
Abstract
Base editors mediate the targeted conversion of single nucleobases in a therapeutically relevant manner. Herein, we present a hypothetical taxonomic and phylogenetic framework for the classification of more than 200 different DNA base editors, and we categorize them based on their described properties. Following evaluation of their in situ activity windows, which were derived by cataloguing their activity in published literature, organization is done hierarchically, with specific base editor signatures being subcategorized according to their on-target activity or nonspecific, genome- or transcriptome-wide activity. Based on this categorization, we curate a phylogenetic framework, based on protein homology alignment, and describe a taxonomic structure that clusters base editor variants on their target chemistry, endonuclease component, identity of their deaminase component, and their described properties into discrete taxa. Thus, we establish a hypothetical taxonomic structure that can describe and organize current and potentially future base editing variants into clearly defined groups that are defined by their characteristics. Finally, we summarize our findings into a navigable database (ShinyApp in R) that allows users to select through our repository to nominate ideal base editor candidates as a starting point for further testing in their specific application.
Introduction
Genome engineering entails the curated manipulation of nucleic acids using bespoke, directed chemistry. Modular gene editing platforms with programmable DNA-binding elements such as CRISPR-Cas, 1 zinc finger nucleases,2,3 and transcription activator-like effectors4,5 mediate the calculated editing of the genome by exploiting host-dependent DNA repair pathways.6–10 Their design, rationale, and development have been principally focused upon minimizing unintended off-target gene editing events while improving on-target product purity, thereby enabling precision medicine.
In this context, we define on-target activity as a measure of the overall gene editing efficiency of a given tool specifically at a user-defined loci, whereas off-target effects are defined as undesired editing outcomes outside of their intended target scope.
Canonical CRISPR-Cas base editors are gene editors that facilitate a site-specific chemical conversion of nucleobases via hydrolytic deamination.11,12 Through extensive protein engineering and evolutionary campaigns, classical base editors have diversified beyond the pioneering cytosine-to-thymine targeting effectors (cytosine base editors [CBEs]) to encompass adenine-to-guanine (adenine base editors [ABEs]), 13 and, later, cytosine-to-guanine (cytosine-to-guanine base editors [CGBEs])14,15 and RNA-modulating variants.16,17
As such, the broad gamut of available base editors can facilitate therapeutically relevant correction of a significant number of tractable, monogenic single-nucleotide polymorphisms (SNPs).18,19 This has sparked an intense interest to enhance their efficacy further, expand their targetable scope, and improve their overall biosafety by focusing on aspects of their architecture and molecular signatures in the cell. 20
At the beginning of 2016, only three significantly different variations on the base editor were described.11,12,21 Briefly, the prototypic base editor architecture comprised a deaminase component that mediated cytosine-to-thymine transitions, and a nickase Cas9 (D10A) component that enabled site-specific localization and induced host-mediated base excision repair pathways. Variations to the base editor architecture was based on the linker type, arrangement (N- or C- termini), and length adjoining the cytosine deaminase and Cas endonuclease components, as well as the presence and positioning of an accessory uracil-DNA-glycosylase inhibitor.
By 2017, the development of adenosine-targeting variants16,22 would increase the number of base editors by 29, with a further 61 editors being described over the next 2 years. They included architectural adjustment 23 (Fig. 1) and codon optimization for improved on-target editing efficiencies, 24 engineered ablations to aberrant RNA-targeting domains,25–27 as well as overhauling domain rearrangements and directed evolutionary campaigns for broadened editing scope 28 and motif recognition (Fig. 1).28,29
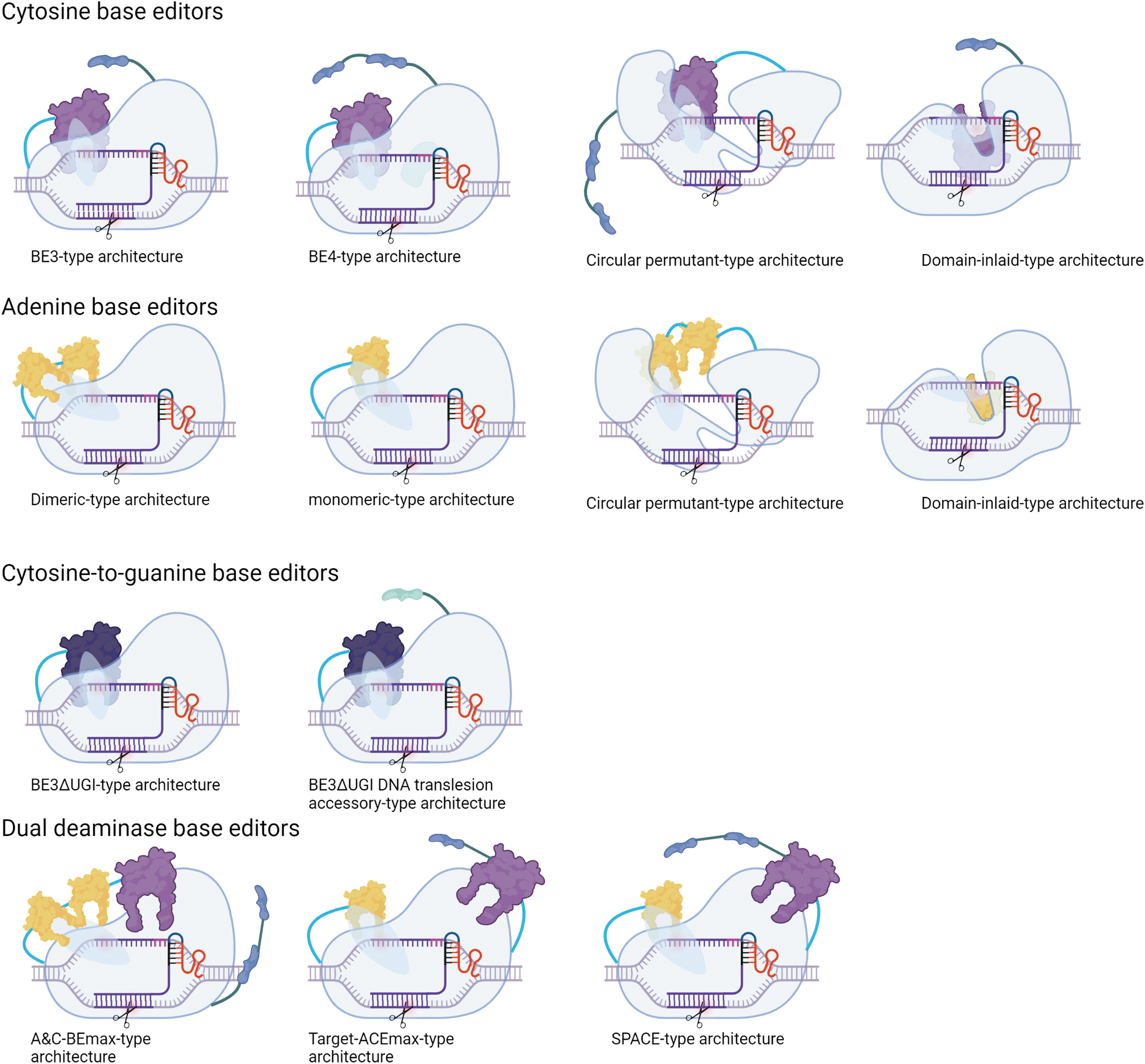
Architectural arrangements of current DNA base editors. DNA base editors broadly exist as three general subclasses: cytosine, adenine, and cytosine-to-guanine acting variants. For cytosine base editors (CBE), most CBEs are typified by a BE3-like architecture in which they have a single N-terminal, cytosine deaminase domain (purple), and usually a uracil glycosylase inhibitor (UGI; navy) adjoined by intervening protein linkers of varying length (cyan and teal). The BE4 architecture is characterized by a dimeric UGI component and is sometimes inclusive of an additional accessory domain embedded in the linkage between the deaminase component and the Cas protein (such as RAD51; light green). Circular permutant variants have their N- and C-terminal domain rearrangements represented by the intervening linker separating the two domains, whereas domain-inlaid variants are distinguished by the embedded portion of the deaminase inside the Cas protein. For adenine base editors (ABEs), their architectures are composed of either a monomeric or dimeric deaminase domain (yellow). As with CBEs, ABEs can also be circularly permuted or formatted as domain-inlaid editors. Cytosine-to-guanine base editors (CGBEs) are BE3-like in structure, but lack the UGI domain, which is replaced by DNA translesion-generating accessories such as uracil DNA glycosylase or other factors (aquamarine). For dual deaminases, there are three general arrangements of the ABE and CBE components, wherein both the ABE and CBE can be at the N-terminus of the Cas protein, or the ABE could be placed at the N-terminus with the CBE component placed at the C-terminus, as well as a general arrangement in which there may be one or two covalently linked UGIs. In general, the Cas component is shown as a prototypic Cas9 variant in which only the HNH domain remains active (represented by scissors targeting the complementary DNA strand).
By 2021, the number of DNA base editors alone would balloon to well in excess of 200 (Fig. 2). Accordingly, meaningful head-to-head characterization of each variant has been stymied by the increasingly complex nature of their profiling. On-target editing efficiencies, activity windows, off-target RNA deamination profiles, 30 off-target guide-dependent 31 and guide-independent 32 DNA deamination profiles, as well as cryptic sequence-specific editing events 33 are significant factors that must be considered. The field is nevertheless guided by the overarching principle of developing a “model” base editor toward clinical translation, and although broad consensus is unlikely owing to the Pareto frontier, a more generalizable and simplified approach toward base editor choice is needed.
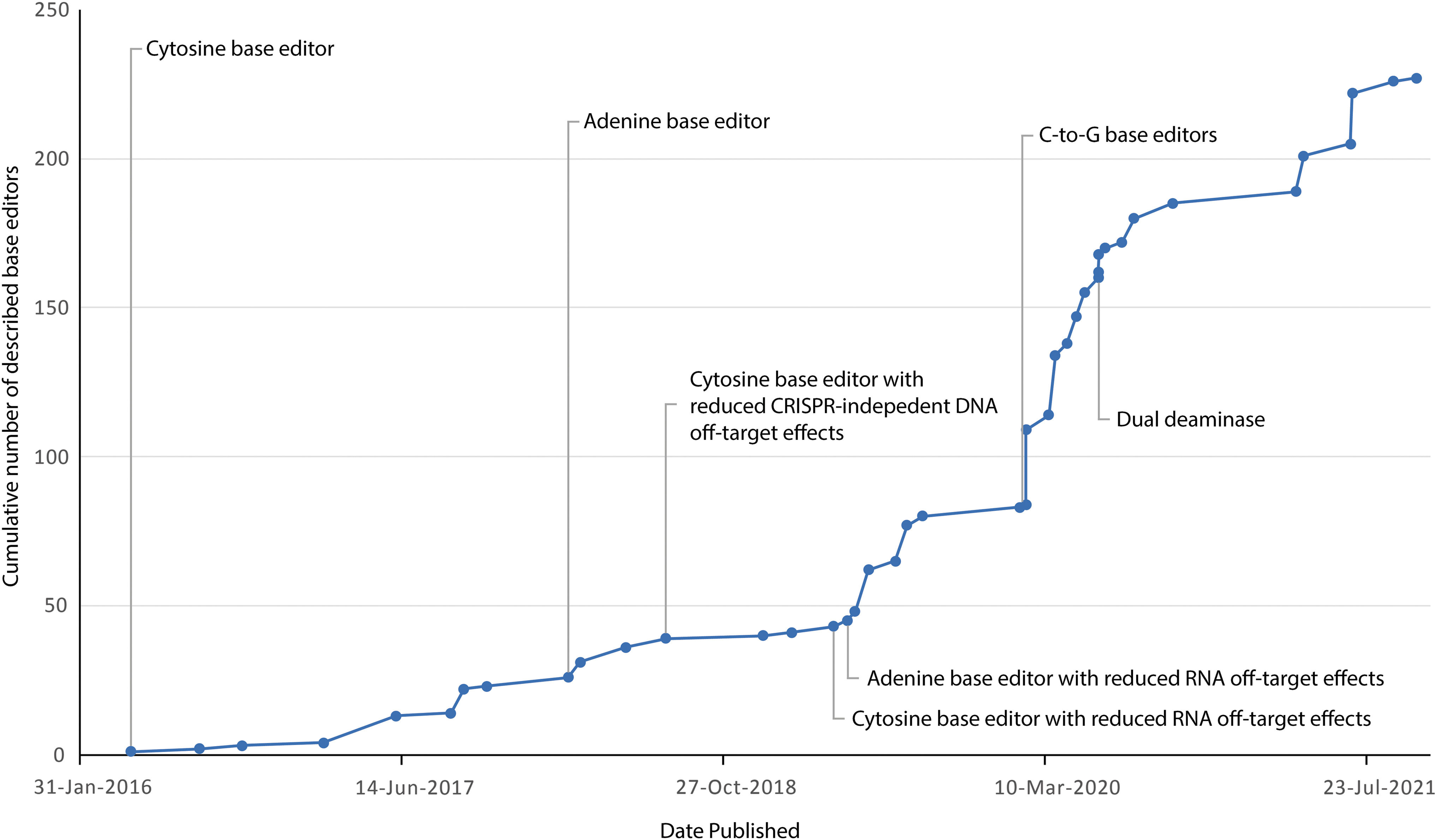
Development of base editors over time. Major milestones in the engineering of DNA base editors are highlighted.
The rich diversity of currently cataloged base editors shows distinguishable but ultimately related variation in their described properties. Thus, a proposed phylogenetic classification scheme for base editors attempts to describe and organize these nuanced variations within the context of end use-case nomination and application. We sought to establish an overarching framework that captures this variation for both current and future base editors. While delineating and partitioning variants into discrete taxa potentially obviates consensus on the model base editor, it is worth noting that currently, a variant that unambiguously outperforms all other variants in metrics for on-target scope and fidelity is lacking.
Even then, there would naturally arise competing variants that present the user with a paradox of choice, thereby requiring some organizational basis to establish either that both hypothetical base editors achieve sufficiently excellent outcomes or that one is differentiated from the other. Thus, we believe that this framework functions to organize the current assortment of base editors into broad themes, as described by the type of chemistry that they facilitate, or their targeting scope, as determined by their endonuclease or deaminase component, or their fidelity. An end-user could benefit by having a thematic structure for base editor nomination, depending on the specificity of their question for their end application.
Phylogenetic clustering in this case serves to highlight, document, and provide a description of the relationships between variants that an experienced base editor user would otherwise be intrinsically aware of but that would not be apparent to a more novice user as the field further matures and clear entry points into the field become more ill-defined. Therefore, given the number of candidates, we believed that an interactive ShinyApp that could aid with base editor choice based on this principle of taxonomic distinction would serve as a useful addendum to the wider base editing community.
Methods
Determination of the on-target activity estimation for each base editor variant
Specifically, the editing efficiency of a base editor variant in the BEERS ShinyApp (Supplementary Table S1) is only listed if preliminary testing for the editor variant was performed in a HEK293 cell line with comparable transfection efficiencies. In general, most studies provide a composite value for editing efficiency, which was subsequently recorded. The “composite” value reflects the average editing efficiency across multiple endogenous testing sites for a particular study. If there were multiple composite values from two different studies, then we averaged the two composite values to achieve the final editing efficiency.
In the absence of a composite value, we first searched for the editing efficiency for the following two positive control sites: HEKsite3 and HEKsite4. The editing efficiencies for these sites were recorded and averaged. Variants that did not show testing on both of these sites but showed testing on at least one of these sites had the following sites considered: RNF2, HEKsite2, FANCF. Specifically for ABEs, we considered the editing efficiencies for site 3 and site 4. In certain cases, only one of these sites was used in a particular study, and so either site 3 or site 4, in addition to the site with the highest editing efficiency, was averaged and recorded.
For protospacer adjacent motif (PAM)-relaxed variants, we used the composite value recorded for their indicated PAM requirement (e.g., the composite value of NGN PAMs was used for NGN variants) or the editing efficiency recorded for the appropriate site containing the appropriate PAM requirement.
Determination of the on-target activity window for base editor variants
For each base editor variant, the highest reported on-target activity (or ceiling value) from its initial publication was recorded (see Supplementary Table S2). Where possible, other references related to the use of that particular base editor variant in HEK293T cells were recorded by performing a PubMed search (input term corresponds to the base editor column in Supplementary Table S2). The activity window for all described base editors were compiled from the listed PMIDs. A grading scale of 0–3 was used. This allowed us to observe the general “shape” and breadth of the activity window.
The ceiling value, which is represented as a percentage derived from published deep-sequencing data, is divided evenly by three, with a value of 1 used where the base editing efficiency fell within lower 33rd percentile, a value of 2 used if the base editing efficiency occurred between the 33rd and 66th percentiles, and a value of 3 used if the editing efficiency fell between the 66th and 100th percentiles of the ceiling value. The ceiling value was only considered for the determination of the activity window and not for the editing efficiency recorded in Supplementary Table S1.
Additionally, it was not used for the ranked nomination of base editors in the ShinyApp, which uses partially normalized values against HEK2 or HEK3, or a combination. In general, the initial publication describing the base editor was usually sufficient to determine the editing window. However, we note that earlier publications may report values for the 3rd, 5th, 7th, and 9th positions, rather than all values in between. If data on a given position were missing, we attempted to search the literature to track down information on that given position, or we assumed that the editing window shape would be parabolic in nature.
Determining the ClinVar correcting scope for base editor variants
PAM counts corresponding to the number of ClinVar targetable SNPs 34 were specified based on the in situ activity window determined above.
SpCas9 variants were specified by an NGGN PAM, xCas9 variants specified by an NGDC PAM, 35 NG-Cas9 variants specified by an NGN PAM,36,37 SpG-Cas9 variants specified by NGN PAM, SpRY-Cas9 variants specified by NRN PAM, SaCas9 variants specified by NNGRRT PAM, SaCas9-KKH variants specified by an NNNRRT PAM, LbCas12a variants specified by a TTTV PAM, 38 NRCH-Cas9 variants specified by an NRSH PAM, NRRH-Cas9 variants specified by an NRRH PAM, NRTH-Cas9 variants specified by NRKN PAM, 39 VQR-Cas9/VRQR-Cas9 variants specified by a NGVG PAM, VRER-Cas9 variants specified by an NGSG PAM, AsCas12a variants specified by an NTTV PAM, 40 and EQR-Cas9 variants specified by an NGAG PAM.
The PAMside field (location of PAM relative to protospacer) was specified as 3′ for all variants except AsCas12a and LbCas12a, whereas the Spacer_len was specified as 20. Edit_window_start and Edit_window_end were specified by all positions within a given protospacer for a particular variant that had a value of ≥2 from our characterization of their on-target editing windows. For ABEs, Edit_from and Edit_to were specified as A and G, respectively, whereas for CBEs, Edit_from and Edit_to were specified as C and T, respectively. The number of available PAMs corresponding to each base editor variant was then summed from the summary file output and compiled (Supplementary Tables S1 and S4).
Categorization of ABE and CBE variants as standard window, wide window, and narrow window
Anzalone et al. 41 describe a useful metric for determining the window breadth and categorization of conventional DNA base editor by formally attributing standard-window ABEs and CBEs as having an in situ targeting range of four nucleotides (ABEs; i.e., positions 4–7) and five nucleotides (CBEs; i.e., positions 4–8), respectively. ABE and CBE variants were categorized by taking the absolute value from subtracting the Edit_window_end field (+1 to account for the inclusion of 0) from the Edit_window_start field (Supplementary Table S1).
Therefore, wide-window ABEs are categorized when the in situ targeting scope is a value of +7, and narrow-window ABEs have a targeting scope value of ≤2. For CBEs, wide-window is defined by a targeting scope value of +8, whereas narrow-window CBEs are defined by one that is ≤3. The editors that are characterized by values in between wide and narrow windows are defined as standard-window editors.
Categorization of ABE and CBE variants into other subcategories such as reduced DNA off-target, reduced RNA off-target, increased product purity, and increased on-target activity
Base editors were categorized as having reduced DNA off-target or reduced RNA off-target profiles based on their initial publication, which was supported by a head-to-head comparison with an unengineered or unevolved variant in the published description. For the 29 CBE variants that had confirmed increased on-target activity, categorization was based on the claims made in their initial description and also by comparing their on-target performance against their wild-type variants (BE3, BE4).
Variants that showed an overall statistically significant difference in editing efficiency in their publications (see listed PMID for each variant) compared to their wild-type counterparts were placed in this category. For the 24 ABE variants that had confirmed increased on-target activity, categorization was similarly carried out as above. Similarly, for determining the increased product purity of a variant, categorization was based entirely on the claims made in either their initial or subsequent publications, as well as a manual inspection of deep-sequencing data, which specifically looked at on-target product purities. No statistical analyses were performed outside of the initial published material describing the base editor.
Calculation metric for determining the optimal CGBE for positions 5 and 6 of a given protospacer in CRISPR-BEERS
BEERS generates a cytosine-to-guanine editing metric by multiplying the average product purity, as a percentile, by the average product yield across the HEK2, EMX1, FANCF, and RNF2 loci for position 6, and the HEK3, HEK4, and EMX1 loci for position 5 for current CGBE variants (with the exception of ABE-P48R) from supplementary figure 13 of Koblan et al.'s work (Supplementary Table S3). 42 For the inclusion of ABE-P48R, only position 6 was considered. Similarly, as described, the average product purity for cytosine-to-guanine transversions, as a percentile of the average product yield, was considered from the FANCF, RNF2, ABLM3, CSRNP3, RHPN2, BRME1, and LOC101927151 loci.
Generation of phylogenetic tree for DNA base editor variants
Multiple sequences of 222 base editors were aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) with the default settings. The phylogenetic tree was inferred from 222 base editor sequences with 4,515 aligned positions. The tree was inferred by maximum likelihood (ML) with IQTREE v2.2.3,43 using a JTT protein substitution model, 44 and visualized using iTOL (https://itol.embl.de). Branch support of the phylogenetic groups was computed using nonparametric bootstrap methods, namely ultrafast bootstrap approximation approach (UFboot)43,45 and Shimodaira-Hasegawa approximate likelihood ratio test (SH-aLRT) 46 with 10,000 replicates. For each bootstrap method, the inferred branch support values were mapped onto the ML tree reconstructed by IQTREE.
Results and Discussion
A taxonomic criterion for broadly classifying diverse base editors
Herein, we describe a hypothetical taxonomic and phylogenetic classification system for currently described DNA base editors to group them based on similar characteristics for the prediction of unstudied behaviors. This classification system shares terminology similar to the taxonomic nomenclature used for CRISPR-Cas systems. 47
Broadly, we define current DNA base editors on the following taxonomic hierarchy from broad to niche: class, representing the target chemistry or predominant point mutation induced at the nucleotide substrate (cytosine-to-thymine, adenine-to-guanine, cytosine-to-guanine, or both cytosine-to-thymine and adenine-to-guanine in the case of dual deaminases); type, which refers to the trafficking partner or Cas-effector variant for the base editor; subtype, which describes their targeting constraints, such as their PAM-requirement (relaxed PAM requirement or strict PAM requirement); and variant, which collectively groups base editors based on the identity of their deaminase component.
As a further addendum, subvariant refers to base editor–specific traits. Here, traits can refer to the breadth of their on-target editing scope (in situ activity window), their genome- and transcriptome-wide fidelity, as well as the product yield and purity of the edits (Fig. 3).
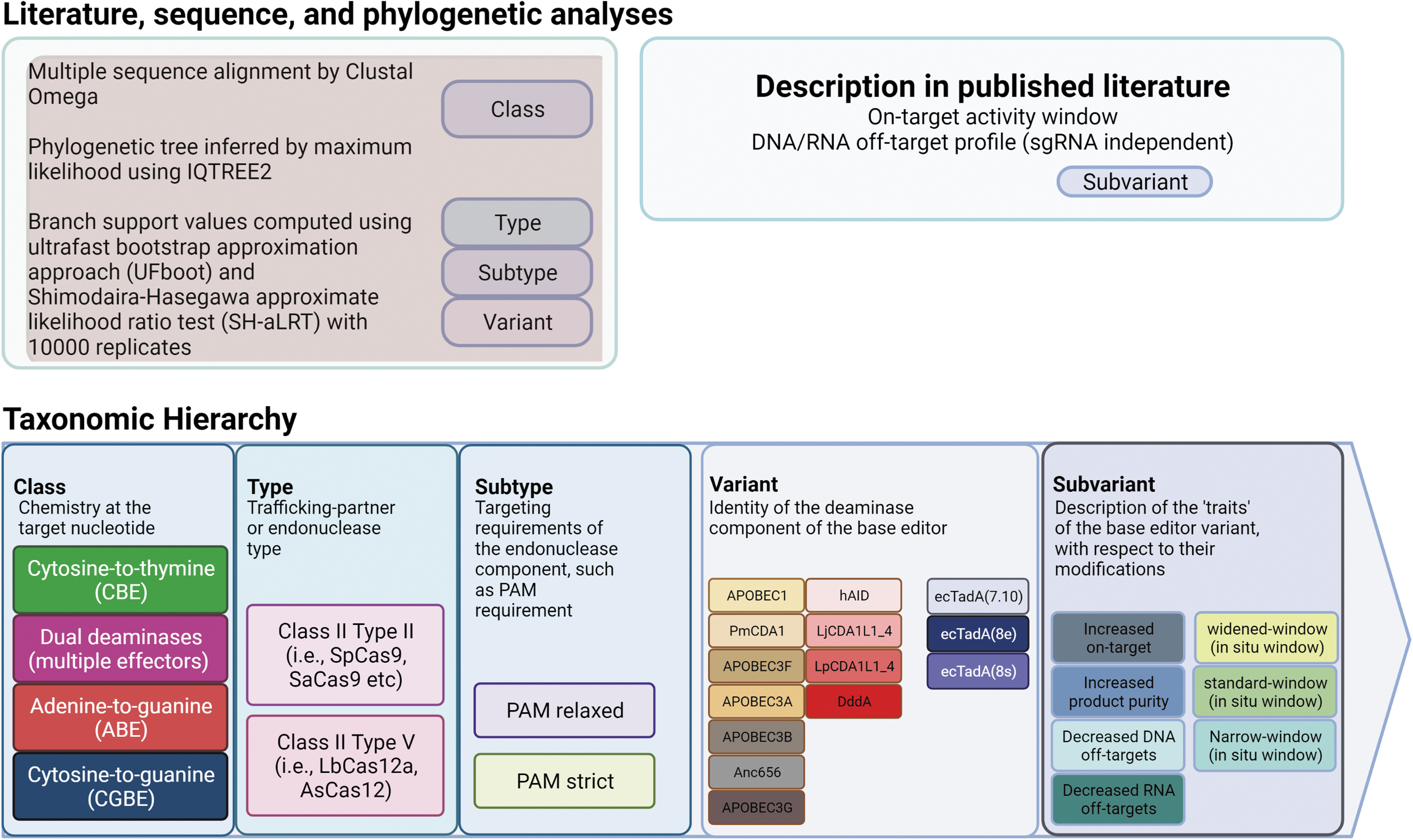
Taxonomic hierarchy for the classification of base editors. Hierarchical organization of variant DNA base editors was based predominantly on preliminary phylogenetic analyses of 222 DNA base editors and description in the literature. Briefly, the nucleotide conversion chemistry at the target substrate forms the basis for taxonomic distinction, with currently four unique classes described (CBEs, dual deaminases, ABEs, and CGBEs). The type of trafficking partner required for localization of the base editor refers to the variant of Cas effector, which follows typical CRISPR-related nomenclature to denote Cas9- or Cas12-type effectors. As a variety of engineering modifications have been made to relax the protospacer adjacent motif (PAM) requirements of certain Cas effectors, the subtype denomination is broadly defined into two categories: PAM strict and PAM relaxed. Next, the base editors are partitioned based on the variant of their unique deaminase component, which can be derived from different host organisms or be synthetically evolved. The subvariant’ denomination refers to their described traits, which can consist of base editor variants that have been engineered for increased on-target activity, increased product purity, reduced RNA off-target effects, reduced DNA off-target effects, wide window, or narrow window.
Establishing a phylogenetic tree as a basis for hierarchical taxonomy
Phylogenetic analysis (Supplementary Fig. S1) guided the development of our classification hierarchy. Briefly, a consensus maximum-likelihood phylogenetic tree was inferred using IQTREE2. 43 Branch support of the phylogenetic tree was assessed using the nonparametric bootstrap approaches UFBoot 45 and SH-aLRT. 46 Here, branch support is defined as the statistical confidence that the model has for clustering a group of base editors that appear phylogenetically related and, in this case, functionally similar.
The inbuilt UFBoot stopping criteria determined a consensus bootstrap tree after 1,700 iterations, reflected by a Pearson's correlation coefficient of 0.991, as determined by the convergence of split support values (Supplementary Fig. S2). The convergence criterion of Pearson's correlation coefficient of the split support values close to 1, (i.e., >0.99) was not met with 1,000 replicates. A generally accepted threshold of 1,000 bootstrap replicates48–50 was not sufficient to achieve bootstrap support convergence.
One possible reason for this might be due to many gap characters in the multiple sequence alignment of large (n = 222) and diverse DNA base editors. 51 Nonetheless, we obtained stable bootstrap estimates at 1,700 iterations, indicating that the resulting split supports from 1,700 iterations did meet the convergence criterion.
For ABEs, we observe that there is now good branch support for the internal nodes comprising domain inlaid base editors (IBEs) 52 such as IBE23, IBE13, IBE12, IBE11, IBE16, IBE10, IBE02, and IBE15 (Supplementary Fig. S3A). IBEs are a group of base editor variants whereby the deaminase component of the base editor is inserted at a region other than the N- or C- termini of the Cas endonuclease component.
In this context, current generation IBEs were described by Chu et al., 52 and hence represent a family of related variants, despite variation to their domain placement. The strong bootstrap support underpinning this clade suggests a common evolutionary tree, which was consistent with their development as a set of tools. 52 Similarly, we observe that there is branch support for ABE8s variants, with ABE8.8 to ABE8.20 occurring within the same clade (Supplementary Fig. S3B).
Here, the clustering for these internal nodes reflects that there are mutually nonexclusive mutations occurring within the deaminase portion of these variants, and this provides us with a strong basis for taxonomic classification as shared variants. The monomeric TadA or miniABEmax variants also share a supported node and correctly have the shared characteristic of a reduced RNA off-target profile (Supplementary Fig. S3C). Interestingly, the ABE8e leaflet did not share branch support with its ABE8e-dimer counterpart. Instead, it was separately clustered among its monomeric permutations, although we note that branch support was also lacking for this clade.
For CBEs, phylogenetic analyses show appropriate branch support between variants such as VQR-BE3 and VRER-BE3, or SECURE-BE3 variants and their related DNA-fidelity altering mutations (Supplementary Fig. S3D). Here, we believe that the clustering of these internal nodes logically captures their common background, which were originally described within the same publication.
Similar to ABEs, CBE variants of IBEs were also branch supported at their internal nodes, with branch support also evident for evolved and their nonevolved counterparts such as FERNY-BE4max and evoFERNY-BE4max. Strong branch support was also obvious for next-generation CBEs such as BE4-AmAPOBEC1 and their related variants, with multiple internal nodes within this clade mirroring their occupation of related sequence space (Supplementary Fig. S3E). 53
While good separation was evident between CBEs, ABEs, and CGBEs, we observed distinct branching for the type of endonuclease variant as well. It was apparent, however, that the partitioning of base editor variants based on their endonuclease component was disproportionately weighted during protein alignment, as both ABEs and CBEs were often clustered together for SaCas9 or Cas12a variants.
Nonetheless, finer clustering and apparent monophyly between base editors that comprised the same deaminase component was obvious, although it was notable that distinction based on protein sequence homology alone was insufficient. One such example was the inaccurate clustering of dual deaminases in the ABE clade, which should have been separated based on their relationship to CBEs (Supplementary Fig. S1).
Therefore, we propose a hypothetical phylogenetic tree in which some nodes were rebranched (Fig. 4). We decided that a minimally interventionist approach should be undertaken when rearranging aspects of the phylogenetic tree—namely that rebranching of nodes should only occur to delineate further between obviously ABE, CBE, and CGBE clades, as well as to establish a distinctive clade for dual deaminase variants.
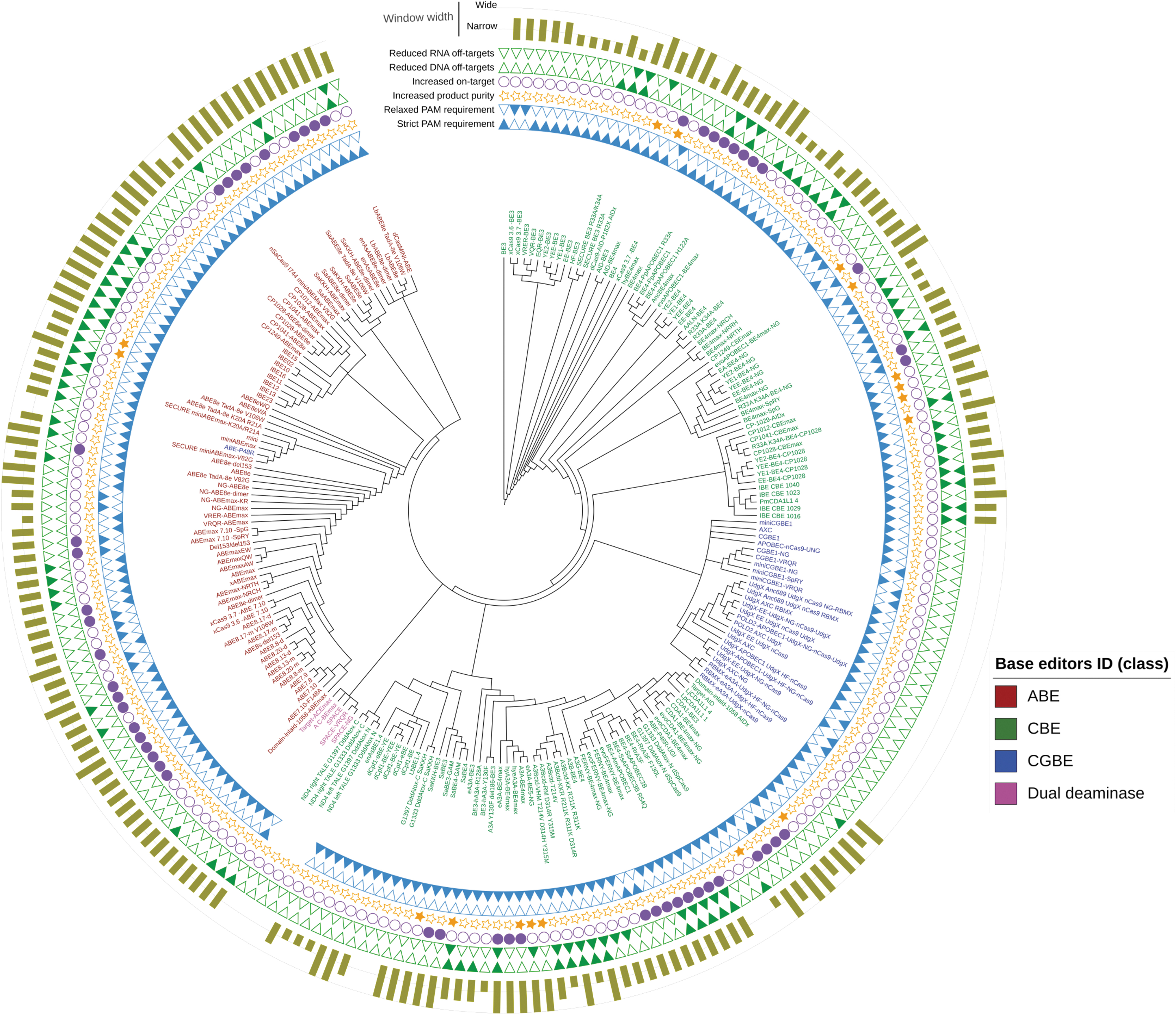
Hierarchical classification tree of 222 DNA base editors guided by preliminary phylogenetic relationship (Supplementary Fig. S1) and inference of chemical properties and biological activities of base editors (Fig. 3). ABEs, CBEs, CGBEs, and dual deaminases are highlighted in red, green, blue, and magenta, respectively. Each annotation corresponds to subcategorization of base editors based on targeting constraints (strict or relaxed PAM requirement); on-target activities (increased product purity and on-target deamination); off-target profiles (reduced off-target deamination on DNA and RNA); and width of the editing window (narrow or wide). This figure can be used to highlight specific base editors with their defined activity features.
To facilitate this rearrangement, IBE and circularly permuted clades were merged 54 and then rebranched at their respective most ancestral CBE and ABE phylogenetic backbones. Next, internal nodes wherein a mixture of both CBE and ABE variants for SaCas9 and Cas12a endonuclease variants were separated based on their target chemistry (i.e., separating LbABE8e-related nodes from dCpf1-BE-related nodes into ABE and CBE branches, respectively). The CGBE1 node was moved to the miniCGBE1 clade, which was previously the most ancestral basis for CGBEs (Supplementary Fig. S1), with other CGBE variants rebranched at this node. Finally, dual deaminase variants such as SPACE, Target-ACEmax, and A&C-Bemax leaflets were sequestered from the ABE clades and rebranched off the central phylogenetic backbone that distinguishes CBEs, ABEs, and CGBEs (Supplementary Note S1).
Based on the strong branch support for the distinct partitioning of ABEs, CBEs, and CGBEs from protein homology alignment, we infer that a hierarchical classification for DNA base editor should prioritize base editor chemistry at the target substrate at the highest level of taxonomic distinction. This effectively establishes three broad classes: ABEs, CBEs, and CGBEs.
While earlier phylogenetic analyses had difficulty partitioning dual deaminases, based on our reorganized phylogenetic tree, we establish that dual deaminases should be classified as a separate class from both ABEs and CBEs to reflect their capacity to mediate both adenine-to-guanine and cytosine-to-thymine base transitions. Here, we propose that the key criterion for the distinguishment of a dual deaminase is the possession of multiple, unique effector deaminase components capable of independent chemistries.
Our phylogenetic analysis broadly covers three endonuclease types—Cas12a-type effectors, SpCas9-type effectors, and SaCas9-type effectors—although other types such as SauriCas9-, 55 ScCas9-, 56 or iSpymac-type 57 base editor variants could naturally be included. Additionally, some of these variants have altered PAM specificities, such as xCas9-, SpG-, SpRY-, and NG-Cas9-type effectors. 35 For example, good branch support was evident for the clustering of YE2-BE4-NG, YE1-BE4-NG, YEE-BE4-NG, and EE-BE4-NG, which were partitioned from their NGG PAM variants such as YE2-BE4, YE1-BE4, YEE-BE4, and EE-BE4.
Additionally, distinct partitioning with branch support was also evident between CGBE1 and CGBE1-NG. Thus, we specify that the next highest taxonomic ranking should use the endonuclease component of the base editor for categorization, with altered PAM specificities for a given endonuclease type being a subtype classification.
The variant subcategory, which now specifies the deaminase component of the base editor, is a subcategory of the endonuclease type. For our description of variant and subvariant, the variant is defined by the identity of the deaminase protein, such as APOBEC1 or ecTadA, whereas the subvariant of the base editor is defined foremost by its initial description in the literature.
While general branch support was observed for ABEs and CBEs that were characterized by either the prototypic APOBEC1 or ecTadA deaminase domains, we also observed supported clustering for the internal nodes wherein the deaminase portion of the base editor varied. For example, the eA3A-BE3 and A3A-BE4max (in addition to other variants such as hyA3A-BE4max, BE3-hA3A-Y130F, etc.), CDA1 and AID (i.e., CDA1-BE4max, CDA1-BE3, LpCDA1L1.1, etc.), A3B (i.e., A3B-BE4- and A3B-ctd-related variants), and A3G variants appear phylogenetically distinct and well clustered, with good branch support (Supplementary Fig. S1).
Conversely, for ABEs, we observe that the phylogenetic distinction is less apparent, with the branching pattern clustered in a fan-like array from the ABE8e-NG node. This clustered arrangement is reflective of the fact that current ABEs are derived from an evolved ecTadA basis and thus share closely related deaminase variants, despite laboratory evolved or engineered modifications. On closer observation, however, we see that the ABE8s clade is branch supported, as are the prototypic ABE7.10 variants. Interestingly, however, ABE8e is the last common branch supported node prior to this broader fan-like clade. Therefore, we define that current-generation ABEs are either derived from ABE7.10, ABE8s, or ABE8e variants, which is apparent from the literature and is now supported by the phylogenetic analysis (Fig. 3).
Nonetheless, while our inferred and rebranched phylogenetic trees can be used as a guide for taxonomic organization at this level, it is very clear that there are key limitations that have been incorrectly inferred, such as the fact that ABE7.8 and ABE7.10 variants are not rooted at the base of the ABE clade.
Collectively, the different base editor subvariants can be further organized and described in reference to traits that include increased on-target activity, increased product purity, decreased DNA off-target, and decreased RNA off-target modifications (Fig. 4) based on their description in the literature. More contemporary base editors appear to share multiple characteristics, 58 and so classification at this taxonomic depth usually refers to the described traits of the editor rather than acting as a strict method for categorization.
Definition of the in situ activity window and key assumptions underpinning its characterization
A significant pillar to our taxonomic model is contingent on base editor–specific traits, such as the in situ activity window. While the initial description of the activity window of each editor is considered, we attempt to update this profile by compiling the profiles of more recently published head-to-head comparisons (Fig. 5). As information about some base editor variants may be incomplete to generate an unbiased profile sufficiently, we resort to using three assumptions to generalize their predicted editing range logically (see Methods).
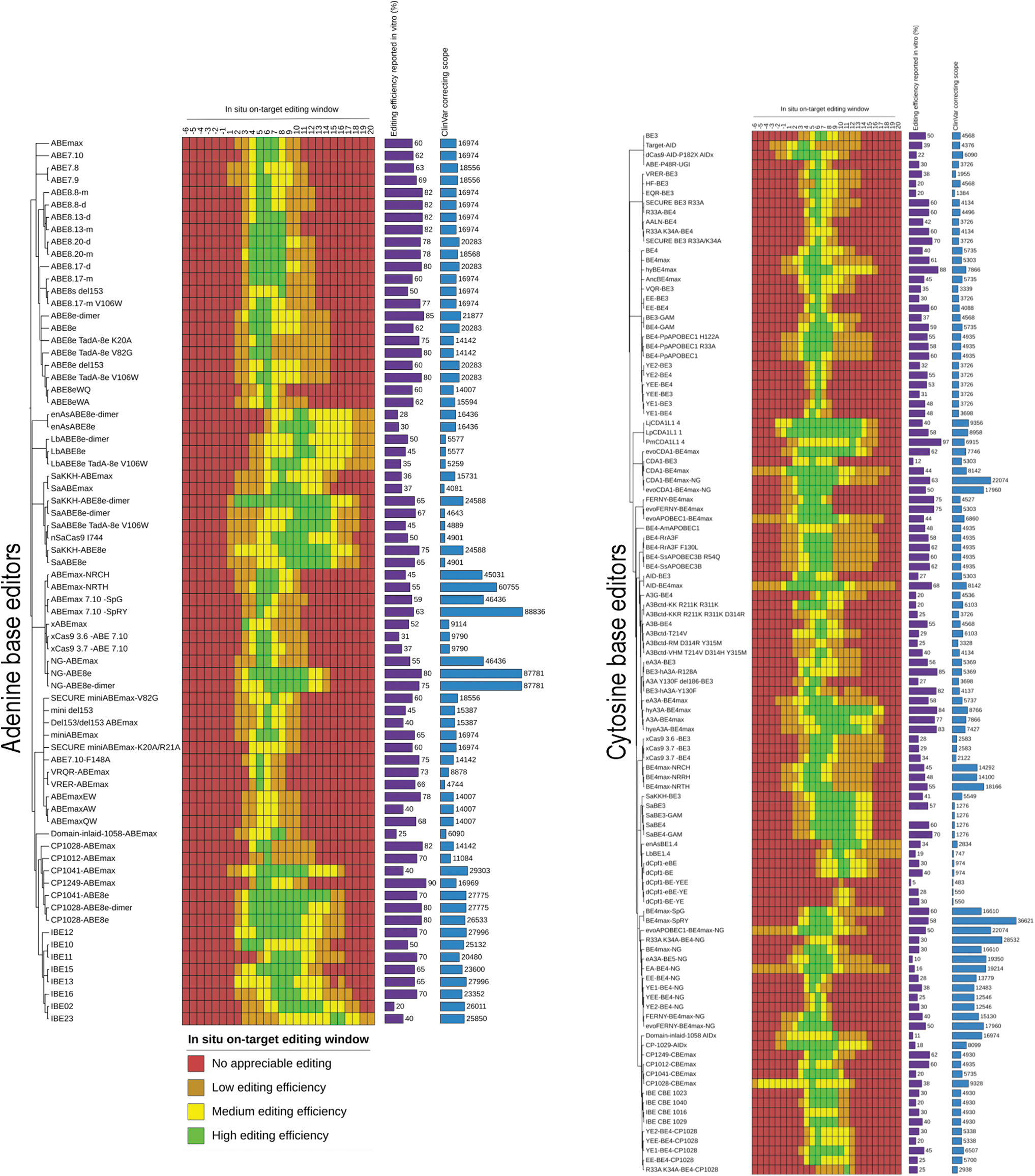
Characterization of the in situ editing window and ClinVar correcting scope of diverse ABEs and CBEs. Breadth of the editing windows were determined based on a review of published experimental data and graded on a scale from 0 to 3, presented in a heatmap. Grading was based on a fraction of the highest reported in vitro editing efficiencies (presented as a bar graph; Supplementary Table S2) for ABEs and CBEs, where 0 (no appreciable editing), 1 (low editing efficiency), 2 (medium editing efficiency), and 3 (high editing efficiency) represent 0%, >0% and ≤33%, >33% and <66%, and ≥66% of the highest reported editing efficiencies, respectively. Data were fitted based on the assumption that the editing window follows a parabolic shape with a central maxima and tapering activity on either side. A simple bar graph represents ClinVar correcting scope of each base editor based on PAM counts corresponding to the number of ClinVar targetable SNPs (Supplementary Table S4).
Base editor variants are then binned into three categories, which aptly describe their on-target activity window: wide-window, standard-window, and narrow-window editors.41,59 The three assumptions that underpin our model are as follows, and only apply when there is an explicit absence of actual deep-sequencing data:
(1) The in situ activity window of the base editor is likely to be symmetrical in shape with a single maxima flanked on either side by tapering on-target editing efficiencies.
(2) Where published data are unavailable for a given position, the editing efficiency is considered with respect to the first assumption; for example, if direct data are only given for positions 3 and 5 within a 20 nt protospacer, then position 4 is logically determined to be an intermediate of the two.
(3) Where published data are unavailable for a given base editor that varies only slightly compared to its unengineered counterpart (or wild type), their activity windows are expected to be similar. The following criteria is used to determine its distinctiveness to its wild-type reference:
a. The base editor does not have a significantly different window if it shares the same deaminase component as its unengineered counterpart and/or only the Cas component of the base editor is different in a noncircularly permuted or domain-inlaid manner (e.g., it would not be expected that the editing range of position 4–8 nt for VQR-BE3 would be changed for VRER-BE3).
b. The base editor is likely to have a similar editing breadth to its wild-type reference where mutations unrelated to the broadening or narrowing of the in situ activity window are engineered.
CRISPR-BEERS: ShinyApp for the selection of an ideal base editor for tailored applications
To consolidate these observations into a useable tool, we present our CRISPR-BEERS ShinyApp (CRISPR Base Editor Exchange Repository Server; https://github.com/hewittlab/CRISPR-BEERS/) based in R to aid users in the navigation of our taxonomic classification system, which uses the same principles of hierarchical taxonomy as outlined above. We have omitted the subclassification of variant as a selectable field in lieu of the subvariant field, which is specific to the end application for the user. This repository of 222 CRISPR base editors can be used to rank subvariants against a protospacer sequence of interest, given a target adenine or cytosine position. Users define the desired base editor traits as well as the position of the target nucleotide within their protospacer as an input to produce a ranked output that suggests the most ideal base editor for that particular application.
Briefly, upon loading the dependencies associated with the script, the ShinyApp is launched, and users can then filter through the database by first specifying the singular or nonsingular effector status of the base editor. This partitions the selection of dual deaminases from single effector base editor subvariants. Next, the endonuclease can be specified, and further specification for the subtype can be made to indicate the PAM requirement, such as strict or relaxed. Traits such as reduced single-guide RNA (sgRNA)-independent DNA off-target or reduced RNA off-targets effects, increased product purity, or increased on-target editing can also be specified, whereas in situ target windows are defined by indicating target cytosine or adenine bases by using either a the slider function for “Editing Window to display” or the indicating the position via the “Column sort by” function.
The database has been manually curated, and the scoring metrics for the ranked comparison of the in situ target window was derived from the published literature. While other webtools may provide information about the best sgRNA, 60 none so far provide a specific recommendation about which particular base editor to use.
In addition to ABEs and CBEs, BEERS also incorporates dual deaminases as well as CGBEs. For CGBEs, only positions 5 and 6 of a given protospacer are considered, and the data for CGBE nomination considered a mixture of good and bad target sites based on only position 6. As such, given that different CGBEs may be more appropriate for different sites, we strongly recommend that BE-HIVE (https://www.crisprbehive.design/) be consulted for a more detailed breakdown of CGBE editing outcomes and selection, with BE-HIVE being able to provide context-specific editing outcomes as well as information on data points beyond position 6.
We tested the BEERS ShinyApp against eight previously reported examples, specifically choosing a target site of interest and a target nucleotide position. For each example, we chose a key hypothesis to determine if the ShinyApp could appropriately select the correct base editor variant out of the database and if the ShinyApp could make additional suggestions. While it is possible that the originally cited studies and examples themselves may not use the most ideal base editor variant to begin with, owing to either date of publication or experimental limitations, this database was nonetheless compiled from a composite of these studies, and so it would be expected that it should be able to suggest at least some of the variants previously cited.
We noted that the database tended to err generously on the number of suggestible base editing variants, and so for each use case, we apply the following filters: activity for a given position being ≥2, or “Column to sort by” as “MAX” with “Descending” option (Table 1). Overall, in eight out of eight cases, BEERS was able to suggest at least one base editor variant that was used in the original case example. Here, we observe that the ShinyApp was able to select base editor variants appropriately when specified with a criterion such as reduced RNA off-targets or increased on target, with the selection of multiple filters allowing for appropriate nomination of a variant that possesses multiple characteristics.
Base editors in common between those in the reference study/source and those nominated in the BEERS database are displayed in bold.
Understandably, some variants do not possess all of the desired characteristics for a given target position, with the ShinyApp producing no output in these scenarios. While BEERS produces a ranked output listing variants with high activity to those with lower activity, this listing was produced based on a comparison of base editor activity on largely two sites only: HEK2 and HEK3. In these scenarios, the output of the BEERS ShinyApp is intended to provide only general guidance for DNA base editor consideration and should not be viewed as an absolute comparison.
Additionally, we present two further examples of how one would navigate our classification system using one hypothetical scenario and one previously reported example that was published when only the ABE7.10 variant was available. 61 To begin, BEERS requires prior knowledge of the PAM and protospacer, and so we would highly recommend that other online web tools should be consulted first (https://sgrnascorer.cancer.gov/sgRNAScorerV2/). 60
Introducing an in-frame STOP codon into an open reading frame
Hypothetically, if the installation of an in-frame stop codon within the open reading frame of a green fluorescent protein (GFP) at amino acid 70 (Q70X) were desired, the following factors are considered in our taxonomic hierarchy and by BEERS (Fig. 6).
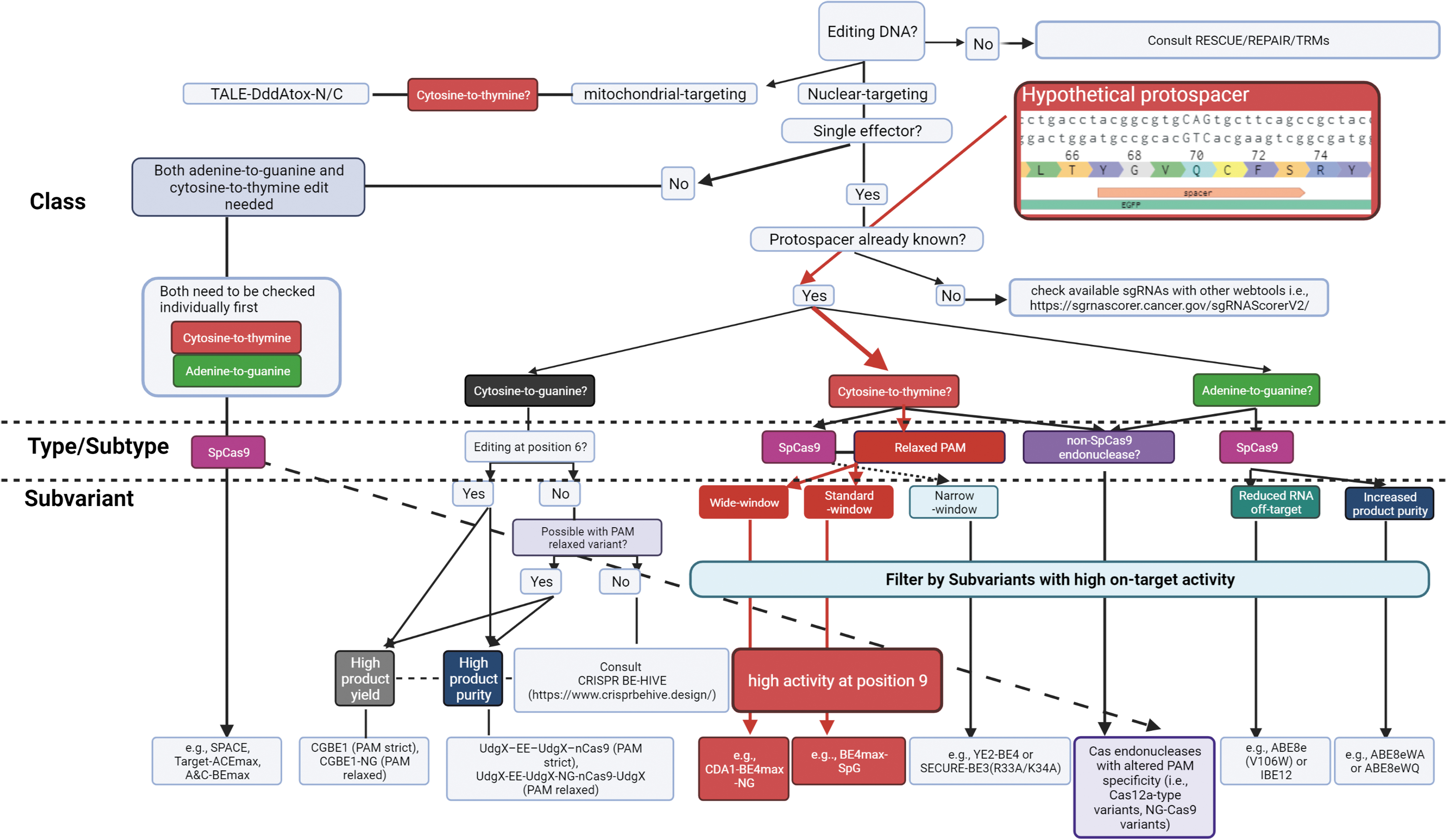
Worked example of a decision tree for considering base editors based on taxonomic classification. In a hypothetical scenario, a stop codon is introduced in the open reading frame of enhanced green fluorescent protein. Here, the protospacer sequence is already known, and the desired target cytosine is defined (position 9 in a 20 nt protospacer). Using the BEERS ShinyApp in R, recommendations are suggested for the relevant base editors as a starting point for experimental consideration based on specific input criteria (Table 1).
Here, glutamine (
Therefore, this indicates a preference for the use of a PAM-flexible Cas9 variant. Optimal editing at position 9 with negligible effect incurred by the bystander cytosines at positions 2 and 5 enables the selection of only position 9 for consideration by the slider function. Other traits such as increased product purity or editing efficiency can be specified as categories as the desirable traits of the base editor. Based on these characteristics, BEERS predicts that the ideal CBE would be either CDA1-BE4max-NG or BE4max-SpG (see Methods).
Introduction of the S683G mutation into mouse androgen receptor (Ar)
Liu et al. 61 generated a mouse model of androgen insensitivity syndrome by introducing the S683G and I878T mutations into the Ar gene using ABE7.10. At the time of publication, only one ABE variant was available.
Therefore, we were interested to see if BEERS could suggest a better ABE variant that addresses many of the issues that affected the initial publication. The two most notable issues were the incidences of aberrant cytosine-to-guanine editing at position 5, and the introduction of a silent mutation at position 3. Their target adenine was at position 7, at the S683 position (sgAr-1). Using BEERS, we would recommend that ABE8eWA and ABE8eWQ would be the most suitable ABE candidates for this particular application, noting that both have been specifically engineered for reduced aberrant cytosine transversion and have optimal editing at positions 7 and 4 but not at position 3 (Table 1). In this case, the slider should be adjusted to include the editing range from 3 to 7.
Limitations
Unfortunately, the data for our model are predominantly based on the base editor's initial characterization and thus suffer significantly from uneven comparisons, such as editing efficiencies across the number of different sites, transfection conditions (whether enrichment for the edit has been applied), varying cell lines (although almost all base editors were characterized in HEK293T cells, on which our data were based), and outdated or entirely absent tranches of biochemical evaluation (earlier base editing studies did not specifically screen for sgRNA-independent DNA or RNA off-target events). Here, we attempt to normalize the data by considering only a handful of positive control sites that are commonly used to characterize base editing efficiency. Some of these common sites included HEKsite 2, HEKsite 3, HEKsite 4, RNF2, FANCF, site3, and site4.
In addition, we present data for editing efficiency on a given variant only if it has been characterized in a HEK293 cell line or if the transfection conditions adhered to either a 3- or 5-day period. Overall, we found that most studies (Supplementary Table S1) used similar transfection conditions and base editor: sgRNA plasmid ratios, with no selection or enrichment methods employed. In general, past literature has suggested that increasing the amount of transfected base editor also increased editing efficiency, 24 but we noted that each study scaled up or scaled down the amount of transfected base editors based on well size. Thus, we deemed that transfection conditions were mostly comparable, provided that similar harvest timing was used. In all cases, each experiment ran for either 3 or 5 days.
Nonetheless, while great lengths have been made to update our model with new information based on more recent publications, we apply assumptions to compensate for missing data, which in itself may bias against those variants that have more in-depth characterization compared to others. Additionally, the use of only a handful of positive control sites to determine base editing efficiency can only go so far in providing an approximate, relativistic estimate that requires obvious experimental follow-up to adjust for batch effects and truly normalized transfection conditions.
Our initial phylogenetic characterization of base editors did not result in immediately intuitive groupings of taxa. While some subvariants, particularly broad window CBEs, clustered modestly well, we found that in most instances, protein alignment of the base editor sequence was insufficient to group all base editors into clearly defined taxa. For example, ABE8e-dimer was separated out from ABE8e, while NG-ABE8e was grouped with variants that had reduced RNA off-target profiles. This was most likely attributable to the overrepresentation of the Cas endonuclease sequence during protein alignment.
Nonetheless, another limitation of this model is that it focuses mainly on evolved or engineered variants, as well as popular variants, and does not capture all DNA base editor variants currently published. Future directions for this work would include wet lab validation comparing base editor variants against specific target sites to show that the ShinyApp is able to nominate base editors accurately in an arrayed setting, based on key parameters that could include a head-to-head comparison of on-target editing efficiency, measurement of bystander editing, or assessment of the transcriptome- and genome-wide fidelity of nominated variants.
Conclusion
Herein, we present a hypothetical taxonomic classification system and describe a phylogenetic framework for base editors based on protein sequence alignment and their described and characterized traits in the literature. Using phylogeny as a basis for analyses, we illustrate the iterative and interrelated nature of base editor development. We highlight how this interrelated distinction has a statistically supported basis used to infer taxonomic categorization, basing our phylogenetic approach toward hierarchical classification on the work described by Makarova et al. 47
Our model is based on key assumptions that have been used to compensate for gaps in the literature. Although some of these assumptions are merely close approximations to their experimental description, we find that our classification system should be sufficient to describe DNA base editors in a manner consistent with established literature. Finally, we apply the principles of our taxonomic classification system to develop a ShinyApp that allows for the nomination of potentially ideal base editor candidates for a given application as a starting point for further in vivo or in vitro testing via CRISPR-BEERS (https://github.com/hewittlab/CRISPR-BEERS).
Footnotes
Acknowledgments
We are thankful for the design inputs of Pratikshya Pandey and design comments of Syed Hammad Ali Bokhari for Figure 5. The data set supporting the conclusions of this article are included within the article as Supplementary Tables S1–S4. CRISPR BEERS is accessible via GitHub (![]() ).
).
Author Disclosure Statement
All authors declare no competing interests.
Funding Information
This work was supported by a Fellowship and Program Grant from The Australian National Health and Medical Research Council (AWH, GNT2009079 and GNT1132719), the Australian Research Council Special Research Initiative in Stem Cell Science (Stem Cells Australia) and Retina Australia.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.