
Abstract
The ability of CRISPR-Cas12a nucleases to function reliably in a wide range of species has been key to their rapid adoption as genome engineering tools. However, so far, Cas12a nucleases have been limited for use in organisms with growth temperatures up to 37 °C. Here, we biochemically characterize three Cas12a orthologs for their temperature stability and activity. We demonstrate that Francisella novicida Cas12a (FnCas12a) has great biochemical potential for applications that require enhanced stability, including use at temperatures >37°C. Furthermore, by employing the moderate thermophilic bacterium Bacillus smithii as our experimental platform, we demonstrate that FnCas12a is active in vivo at temperatures up to 43°C. Subsequently, we develop a single-plasmid FnCas12a-based genome editing tool for B. smithii, combining the FnCas12a targeting system with plasmid-borne homologous recombination (HR) templates that carry the desired modifications. Culturing of B. smithii cells at 45°C allows for the uninhibited realization of the HR-based editing step, while a subsequent culturing step at reduced temperatures induces the efficient counterselection of the non-edited cells by FnCas12a. The developed gene-editing tool yields gene-knockout mutants within 3 days, and does not require tightly controllable expression of FnCas12a to achieve high editing efficiencies, indicating its potential for other (thermophilic) bacteria and archaea, including those with minimal genetic toolboxes. Altogether, our findings provide new biochemical insights into three widely used Cas12a nucleases, and establish the first Cas12a-based bacterial genome editing tools for moderate thermophilic microorganisms.
Introduction
A wide range of CRISPR-Cas immune systems from bacteria have been repurposed to universal genetic engineering tools.1–3 Many of the current tools exploit the RNA-guided DNA endonuclease Cas9 from the type II CRISPR-Cas system of Streptococcus pyogenes (SpCas9). SpCas9 can generate double-stranded DNA breaks (DSBs) at the target sequence, referred to as the protospacer, when guided either by two RNA (crRNA and tracrRNA) molecules or by a synthetic chimera of the two, called single guide RNA (sgRNA). 4 The guide RNA-target DNA binding, which precedes the Cas9 cleavage activity, is dependent on the initial recognition of a protospacer adjacent motif (PAM), a short (3–5 bp long) DNA sequence at the 3′ end of the protospacer.5,6 The facile programmability of CRISPR-Cas-derived DNA targeting systems have greatly facilitated efficient genetic manipulation of model and non-model bacteria. Nevertheless, the use of SpCas9 is limited in some organisms due to reported toxicity in the recipient cells.7–9
Cas12a (formerly Cpf1), a type V-A endonuclease of the class 2 CRISPR-Cas system, is a dual nuclease that is involved in crRNA processing, target-site recognition, and DNA cleavage.10–12 Cas12a recognizes T-rich PAM sequences and can be guided by a single crRNA without an additional tracrRNA. 10 Similar to Cas9, Cas12a has successfully been used for genome editing, not only in mammalian cells10,12 but also in a repertoire of industrially relevant microorganisms, including several bacteria and yeast.7,8,13–16
Bacterial cell factories are being developed that convert renewable feedstocks, such as plant biomass-derived sugars, into green fuels or chemicals. Using such microbial fermentations, however, production costs of low-value products are still relatively high and not competitive with traditional fossil fuel–based refinery processes. Compared to the mesophilic organisms that are generally used in industrial bio-refineries, thermophilic organisms are an attractive alternative for several reasons: (1) lower operational costs (cooling and contamination risks); (2) higher substrate and product solubility; (3) higher reaction and production rates, leading to reduced dosage of hydrolytic enzymes; and (4) reduced risk of enzyme product inhibition.17,18
Traditional genetic manipulation methods, based on recombination systems, are usually laborious and time-consuming, especially for non-model organisms such as most thermophilic bacteria. This is a major bottleneck both for fundamental studies of these organisms and for their development into industrial production hosts. We have previously developed an efficient SpCas9-based engineering tool for the facultative thermophile Bacillus smithii ET 138, combining homologous recombination (HR) at elevated temperatures (55°C) and SpCas9-based counterselection at moderate temperatures (37°C). 19 In addition, after discovery of a thermostable Cas9 variant, we developed a ThermoCas9-based engineering tool for B. smithii ET 138, combining HR and ThermoCas9-based counterselection at 55°C. 20 However, SpCas9 and ThermoCas9 require G-rich and C-rich PAM sequences (5′-NGG-3′ and 5′-NNNNCCAA-3′, respectively), which may not always be available at the desired site of engineering, more particularly in AT-rich regions of genomes. On the other hand, Cas12a recognizes a T-rich PAM sequence (5′-TTTV-3′) for target cleavage. 10 Therefore, the addition of thermostable Cas12a-based tools would further extend the genome editing toolbox for thermophilic organisms.
To this end, we assessed the in vitro temperature tolerance and stability of three different Cas12a orthologs: FnCas12a from Francisella tularensis subsp. novicida U112, AsCas12a from Acidaminococcus sp. BV3L6, and LbCas12a from Lachnospiraceae bacterium ND2006. The FnCas12a nuclease was selected for subsequent in vivo analyses, including the development of a genome editing tool for facultative thermophiles. We combined HR-based editing at elevated temperatures with FnCas12a-based counterselection at lower temperatures for rapid and efficient and accurate gene deletions in B. smithii, further reducing the time of the editing process to 2–3 days. Overall, this study describes new biochemical insights into three widely used Cas12a nucleases, and establishes the first Cas12a-based genome editing tool for moderate thermophiles.
Methods
Protein expression
FnCas12a and AsCas12a were expressed and purified as previously described. 21 The FnCas12a and AsCas12a expression constructs were synthesized by GenScript. FnCas12a and AsCas12a were tagged with a N-terminal SV40 NLS, and C-terminal SV40 NLS and 6xHis-tag. Proteins were expressed in NiCo21 (DE3) Escherichia coli cells (NEB #C2529) containing the respective expression plasmid by growing in LB-Kan (40 μg/mL) at 37°C followed by inducing at 23°C for 16 h using 0.4 mM IPTG. Cells were lysed using sonication, and recombinant Cas12a proteins were purified using HiTrap DEAE FF, HisTrap HP (Ni-NTA), and HiTrap Heparin HP columns from GE Healthcare (Pittsburgh, PA). The purified proteins were dialyzed and concentrated into 20 mM Tris-HCl (pH 7.4), 500 mM NaCl, 1 mM DTT, 0.1 mM EDTA, and 50% glycerol.
In vitro activity assays
Buffers and chemicals were obtained from New England Biolabs (Ipswich, MA), VWR (Radnor, PA), and Sigma–Aldrich (St. Louis, MA). NEBuffers were from New England Biolabs (Ipswich, MA). The oligonucleotides (IDT, Coralville, IA) and the plasmids used for the enzyme activity assays are listed in Supplementary Tables S1 and S2, respectively. The 515 bp linear DNA substrates were amplified (using NEB1 and NEB2) by polymerase chain reaction (PCR) using Q5 2 × master mix (NEB #M0492S) from the pMiniT-WTAP Exon 8 plasmid, which contains a fragment of the human WTAP gene. The substrate contains the target sequence 5′-TTTCCCACTCACTGCTTTCTCCTC-3′, where TTTV is a generic PAM sequence for Cas12a. PCR products were purified using a Monarch PCR and DNA Cleanup Kit (NEB #T1030S). crRNA for Cas12a was synthesized (NEB3), and the guide for Cas9 was generated using the EnGen sgRNA synthesis kit S. pyogenes (NEB #E3322S) using the template for top strand (NEB4). Recombinant SpCas9 and LbCas12a were obtained from New England Biolabs.
Activity of the Cas12a ribonucleoproteins (RNPs) was determined by monitoring the double-stranded DNA (dsDNA) cleavage of 5′-FAM labeled PCR products. To mimic the biological conditions, the respective crRNA and the nucleases were separately pre-equilibrated at the designated temperature in 1 × NEBuffer2.1 (10 mM Tris-HCl [pH 7.9], 50 mM NaCl, 10 mM MgCl2, 100 μg/mL BSA) freshly supplemented with 1 mM DTT. RNP loading was performed for 10 min prior to the addition of the FAM labeled DNA substrate. Final reactions contained 100 nM Cas12a protein, 200 nM crRNA, and 10 nM DNA. DNA substrate was digested at varying temperatures as indicated for 10 min in a T100 Thermal Cycler (Bio-Rad, Hercules, CA). Reactions were quenched and then subjected to capillary electrophoresis on a 3730xl Genetic Analyzer (Applied Biosystems, Foster City, CA). 22 The cleavage products were quantified using Peakscanner (Thermo Fisher Scientific, Waltham, MA).
Thermal stability of Cas12a proteins
The thermal stability of the nucleases was determined in the presence and absence of their respective crRNAs using a Prometheus nano differential scanning fluorimetry (NanoDSF) instrument using standard 10 μL capillaries (NanoTemper Technologies, Munich, Germany) Cas12a proteins were analyzed at a concentration of 10 μM in 1 × NEBuffer2.1 (10 mM Tris-HCl [pH 7.9], 50 mM NaCl, 10 mM MgCl2, 100 μg/mL BSA) freshly supplemented with 1 mM DTT. Cas12a RNP were analyzed as mixtures of 10 μM Cas12a protein and 20 μM crRNA in the same buffer. RNP formation was performed for 10 min at 25°C prior to loading in capillaries. Unfolding of the protein or complex was monitored from 20°C to 80°C at a rate of 1°C/s according to the manufacturer's instructions, and data are reported as the inflection point of the melting curve of the protein or RNP complex.
Strains, transformations, and growth media
Bacterial strains used in the current study are E. coli DH10β and B. smithii ET 138 ΔsigF ΔhsdR. 19 The E. coli and B. smithii strains were routinely cultured at 37°C and 55°C, respectively, unless stated otherwise. E. coli strains were grown in 10 mL Luria-Bertani broth (LB) in 50 mL Greiner tubes or plated on LB with 15 g/L agar (Difco) plates containing 15 μg/mL chloramphenicol. B. smithii ET 138 ΔsigF ΔhsdR cells were made competent and transformed, as described previously. 23 B. smithii strains were cultured in 10 mL LB2 medium (10 g/L tryptone, 5 g/L yeast extract, 100 mL/L 10 × ESS, pH 6.9–7.0) 23 in 50 mL Greiner tubes at 150 rpm or plated on LB2 with 30 g/L agar (Difco) plates supplemented with 7 μg/mL chloramphenicol, vitamins, and metals. 23 For all the gene editing experiments, glucose (0.5 g/L), xylose (0.5 g/L), and uracil (50 mg/L) were added to the media to induce expression of Cas12a and to provide for the uracil auxotrophic pyrF deletion mutants, respectively. Additionally, thermophile vitamin medium with yeast extract (TVMY; 0.5 g/L yeast extract, 8.37 g/L MOPS, 100 mL/L 10 × ESS, pH 6.9–7.0) 23 or plates (made with 30 g/L agar) with the same supplements was used in the editing experiments. For the evaluation of 5-fluorootic acid (5-FOA) sensitivity of B. smithii ET 138 ΔsigF ΔhsdR ΔpyrF cells, TVMYxgu and LB2xgu agar plates supplemented with 1 g/L 5-FOA were used.
Plasmid construction
The plasmids constructed and the primers (IDT) used for cloning and sequencing are listed in Supplementary Tables S1 and S2, respectively. The fragments for assembling the plasmids were amplified by PCR with Q5® High-Fidelity 2 × Master Mix (New England Biolabs). The amplicons were run on a 1% agarose gel electrophoresis and purified using a Zymogen gel DNA recovery kit (Zymo Research, Freiburg, Germany). The HiFi DNA assembly mix was purified using the DNA Clean and Concentrator-5 (Zymo Research), and electroporated into competent E. coli DH10B cells using the following settings: 2.5 kV, 200 Ω, and 25 μF. Colony PCR with OneTaq® 2 × Master Mix with Standard Buffer (New England Biolabs) on single colonies was used to screen for the right transformants. Plasmid extractions were performed using the GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific), and verified by Sanger sequencing (Macrogen Europe B.V., Amsterdam, The Netherlands).
Colony PCR, genomic DNA isolation, and sequencing
Potential B. smithii ET 138 ΔsigF ΔhsdR ΔpyrF colonies were randomly selected and subjected to colony PCR using the Q5® High-Fidelity 2 × Master Mix (New England Biolabs) with the genome specific primers BG15752 and BG15559. Genomic DNA from B. smithii strains was isolated using the MasterPure™ Gram Positive DNA Purification Kit (Epicentre Biotechnologies, Madison, WI). Purification of PCR products was performed using the DNA Clean and Concentrator-5 Kit (Zymo Research). The DNA fragments were subsequently sent for Sanger sequencing (Macrogen Europe B.V.) for confirmation of the gene deletion.
Results
Biochemical characterization of Cas12a nucleases
Initially, we performed bioinformatic analysis on the publicly available genomes of thermophilic bacteria, aiming to find novel, possibly thermotolerant or thermophilic Cas12a orthologs. Unfortunately, this analysis was not fruitful. Hereafter, we biochemically assessed the three best characterized and frequently used Cas12a variants—Francisella tularensis subsp. novicida U112 (FnCas12a), Acidaminococcus sp. BV3L6 (AsCas12a), and Lachnospiraceae bacterium ND2006 (LbCas12a)—for their temperature stability and activity. To this end, we expressed the selected genes in E. coli, and purified the corresponding Cas12a proteins (FnCas12a, AsCas12a, and LbCas12a). As reference, the Cas9 nuclease from S. pyogenes (SpCas9) was used.
To assess the activity of the selected Cas12a and Cas9 nucleases at elevated temperatures, we executed in vitro target cleavage assays between 20°C and 68°C. To facilitate easy monitoring of DNA cleavage, we generated a linear dsDNA substrate with the 5′ end labeled with 6-carboxyfluorescein (FAM) using PCR. The loading of the crRNA to the protein to form the RNP complex was performed at the assay temperature, and the cleavage reactions were initiated by the addition of the FAM-labeled DNA target, which was preincubated at the designated incubation temperatures for 3 min. We observed high DNA cleavage activity by SpCas9 from 15°C to 37°C, and intermediate DNA cleavage activity at 41°C (Fig. 1A); SpCas9 activity rapidly decreased to undetectable levels when incubated at ≥44°C, corroborating previous findings.19,24 The widely used LbCas12a nuclease exhibited complete DNA cleavage activity at temperatures from 20°C to 33°C, moderate DNA cleavage activity at 37°C, and no DNA cleavage activity at >41°C (Fig. 1A). AsCas12a, the other extensively studied Cas12a nuclease, exhibited complete DNA cleavage activity only at 30°C; above and below that temperature, its DNA cleavage activity rapidly decreased to undetectable levels at 41°C and to very low levels at 20°C. In the case of FnCas12a, we observed complete cleavage of DNA substrates at temperatures from 20°C to 41°C, while FnCas12a was the only tested nuclease that remained active—albeit weakly—at 44°C (Fig. 1A). These data indicate the potential to use FnCas12a as a genome editing tool for both mesophilic and moderately thermophilic organisms.
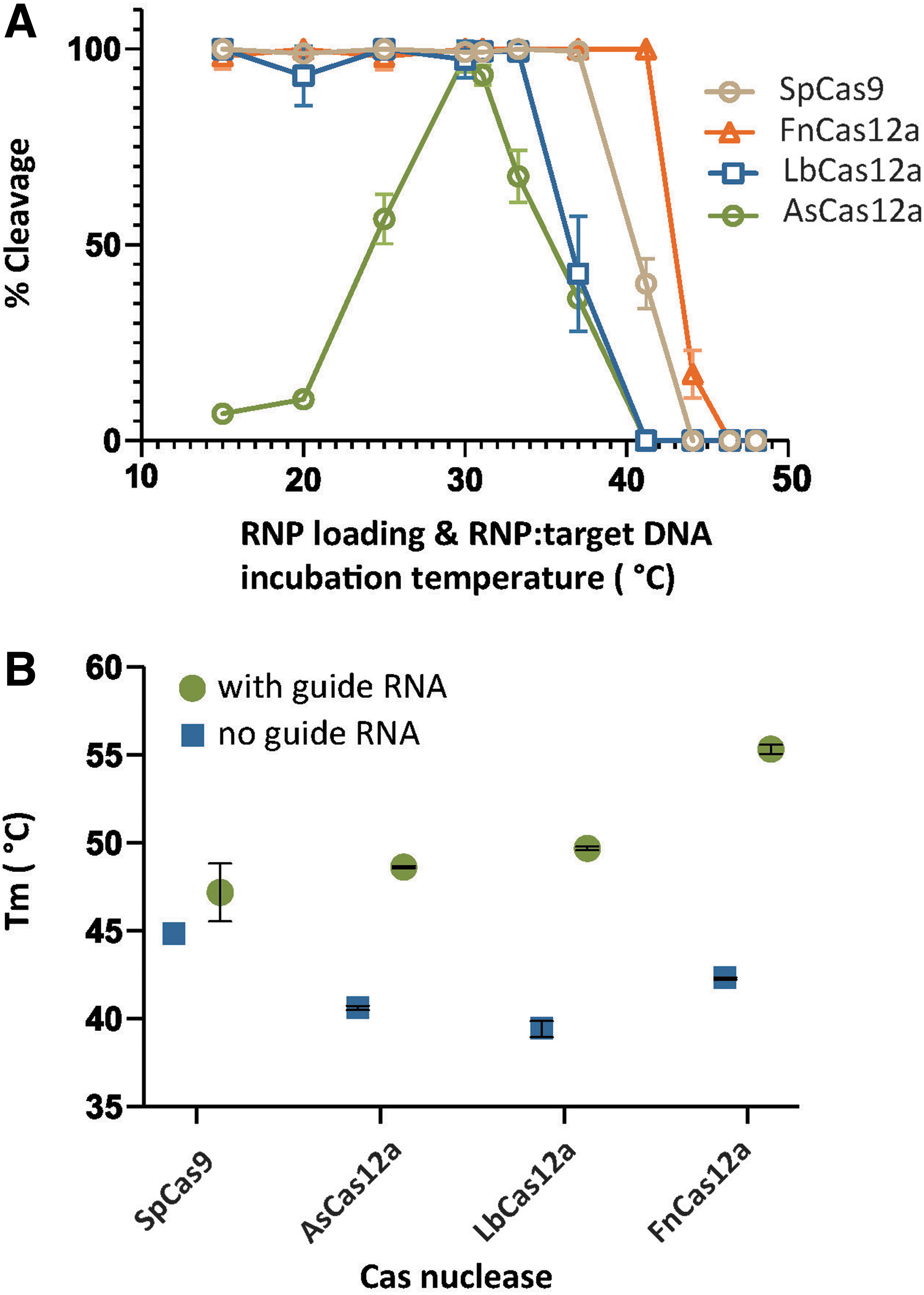
Activity of Cas12a orthologs at varied temperatures.
In cells, the Cas12a proteins may be present with and without their crRNA guide. Therefore, it was decided to compare the thermostability of the recombinant proteins as well as the RNP complexes using nano differential scanning fluorimetry (nanoDSF). 25 We observed that SpCas9 has a melting temperature (Tm) of 45°C for the gRNA-free recombinant protein and 47°C for the RNP complex (Fig. 1B and Supplementary Fig. S1A). In the case of LbCas12a and AsCas12a, the crRNA-free proteins had Tm values in the range of 40°C, and the RNPs of 49°C (Fig. 1B and Supplementary Fig. S1B and C). The FnCas12a RNP had a Tm of 55°C compared to the crRNA-free recombinant protein, which had Tm of 47°C (Fig. 1B and Supplementary Fig. S1D). We observed that LbCas12a and FnCas12a proteins exhibited a change in their unfolding profile upon addition of guide RNA (Supplementary Fig. S1C and D). In the case of SpCas9 RNP and AsCas12a RNP, the melting curve fluorescence profiles remained constant (Supplementary Fig. S1A and B). This indicates that guide RNA loading induces a conformation change in LbCas12a and FnCas12a that causes a shift in exposed tyrosine or tryptophan residues, leading to a different unfolding profile direction in nanoDSF. The overall trend that the thermal stability of the Cas12a orthologues (and SpCas9) is strongly enhanced after the association of an appropriate crRNA is similar to our previous findings for ThermoCas9, a thermotolerant Cas9 orthologue. 20
To conclude the biochemical characterization of the Cas12a orthologs, we performed the above-described in vitro cleavage assays over a range of NaCl concentrations and pH values (Supplementary Fig. S2). SpCas9 and AsCas12a were tolerant to a wide range of salt concentrations, from 50 to 500 mM NaCl (Supplementary Fig. S2A). On the other hand, LbCas12a and FnCas12a lost their activity above 300 mM NaCl (Supplementary Fig. S2A). All the tested nucleases, except AsCas12a, were highly active between a wide range of pH (6.0–9.0). Under the tested conditions, AsCas12a displayed reduced activity above pH 7.5 (Supplementary Fig. S2B).
FnCas12a is capable of targeted DNA cleavage in B. smithii genome
The thermal stability and activity of FnCas12a RNP complex motivated us to test the utility of FnCas12a as a genome editing tool for moderate thermophilic bacteria. To this end, we employed B. smithii ET 138, a facultative anaerobic, facultative thermophilic bacterium that grows between 37°C and 65°C, with an optimum at 55°C. 26 We used B. smithii ET 138 ΔsigF ΔhsdR, a strain that was previously made sporulation and restriction deficient. 23
Initially, to detect any potential toxicity of FnCas12a in the B. smithii cells, we designed a single plasmid approach, and constructed a non-targeting (NT) pFnCas12a_NT plasmid (Fig. 2A) by cloning the Francisella novicida U112 cas12a (fncas12a) gene and the crRNA encoding gene (comprised of 36 nt repeat and a 25 nt spacer sequence) in the Bacillus–E. coli shuttle vector pNW33n. The target sequence is flanked by a non-canonical PAM (5′-CTCG). The expression of the fncas12a gene and the crRNA encoding gene is under the control of the promoter of the xynA gene (P xynA ) from Thermoanaerobacterium saccharolyticum and the promoter of the phosphotransacetylase gene (P pta ) from Bacillus coagulans,27,28 respectively.
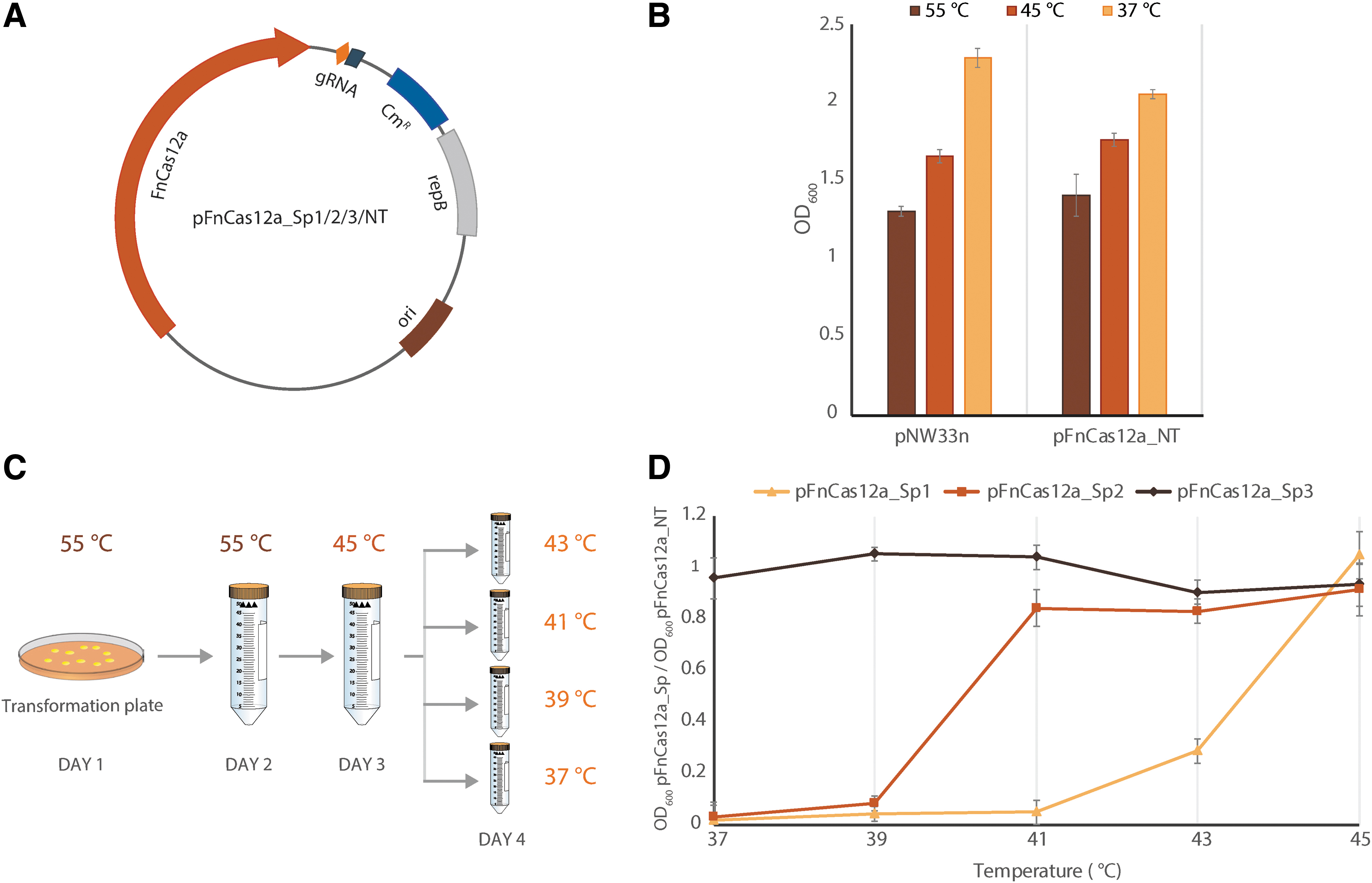
Transformation of FnCas12a-mediated targeting plasmids in Bacillus smithii ET 138.
The pFnCas12a_NT plasmid and an empty pNW33n control plasmid were separately electroporated into competent B. smithii cells plated on LB2 plates supplemented with chloramphenicol and incubated overnight at 55°C. The following day, four single colonies per transformation were used for sequential transfers in fresh LB2xg liquid media supplemented with chloramphenicol from 55°C to 37°C every 24 h, with a mandatory for growth intermediate step at 45°C. No major difference in growth was observed between the pFnCas12a_NT cultures and the pNW33n control cultures (Fig. 2B), indicating that the expression of FnCas12a is not toxic for the B. smithii cells at any of the above tested temperatures.
Next, we evaluated the in vivo targeting activity of FnCas12a, correlating it with the induction of lethal DSBs in the B. smithii genome. To this end, we designed and constructed three targeting plasmids, pFnCas12a_Sp1–3, each containing a crRNA with a unique spacer (Spacer 1 [Sp1], Spacer 2 [Sp2], and Spacer 3 [Sp3]), corresponding to a different target sequence (Supplementary Table S3) within the pyrF gene in the B. smithii genome. The pyrF gene encodes for orotidine 5′-monophosphate (OMP) decarboxylase, which is required for pyrimidine biosynthesis. This enzyme is also able to convert the uracil analogue 5-FOA to the toxic product 5-fluorodeoxyuridine 5′-monophosphate. 29 Therefore, ΔpyrF strains are uracil auxotrophs and, unlike the wild type, resistant to 5-FOA.30–32
The three targeting plasmids and the control plasmids were individually electroporated into a single batch of B. smithii competent cells. Four single colonies per transformation were subjected to sequential transfers at different temperatures: 37°C, 39°C, 41°C, and 43°C (Fig. 2C). No significant differences in growth were observed for all the cultures incubated at 55°C (not shown) and 45°C. Conversely, the growth of the cultures harboring the pFnCas12a_Sp1 and pFnCas12a_Sp2 plasmids were significantly inhibited at ≤43°C and ≤41°C, respectively (Fig. 2D). However, no difference in growth for the cultures harboring the pFnCas12a_Sp3 plasmid was observed (Fig. 2D). The observed fluctuation is most likely due to the reported crRNA-dependent variation in CRISPR-Cas12a targeting efficiency,33–35 similar to that of the CRISPR-Cas9 system.36,37
Marker-less chromosomal gene deletion using FnCas12a and plasmid-borne editing template
HR-Cas12a mediated genome editing requires three parts: (1) a dsDNA fragment that carries the desired modifications and includes the HR flanks, allowing for the desired genomic recombination through double crossover; (2) the Cas12a endonuclease; and (iii) the crRNA that leads Cas12a to the target DNA site, enriching mutations introduced through HR by counterselecting (eliminating) wild-type clones. Based on our previous research 19 and by employing the here-developed FnCas12a targeting system, we set out to develop a HR-FnCas12a based counterselection system for genome editing in B. smithii. A sequential culturing process of first equipping the cells with an appropriate plasmid-borne template for HR at 55°C, and then inducing the expression of active FnCas12a at 37°C, is expected to lead to counterselection against the wild-type genotype.
As a proof of principle, we programmed the designed HR-FnCas12a system for deletion of the pyrF gene in B. smithii. Two editing plasmids (pFnCas12a_ΔpyrF-HR_Sp1 and pFnCas12a_ΔpyrF-HR_Sp2) were constructed, both containing the previously described Sp1 and Sp2 for efficient pyrF targeting, together with a 2 kb HR template encompassing 1 kb of both the up- and downstream genomic regions flanking the selected pyrF gene fragment (Fig. 3A). A NT plasmid (pFnCas12a_ΔpyrF-HR_NT) containing the same HR template as the editing plasmids but with a NT spacer and (non)targeting plasmids without the HR template were taken as a control for recombination without counterselection.
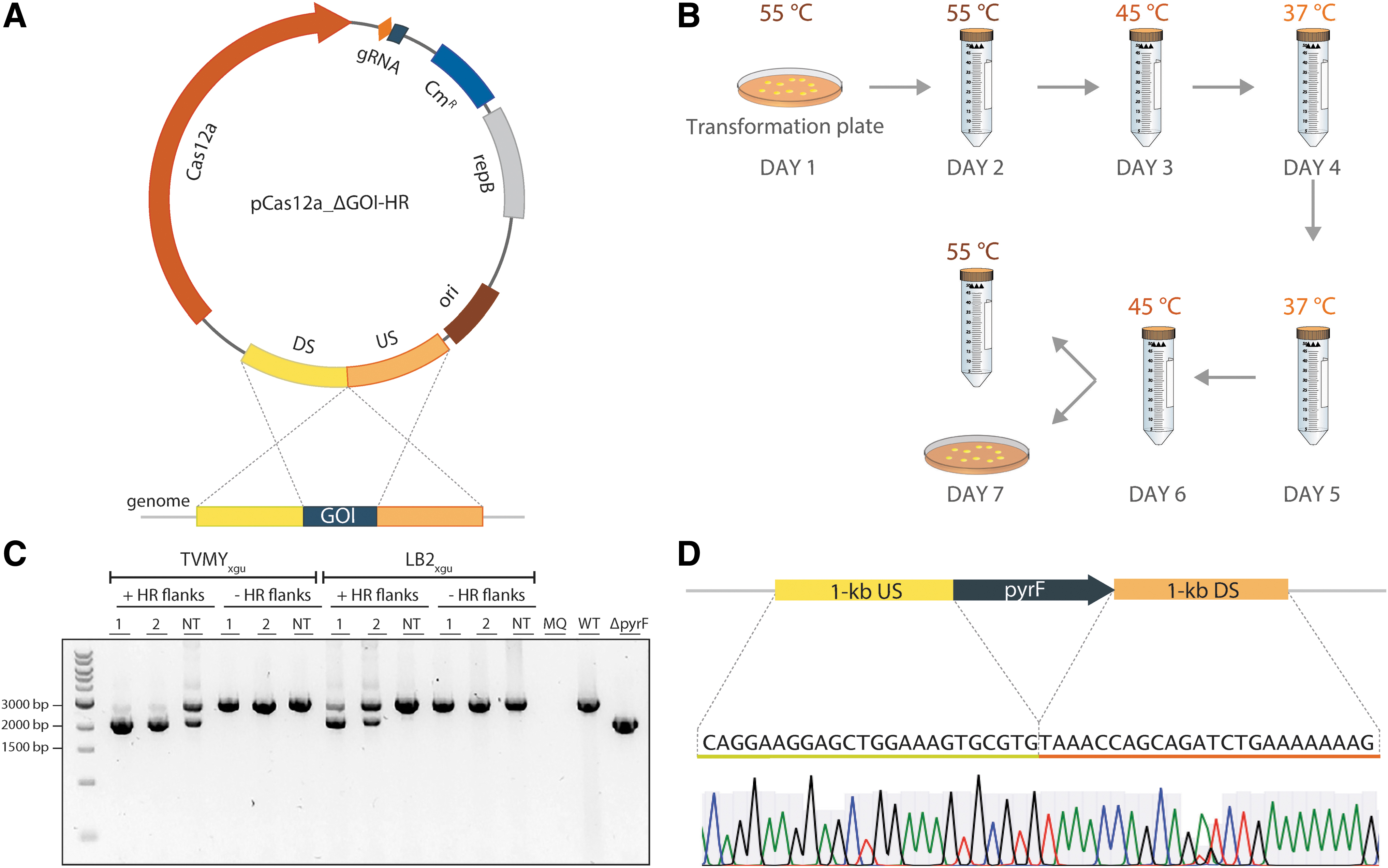
Transformation of HR-FnCas12a mediated editing plasmids in B. smithii ET 138.
After transforming the constructs into B. smithii ET 138 ΔsigF ΔhsdR at 55°C, one verified transformant per construct was used to inoculate two different selection media supplemented with uracil (rich LB2xgu and minimal TVMYxgu) to complement the auxotrophy in the case of a successful pyrF gene deletion. After growth at 55°C for 24 h, cells were transferred stepwise every 24 h to fresh media while gradually lowering the culturing temperature from 55°C to 45°C to 37°C in order to allow for gradual adjustment of the cellular metabolism (Fig. 3B).
As expected, PCR analysis of the genomic DNA extracted from all the cultures of cells harboring the pFnCas12a_Sp1/Sp2/NT, but lacking the HR flanks, showed the presence of the pyrF wild-type bands (Fig. 3C). In the presence of the HR flanks, however, population PCR analysis of the genomic DNA extracted from the TVMYxgu liquid cultures of cells harboring the pFnCas12a_ΔpyrF-HR_Sp1 or the pFnCas12a_ΔpyrF-HR_Sp2 editing plasmids clearly indicated the presence of mostly (∼90%) mutant amplicons (Fig. 3C). In addition, despite the lack of counterselective pressure, PCR analysis of the genomic DNA extracted from the minimal TVMYxgu liquid cultures of cells harboring the pFnCas12a_ΔpyrF-HR_NT plasmid after incubation also showed the presence of both wild-type and mutant bands (Fig. 3C). In contrast, PCR on genomic DNA extracted from the rich LB2 liquid medium cultures harboring the pFnCas12a_ΔpyrF-HR_NT plasmid did not show the presence of any pyrF knockout bands, as was to be expected (Fig. 3C). On the other hand, PCR on genomic DNA extracted from the LB2-grown cultures harboring the pFnCas12a_ΔpyrF-HR_Sp1 or pFnCas12a_ΔpyrF-HR_Sp2 editing plasmids showed the presence of mixed genotype bands (Fig. 3C). The striking difference in the density of the knockout bands between the cultures harboring the targeting and the NT plasmids underlines the significant contribution of FnCas12a-based counterselection to the efficiency of the genome editing tool.
The above-mentioned cultures were plated on corresponding media and incubated overnight at 55°C to screen for clean deletion mutants. We randomly selected 10 colonies from each plate, and used colony PCR to confirm the desired pyrF deletion. It was found that 9/10 of the tested pFnCas12a_ΔpyrF-HR_Sp1 colonies and 6/10 of the tested pFnCas12a_ΔpyrF-HR_Sp2 colonies from the TVMYxgu medium plates were clean pyrF gene deletion mutants (Supplementary Fig. S3). In addition, 7/10 of the tested pFnCas12a_ΔpyrF-HR_Sp1 colonies and 8/10 of the pFnCas12a_ΔpyrF-HR_Sp2 harboring colonies from the LB2xgu medium plates were clean pyrF gene deletion mutants (Supplementary Fig. S3). As expected, editing efficiencies were significantly lower without counterselection (i.e., in the TVMYxgu cultures harboring the pFnCas12a_ΔpyrF-HR_NT plasmid). In that case, analysis of 10 randomly selected colonies revealed that 1/10 was a clean ΔpyrF mutant and 2/10 had a mixed genotype. The gene deletion was verified by sequencing using specific primers on the genomic DNA isolated from three representative colonies (Supplementary Fig. 3D). The phenotype of three representative ΔpyrF mutant colonies was further verified by their ability to grow on uracil-containing plates supplemented with 5-FOA (not shown).
To use the HR-FnCas12a tool effectively in B. smithii for iterative genome engineering, a cured plasmid-free strain is desired for gene editing cycles with multiple vectors. For this purpose, a sequence-verified ΔpyrF mutant was grown in TVMYxgu medium without antibiotic for three iterative inoculation cycles of 8–16 h each. The final culture was inoculated on TVMYxgu plates with and without antibiotic (chloramphenicol). Colony PCR analysis with plasmid-specific primers on 6/7 tested colonies confirmed loss of the plasmid (Supplementary Fig. S4). The six plasmid-cured B. smithii ET 138 ΔsigF ΔhsdR ΔpyrF colonies were also verified to be uracil auxotroph and 5-FOA resistant (not shown).
Next, we aimed to optimize the efficiency of the system further in order to reduce the number of culturing steps and the total time for the editing process. To this end, a small amount from the glycerol stock of B. smithii cells harboring the pFnCas12a_ΔpyrF-HR_Sp1 plasmid was plated and incubated at 45°C overnight. Single colonies from the plate were used to inoculate TVMYxgu liquid media and incubated overnight at 37°C. The following day, the cultures were plated on corresponding media and incubated overnight at 45°C. We genotyped 10 randomly selected colonies that survived FnCas12a targeting from each plate, through colony PCR and sequencing. Interestingly, all the tested colonies (10/10) from the TVMYxgu plate were ΔpyrF mutants (Supplementary Fig. S5). Again, the ΔpyrF genotypes were verified by sequencing and the phenotypes for their ability to grow on TVMYxgu plates supplemented with 5-FOA (not shown). Notably, the total time required from the first transformation to successfully obtaining a B. smithii mutant using the system developed here is as short as 3 days.
Discussion
Our work on the temperature and pH stability and salt concentration tolerance of Cas12a nucleases expands our knowledge on the biochemical conditions at which Cas12a can be used. We show that the thermostability of the Cas12a nucleases strongly depends on the association with an appropriate guide RNA. Among the Cas12a nucleases tested, FnCas12a was shown to be most thermostable. Given the relatively high degree of both sequence and structural homology of LbCas12a and AsCas12a with FnCas12a (42% sequence identity with LbCas12a and 35% sequence identity with AsCas12a), it is hard to pinpoint a specific feature that confers the higher thermostability to FnCas12a.
Similar to Cas9 proteins from class 2 type II CRISPR systems, Cas12a proteins from class 2 type V-A CRISPR systems have been used for genome engineering in different bacterial species.7,8,13,16,38,39 Thermophiles have various advantages as production hosts for bio-based industries over current mesophilic model organisms. However, their limited genetic toolboxes often hamper their development into efficient cell factories. Previously, SpCas9- and thermostable ThermoCas9/GeoCas9-based systems have been demonstrated to be suitable Cas9-based tools for engineering thermophilic bacteria.19,20,40,41 To date, all genome engineering applications in thermophilic microorganisms have been based on Cas9.
A Cas12a-based genome editing tool is a complementary addition to the CRISPR-based bacterial genome engineering toolbox, and offers distinct advantages over using a Cas9-based system. First, Cas12a recognizes TTTV PAM sequences, 10 whereas SpCas9 recognizes an NGG PAM sequence. 4 This increases the number of target sites within a genome, allowing for precise selection of the cleavage site. This is particularly handy for generation of point mutations and when targeting short intergenic regions. Second, in some organisms, inherent toxicity to SpCas9 has been reported. In such cases, Cas12a is an excellent alternative. 7 Finally, it is possible to exploit the pre-crRNA processing activity of Cas12a for rapid and facile multiplexing. 42
During our study, we observed guide-dependent differences in FnCas12a targeting efficiency. This could be attributed to sequence and structural features of the spacer-derived flexible part of the crRNA, such as GC content, Tm, and secondary structure. 35
The HR-FnCas12a counterselection-based genome editing tool developed here could potentially also be used for generating gene insertions and single nucleotide substitutions. Furthermore, the HR-Cas12a systems eliminate the need of an antibiotic gene as a selection marker or the presence of chromosome scar sites (e.g., LoxP recognition sites). It is substantially more time-effective compared to the previously established lacZ-based and SpCas9-based counterselection systems.19,23 It only takes 3–4 days from transformation to obtaining a clean deletion mutant, including the plasmid curing step, while maintaining a high efficiency. The efficiency of obtaining mutants is similar to that of the previously developed HR-Cas9 system. 19
Conclusion
We developed a single-plasmid system coupling HR with Cas12a-based counterselection to allow for efficient gene editing in a moderate thermophilic bacterium, B. smithii ET 138. Based on in vitro analyses of temperature stability and activity of the tested Cas12a variants, we selected FnCas12a for use in genome editing. Accordingly, we developed an editing system where mutants are generated via HR events at elevated temperatures (>45°C) before Cas12a-induced counterselection takes place at 37°C for FnCas12a. This tool is potentially applicable to organisms with an active HR pathway and a growth temperature range covering at least 15–45°C, further expanding the range of organisms in which CRISPR-based editing can be applied.
Footnotes
Acknowledgments
We would like to thank Tess Hoogeboom for her technical assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
J.v.d.O. is supported by the NWO/TOP grant 714.015.001. M.M., G.B.R., J.L.C., and R.T.F. are employed by New England Biolabs. R.v.K. is employed by Corbion.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.