program for the different RNP complexes of SpCas9 and sgRNAs. Two days after RNP treatment, we extracted the DNA of the patient-derived iPSCs and evaluated the presence of indels using the Surveyor assay and Sanger sequencing. For the sample treated with the C+ sgRNA, the Surveyor endonuclease digestion of the corresponding PCR product (S+) (Fig. 1D) resulted in an additional band on the agarose gel, suggesting the presence of indels at the SpCas9 cleavage site.
No additional bands were present in the NT cells, nor in C+ sgRNA-treated cells without Surveyor digestion (S-) (Fig. 1D). In contrast, no clear additional bands were present after Surveyor endonuclease digestion for samples treated with sgRNA-1 and sgRNA-2 targeting the patient's GPR143 mutation, suggesting a very low cleavage efficiency (Fig. 1E). To evaluate the percentage of indels in the RNP CRISPR-SpCas9-treated iPSCs, we also used the Sanger sequencing readings to run both the TIDE and ICE analyses. Indels were estimated at ∼96.3% for C+ sgRNA, ∼1.5% for sgRNA-1, and ∼3.25% for sgRNA-2, confirming the trend obtained with the Surveyor assay.
Taken together, these results indicate that we have a solid workflow for in vitro sgRNA screening, CRISPR-Cas transfection, and cleavage efficiency evaluation in patient-derived iPSCs. Nonetheless, the two tested sgRNAs targeting the GPR143 intronic G > A mutation presented a very low efficiency for moving forward with HDR experiments.
AsCas12a's crRNAs are more efficient than SpCas9's sgRNAs in targeting the GPR143 intronic G > A mutation in the OA1 patient-derived iPSCs
Due to the T/A-richness of the targeting sequence, we considered the Cas12a nuclease as an alternative to SpCas9, because it recognizes a TTTV PAM sequence. We first investigated whether AsCas12a or LbCas12a was more efficient in creating indels in patient-derived iPSCs, and we found that AsCas12a, when cotransfected with the Electroporator Enhancer, showed more than two times higher efficiency than LbCas12a (∼88% and ∼40%, respectively, Supplementary Fig. S2). We thus designed four crRNAs targeting the mutated sequence (Fig. 2A) to use in combination with the AsCas12a nuclease.
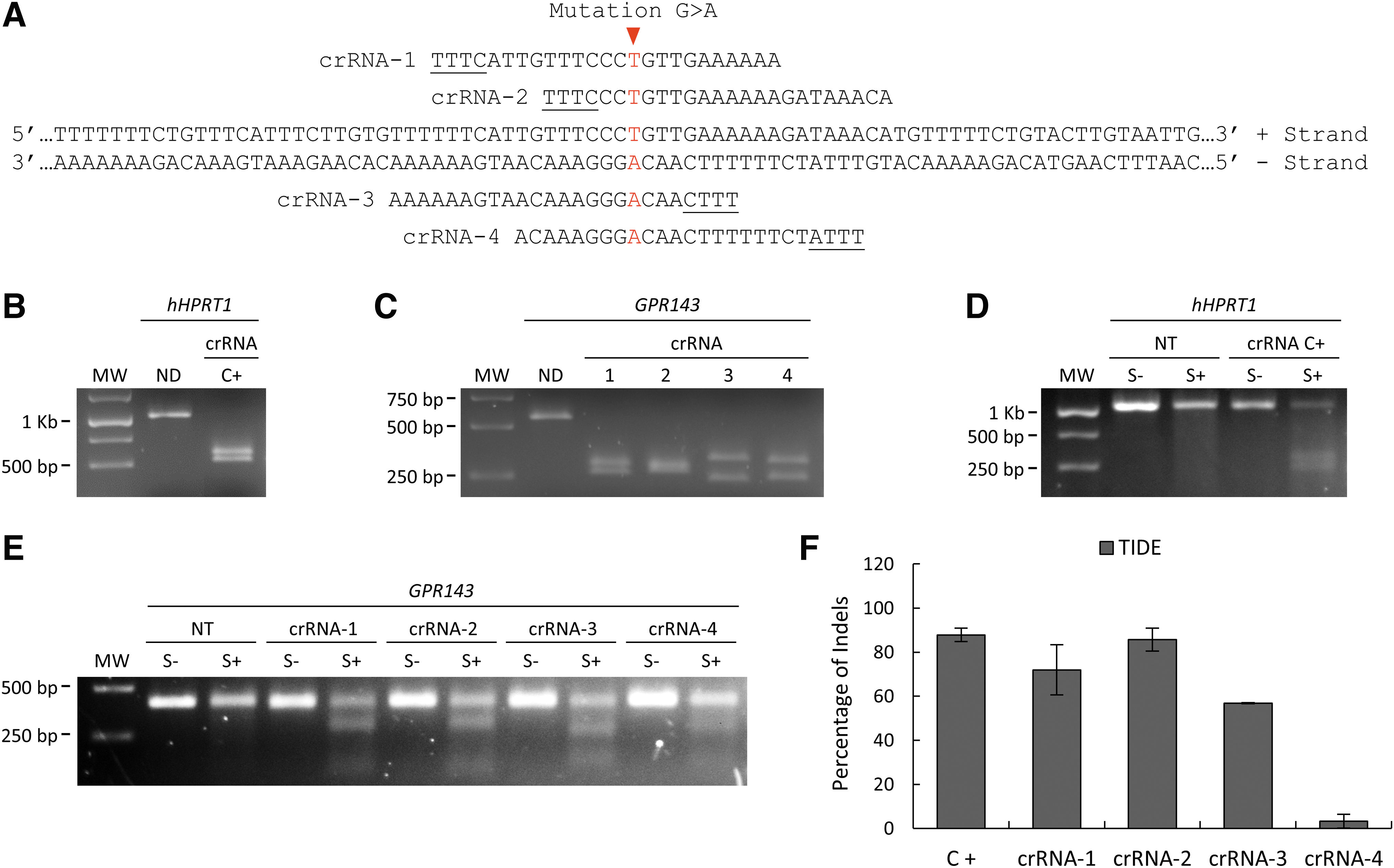
CRISPR-AsCas12a targets and crRNA screening in OA1 patient-derived iPSCs.
In the in vitro digestion assay, C+ crRNA, targeting the hHPRT1 gene, mediated the complete digestion of the PCR product, which resulted in a double band after agarose gel electrophoresis (Fig. 2B). The four GPR143 mutation-specific crRNAs also mediated a complete digestion and resulted in two bands, compared with the ND full-length PCR product (Fig. 2C), suggesting a higher cleavage efficiency than the two previously tested sgRNAs coupled with SpCas9.
The Surveyor endonuclease digestion, performed 48 h after CRISPR-AsCas12a nucleofection of the patient-derived iPSCs, showed additional bands in the agarose gel for the sample treated with the C+ crRNA (S+ in Fig. 2D), as well as for the samples treated with the GPR143 mutation-specific crRNA-1 to -3 (S+ in Fig. 2E). Only fainter additional bands were seen for crRNA-4 (S+ in Fig. 2E) and no additional bands were present for the NT sample or for samples incubated without the Surveyor endonuclease (NT and S- in Fig. 2D, E, respectively). This suggests the presence of indels only in CRISPR-AsCas12a-treated samples. The TIDE analysis, performed with the sequencing reading of the same treated samples, confirmed the trend obtained with the Surveyor assay, and gave an indel percentage of ∼88% for C+ crRNA, ∼72% for crRNA-1, ∼86% for crRNA-2, ∼57% for crRNA-3, and ∼5% for crRNA-4 (Fig. 2F).
Thus, we identified at least three crRNAs, crRNA-1 to -3, that, when coupled with the AsCas12a nuclease, can efficiently cleave the patient's DNA close to the GPR143 intronic mutation.
CRISPR-AsCas12a mediates correction of the GPR143 intronic G > A mutation in the OA1 patient-derived iPSCs and does not result in off-target cleavage
For the design of the donor template, we did not mutate the PAM sequence or add new SNPs for restriction enzyme selection to avoid unpredictable effects on splicing; thus, we only substituted the mutated nucleotide with the correct one. In addition, previous studies had shown that in contrast to SpCas9, 52 when using AsCas12a, the HDR rate is higher if the donor template is complementary to the target strand. 53 Hence, we generated an ssODN that could work with both crRNA-1 and -2, as they target the same DNA strand and showed the highest cleavage efficiency. Thus, our ssODN, complementary to the target strand, carried the correct nucleotide and had 40 nucleotide homology arms from each of the two crRNA cleavage site ends, with a total length of 88 nucleotides (Fig. 3A).
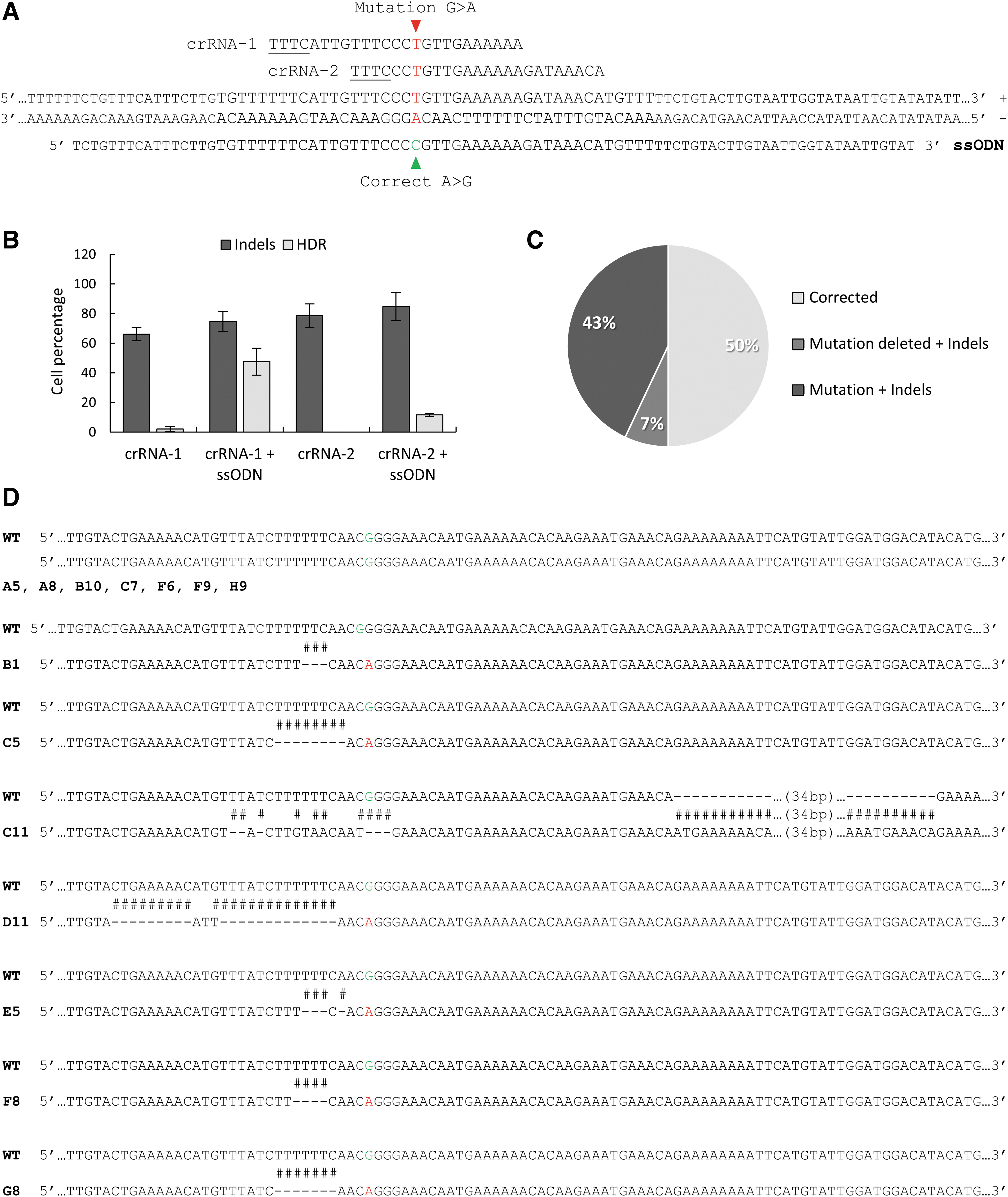
CRISPR-AsCas12a-mediated HDR of the GPR143 intron 7 mutation in OA1 patient-derived iPSCs.
We determined the HDR rate in the patient-derived iPSCs 48 h after CRISPR-AsCas12a ± ssODN nucleofection via the TIDER analysis of the corresponding Sanger sequencing readings. Minimal or no HDR events were observed in samples treated without the ssODN, ∼2% for crRNA-1 and 0% for crRNA-2. However, when the ssODN was present, the HDR rate increased to ∼48% for crRNA-1 and to ∼12% for crRNA-2 (Fig. 3B). Interestingly, even though crRNA-2 presented a relatively higher cleavage efficiency than crRNA-1, ∼80% and ∼70% indel percentage, respectively, the HDR rate obtained with crRNA-1 was four times higher than that with crRNA-2. Thus, we moved forward with the correction experiment using only crRNA-1 coupled with the ssODN.
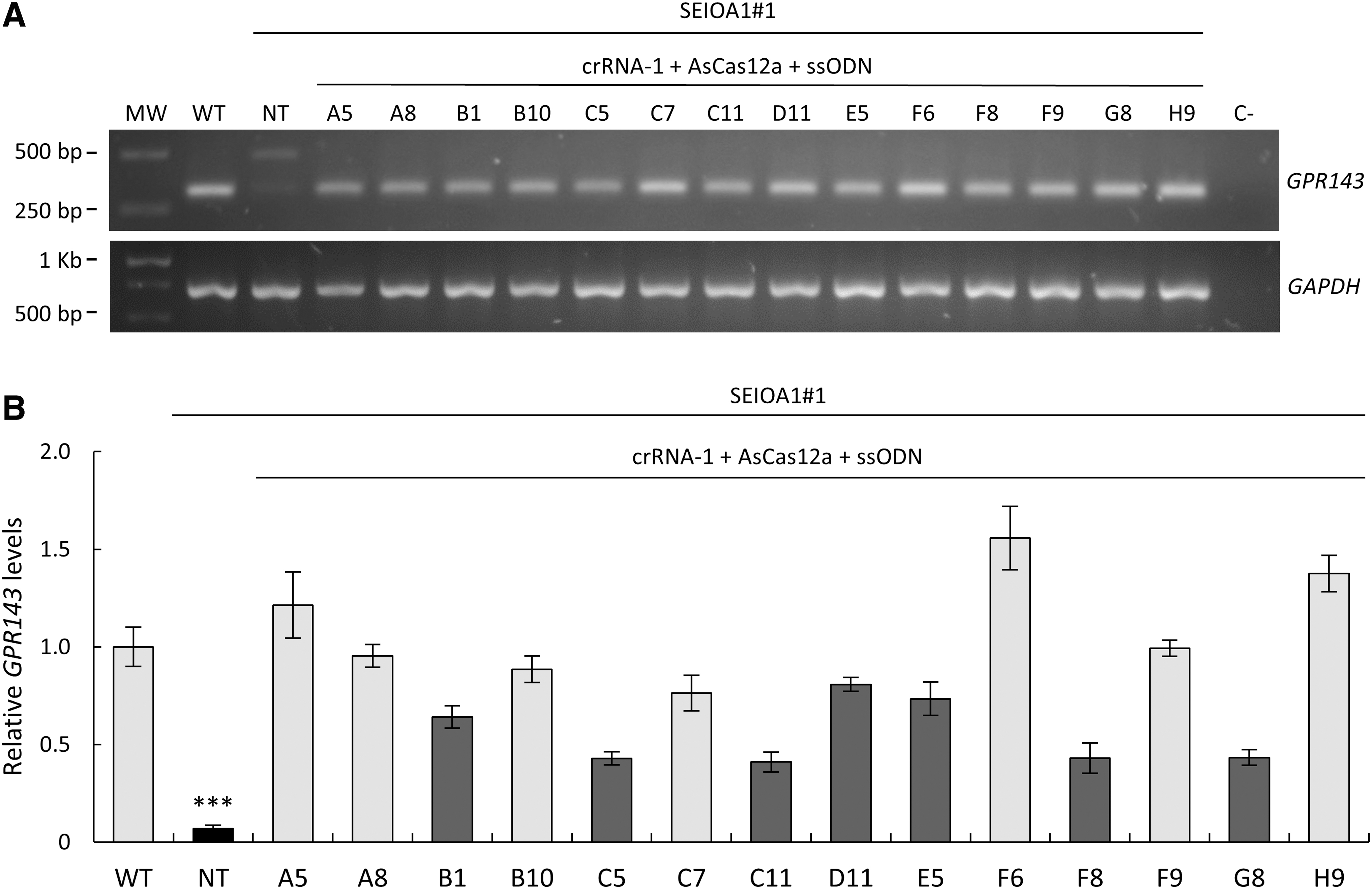
To select genetically identical iPSC colonies originating from a single cell, we harvested crRNA-1 + ssODN-treated cells 48 h postnucleofection and, by fluorescence-activated cell sorting, we seeded one single cell per well of a full 96-well plate. Out of the 96 seeded cells, 14 survived the sorting treatment and generated independent clones. Genotype analysis of the 14 clones revealed that 50% of them, clones A5, A8, B10, C7, F6, F9, and H9, presented a GPR143 intron 7 sequence identical to that of control cells, and thus were fully corrected (Fig. 3C). The patient's mutation was still present in six of the remaining seven clones, B1, C5, D11, E5, F8, and G8, but there were additional indels close to the mutation. Lastly, one clone, C11, had no mutation and a combination of more complex indels. This suggests that the AsCas12a nuclease efficiently cleaved the DNA of the last seven clones, but that the HDR did not occur. The detailed sequences for the 14 clones are shown in Figure 3D.
As the GPR143 mutation creates a cryptic spice site, causing the incorporation of a pseudoexon, we investigated by reverse transcription (RT)-PCR the size of the mRNA using a primer pair amplifying exons 6 to 8 (Fig. 4A). In control cells (wild-type [WT]), only one normal-sized band was present in the agarose gel. In contrast, in NT patient-derived iPSCs, in addition to the faint normal-sized band, a predominant upper band containing the pseudoexon was also present. Surprisingly, only one normal-sized band was present for all the 14 clones isolated post-treatment. Thus, the normal splicing was restored not only in the seven fully corrected clones, but also in the remaining seven clones, suggesting that the indels present around the mutation had efficiently inactivated the alternative splice site.
Effect of the correction of the GPR143 intron 7 mutation in OA1 patient-derived iPSCs on splicing and expression.
To evaluate the GPR143 expression after treatment, we performed a qPCR study (Fig. 4B). All the 14 clones showed a significantly higher GPR143 expression level when compared with the parental NT cells and, for some of the edited clones, GPR143 expression raised to a level similar to the one of control cells (WT).
Finally, we evaluated the potential cleavage of the AsCas12a in the top 10 CRISPOR-predicted off-target sequences, 5 exonic and 5 intronic, listed in Supplementary Table S3. We Sanger sequenced the genomic DNA region surrounding the off-target sites both in patient-derived NT iPSCs and in edited clones, as well as in control cells. We found no differences in any of the sequenced regions, suggesting that AsCas12a cleavage was specific for the GPR143 on-target site.
In conclusion, we successfully corrected the GPR143 intronic G > A mutation in OA1 patient-derived iPSCs and restored the normal mRNA splicing and expression levels, without off-target events.
CRISPR-AsCas12a-corrected OA1 patient-derived iPSC clones are genetically stable and maintain the characteristic stem cell phenotype
To determine whether the corrected patient-derived iPSCs maintained the normal stem cell phenotype as that of the parental non-corrected cells, we examined multiple parameters in two corrected clones, A8 and B10. Both clones conserved the characteristic iPSC morphology, with sharp colony borders and tightly packed cells (Fig. 5A, K). Despite the stressful cell transfection and single-cell sorting procedures, the KaryoStat analysis indicated that no chromosomal alterations were present in the genome of both clones (Fig. 5B, L); the clones were also mycoplasma free (Supplementary Fig. S3). Moreover, immunofluorescence studies showed that the corrected clones maintained the expression of typical pluripotency markers, such as NANOG (Fig. 5C, M), OCT4 (Fig. 5D, N), SOX2 (Fig. 5E, O), and TRA1–81 (Fig. 5F, P).
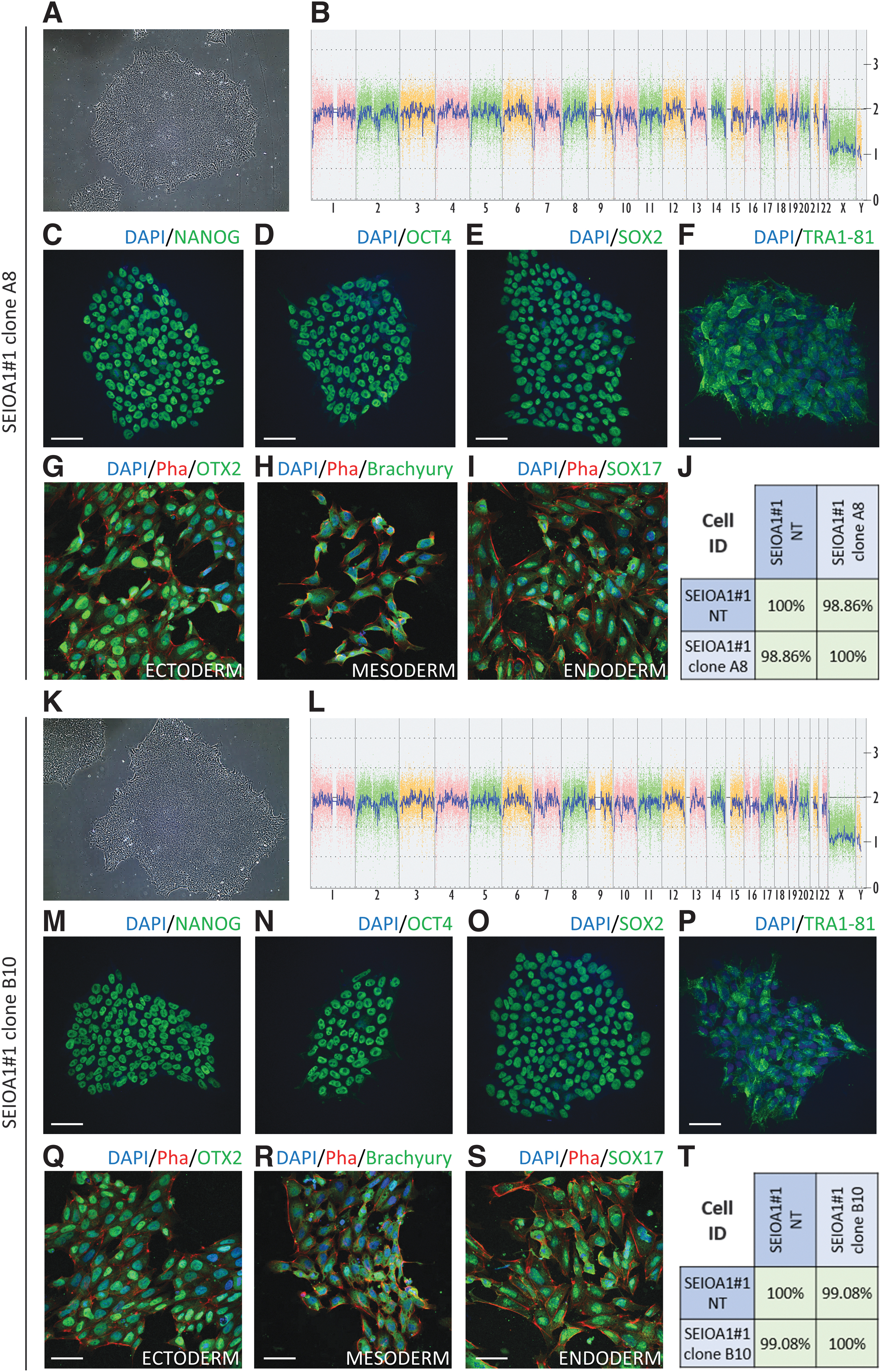
Characteristic stem cell phenotype and genetic stability of the CRISPR-AsCas12a-corrected OA1 iPSC clones A8 and B10. Phase-contrast image of growing colonies for clone A8
In addition, following in vitro differentiation, we confirmed the ability of the A8 and B10 clones to differentiate into the three embryonic germ layers, as shown by immunofluorescence studies that detected the expression of OTX2 for ectoderm (Fig. 5G, Q), BRACHYURY for mesoderm (Fig. 5H, R), and SOX17 for endoderm (Fig. 5I, S). Finally, the Cell ID assay, which analyzed 150k SNPs, indicated that the two tested clones were isogenic lines of the parental non-corrected line (Fig. 5J, T).
In summary, we successfully generated isogenic corrected OA1 patient-derived iPSC clones that are genetically stable and maintain the characteristic stem cell phenotype.
Discussion
Previous technical advances and growing knowledge often lead the way for developing new therapeutic strategies for currently incurable diseases such as OA1. In 2006, Vetrini et al. 18 exploited an AON to mask the same GPR143 intron 7 mutation carried by the SEIOA1#1 line. This AON prevented the recognition of the pathological splice site in patient's melanocytes, avoiding in this way the incorporation of the pseudoexon and premature stop codon formation. However, rescue of GPR143 mRNA levels was only 60% of that of control cells. AON therapy is promising and currently in clinical trials for other diseases 54 (NCT03780257, NCT03140969), but degradation of the AON over time leads to its readministration, estimated at up to four times per year.
As we wanted to achieve a permanent correction of the GPR143 intron 7 mutation, we explored the more recent option of genome editing by using the CRISPR-Cas system to promote HDR, a strategy that has previously been proven feasible by other groups.41,44,55 For its delivery, we opted for an RNP formulation instead of a plasmid-based approach. RNPs are less toxic, ready to act, and not constantly produced in the host cells. These characteristics help to reduce the potential of off-target cleavage of the Cas nuclease,34,38 avoiding the need for Cas inactivation.56,57
We initially designed sgRNAs to use in combination with the most widely studied nuclease, SpCas9. Only two sgRNAs were available in the region close to the patient's mutation and, even though they partially worked in the in vitro digestion assay, they were both dramatically inefficient in mediating the DSB in iPSCs, hindering any concrete chance of HDR. It is known that the sequence of a given sgRNA can drastically influence its activity.58,59 Considering that the nucleofection efficiency was higher than 90% and that the DNA cleavage obtained with the C+ sgRNA targeting the CDC42BPB gene was estimated at ∼96% by ICE/TIDE analyses, we concluded that the problem came from the intrinsic sgRNA-1 and -2 sequences.
Given that the sequence surrounding the mutation is particularly T/A-rich, we used as an alternative the Cas12a/Cpf1 nuclease, which works with the 5′ TTTV PAM. Cas12a is smaller than SpCas9 and works with a shorter gRNA, rendering the complex potentially easier to transfect. Cas12a also produces overhanging ends, which may favor HDR. We first used a C+ crRNA targeting the hHPRT1 gene to compare the LbCas12a and AsCas12a cleavage efficiencies. We found that, when iPSCs were cotransfected with the commercially available Electroporator Enhancer that acts as a specific nuclease carrier, AsCas12a was approximately two times more efficient in creating indels than LbCas12a. Due to this high efficiency, we did not combine the CRISPR-AsCas12a to any fluorescent selection markers.
We then designed and tested four mutation-specific crRNAs for AsCas12a, and showed that all of them mediated a full digestion of the PCR product in the in vitro digestion assay. This assay is a fast and relatively inexpensive technique for a first screening of the selected gRNAs to exclude those that are non-efficient, 60 such as sgRNA-1 and -2 for SpCas9 that only mediated a partial digestion of the PCR product. However, this assay does not guarantee gRNA efficiency in cells, since some of them may still be inefficient, as shown for crRNA-4.
For the evaluation in cells, we observed a discrepancy of the cleavage estimation between the Surveyor assay and the ICE/TIDE analyses. The most striking case is the one of C+ sgRNA for SpCas9, for which a predominant uncut band is seen in the Surveyor assay; whereas the ICE/TIDE analyses estimated an efficiency of ∼96% (Fig. 1D, F, respectively). The Surveyor nuclease has low sensitivity for single-base loops and, as consequence, has low efficiency in detecting single-base indels. 61 Interestingly, the ICE/TIDE analyses estimated that, for the sample treated with C+ sgRNA and SpCas9, ∼73% of the treated cells presented a single-base pair indel, which would explain why the non-cleaved band in the Surveyor assay was the predominant one. Moreover, it has been recently shown that the ICE/TIDE analyses provide similar editing profiles compared with next-generation sequencing (NGS) results, with respect to size and frequency of indels. 61 Thus, because the ICE/TIDE analyses are less expensive and time-consuming than NGS sequencing, as well as more sensitive than the Surveyor assay, we concluded that they are a particular valuable tool for indel estimation and thus for choosing the most efficient gRNAs.
For the design of the donor ssODN, we followed previous reports showing that the use of the sequence corresponding to the non-target strand increases the HDR when combined with AsCas12a, 53 in contrast to the SpCas9 preference for the target strand sequence. 52 As the synthesis of shorter molecules is more accurate, we opted for homology arms as short as ∼40 bp, which proved to be efficient. 53 However, as we wanted to test the same ssODN with both crRNA-1 and -2, we designed a slightly asymmetric 88-bp ssODN. Chemical modifications were also added to increase stability. 62 Interestingly, even if crRNA-2 was more efficient at creating indels than crRNA-1, the latter mediated the highest HDR rate. The patient's mutation corresponds to position 11 of crRNA-1 and to position 3 of crRNA-2. This correlates with the observation that nucleotides located in positions 8 to 16 of the crRNA are easier to edit.
Thus, similar to SpCas9, 63 the position of the target mutation in relation to the gRNA sequence seems to be a crucial factor to consider when planning HDR experiments with AsCas12a. Lastly, we did not add any predicted silent mutation in the ssODN for further selection, because we did not want to take the risk of introducing unexpected alterations in the edited intron. This choice was also justified by the very high transfection efficiency and the promising HDR rate that we had obtained.
For the selection of clones post-CRISPR-AsCas12a+ssODN treatment, we chose the single-cell sorting approach rather than clonal dilution, to avoid the excessive use of culture medium and to reduce the chances of chimeric populations. The survival rate post-sorting was only ∼15%, but this could probably be increased by testing different combinations of dissociating reagents and matrices. 64 Despite this low survival, 7 out of 14 isolated clones were fully corrected, with a final HDR rate of 50%, very similar to the TIDER-estimated ∼48%. The rest of the clones presented a heterogeneous panel of indels very close to the mutation and one clone had the mutation itself deleted. Interestingly, when we investigated the splicing pattern of all the isolated clones by RT-PCR, we found a complete rescue of the splicing defect in all of them, meaning that the indels were sufficient to disrupt the pathological splice site.
This observation has a high impact for therapeutic purposes, as some intronic mutations may be rescued without the necessity of HDR, both in vitro and in vivo. 21 When we looked at the GPR143 expression by qPCR, all the clones showed significantly higher expression than the parental NT patient's cells. For some clones, the level of GPR143 expression was similar to that of control cells. This correlates with the RT-PCR result, in which we showed no pseudoexon insertion and, as a consequence, no premature stop codon and no mRNA nonsense-mediated decay.
However, a heterogeneous pattern of expression was observed between the edited iPSC clones. This could be due to a slight clone-to-clone variability 65 or to the fact that the iPSCs needed more recovery time and a few more passages after the CRISPR-Cas treatment to stabilize. It is also possible that the GPR143 level would be less fluctuating in pigmented cell types expressing the GPR143 protein at a higher level than iPSCs, such as melanocytes and retinal pigment epithelial cells. Nonetheless, the CRISPR-AsCas12a-mediated correction did not alter iPSC pluripotency or genetic stability, despite stressful cell treatments such as nucleofection and single-cell sorting. Lastly, no off-target events were observed in the top 10 predicted loci, confirming the high specificity of the AsCas12a.66,67 However, whole-genome sequencing will be needed to confirm the safety of the approach for translation purposes.
In 2017, Zhang et al. 21 demonstrated that As/LbCas12a can be successfully used to correct mutations causing Duchenne muscular dystrophy (DMD) in patient-derived iPSCs by introducing small indels or deleting a premature stop codon. Moreover, Ma et al. showed in 2018 that LbCas12a was also able to mediate knock-in editing in human stem cells. 68 To the best of our knowledge, our study reports for the first time the use of AsCas12a RNPs to correct a mutation causing a human disease, OA1, via HDR. Thus, our results strongly highlight the translational potential of the AsCas12a nuclease to target other genetic diseases that could benefit from HDR correction.
In summary, the goal of our study was to correct a specific GPR143 intron 7 mutation that causes OA1 in patient-derived iPSCs. We achieved this correction using the CRISPR-AsCas12a system, which efficiently increased the HDR rate. The generation of the isogenic OA1 patient-corrected iPSC lines holds different potentials. First, the differentiation of iPSCs into the RPE will generate a more pertinent human model for the study of OA1 pathophysiology in vitro. Second, the co-culture of iPSC-derived RPE and retinal organoids69,70 could also shed light on how the GPR143-defective RPE influences other retinal layers. 71 Third, the rescue of GPR143 may be beneficial for neurite sprouting of ganglion cells, probably via extracellular vesicle communication.72,73 If this were the case, the autologous transplantation of patient-corrected iPSC-derived RPE 74 (NCT04339764) would be an attractive therapeutic option for OA1 patients, as some synaptic plasticity may still be present after birth. 75
Lastly, the in vivo correction of specific mutations in patient's RPE is now becoming a concrete possibility, 76 as demonstrated by clinical trials for other diseases based on CRISPR-Cas gene editing (NCT03872479, NCT04601051). Moreover, since the constitutive expression of the Cas nuclease still raises safety concerns, the in vivo delivery of CRISPR-Cas RNPs could overcome the issue. 77
Conclusion
In our proof-of-concept study, we demonstrated the feasibility of correcting a splicing mutation in the GPR143 intron 7 by delivering the CRISPR-AsCas12a system into OA1 patient-derived iPSCs. We showed that, when efficient sgRNAs for SpCas9 are not available for targeting a specific genomic region, especially if T/A-enriched, AsCas12a is a promising alternative and it is compatible with HDR experiments. We also highlighted that the very high efficiency achieved in delivering the CRISPR-Cas RNPs to iPSCs, the choice of appropriate gRNAs, and an optimized design of the ssODN all lead to excellent HDR rates. Interestingly, we observed that when the HDR did not occur, the indels generated by NHEJ were sufficient for disrupting the pathological splice site, rescuing the normal splicing and GPR143 expression. Our correction strategy holds the potential of generating a pertinent human cellular model for ocular albinism and for future in vivo therapeutic approaches.
Footnotes
Acknowledgments
A special thanks to Carla Sanjurjo-Soriano for her advice on the methods that we used and for helpful discussions, and to Vasiliki Kalatzis for critical reading of the article. Financial support from The Vision of Children Foundation is gratefully acknowledged.
Authors' Contributions
S.T., E.B., and D.B.F. designed the study. A.G.D. and B.C. provided technical advice. S.T. performed the experiments and analyzed the data. S.T. and D.B.F. wrote the article. E.B. and D.B.F. obtained funding support. Finally, all authors approved the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Funder name: The Vision of Children Foundation.
Associated grant no.: 441482-DF-81002.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.