
Abstract
A multitude of molecular interactions with chromatin governs various chromosomal functions in cells. Insights into the molecular compositions at specific genomic regions are pivotal to deepen our understanding of regulatory mechanisms and the pathogenesis of disorders caused by the abnormal regulation of genes. The locus-specific purification of genomic DNA using the clustered regularly interspaced short palindromic repeats (CRISPR) system enables the isolation of target genomic regions for identification of bound interacting molecules. This CRISPR-based DNA purification method has many applications. In this study, we present an overview of the CRISPR-based DNA purification methodologies as well as recent applications.
Introduction
In the field of chromosome biology, it is an enigma how genomic events are regulated so precisely. A large number of molecular interactions occur with chromatin to modulate gene transcription, DNA replication or repair, and other critical genomic processes.1–3 Identification of these interacting components can advance the understanding of the molecular mechanisms underlying these genomic events.
The most comprehensive and unbiased approach to achieve this goal is biochemical isolation of a specific genome region while retaining the molecular interactions. Several methods have been developed for this purpose. A locus of interest can be isolated by insertion of recognition sequences of DNA-binding molecules, expression of the DNA-binding molecules in the cells, fragmentation of chromatin by sonication or other methods, followed by affinity purification of the DNA-binding molecules bound to the target locus.4–14 Cross-linking can be performed, if necessary. The locus tagged with the recognition sequences of the DNA-binding molecules can also be isolated by cross-linking, if necessary, fragmentation, and incubation with recombinant or synthetic DNA-binding molecules before affinity purification of the DNA-binding molecules bound to the target locus. 15 However, this approach is tedious and requires the production of cells that retain the recognition sequences of exogenous DNA-binding molecules.
To avoid this tedious step, the proteomics of isolated chromatin (PICh) assay, employing locked nucleic acids containing biotinylated oligonucleotides, can be used to isolate telomeres or centromeres for identification of their associated proteins.16–18 Previously, PICh was restricted to genomic regions containing multiple repeats or abundant targets due to a low isolation efficiency. Recent advancements enable the isolation of a more complex target of ribosomal DNAs. 19 However, PICh requires the reoptimization of probe sequences and purification conditions, thereby limiting its adaptability to different genomic targets. Thus, more flexible and efficient techniques are necessary for the purification of target loci.
The advancement of engineered DNA-binding molecules has made it easier to purify a locus of interest. The zinc finger proteins and transcription activator-like (TAL) proteins are pioneering examples of engineered DNA-binding molecules.20–23 More recently, the clustered regularly interspaced short palindromic repeats (CRISPR) system was adapted as a gene editing tool enabling the study of reverse genetics.24,25 Moreover, the development of nuclease-dead Cas9 (dCas9) has expanded the application of the CRISPR system beyond gene editing. 26 Since dCas9 retains its ability to bind to target sequences, the catalytically inactive CRISPR-dCas9 system is a programmable DNA-binding tool.27,28 The first report of a locus-specific CRISPR-based DNA purification method for subsequent identification of interacting molecules was published in 2013. 29 Since then, about 30 articles using the method have already been published (summarized in Fig. 1). The CRISPR-based DNA purification system utilizes the catalytically inactive CRISPR complex to form a tagging complex for isolation of the target locus. Interacting molecules bound to the locus of interest are identified first by isolation of the target locus by affinity purification, followed by subsequent identification of binding molecules. Thus, this is a powerful technique to purify the target locus while retaining the interaction between DNA and its binding components, which provides comprehensive insights into the interacting molecules by use of downstream biochemical applications.
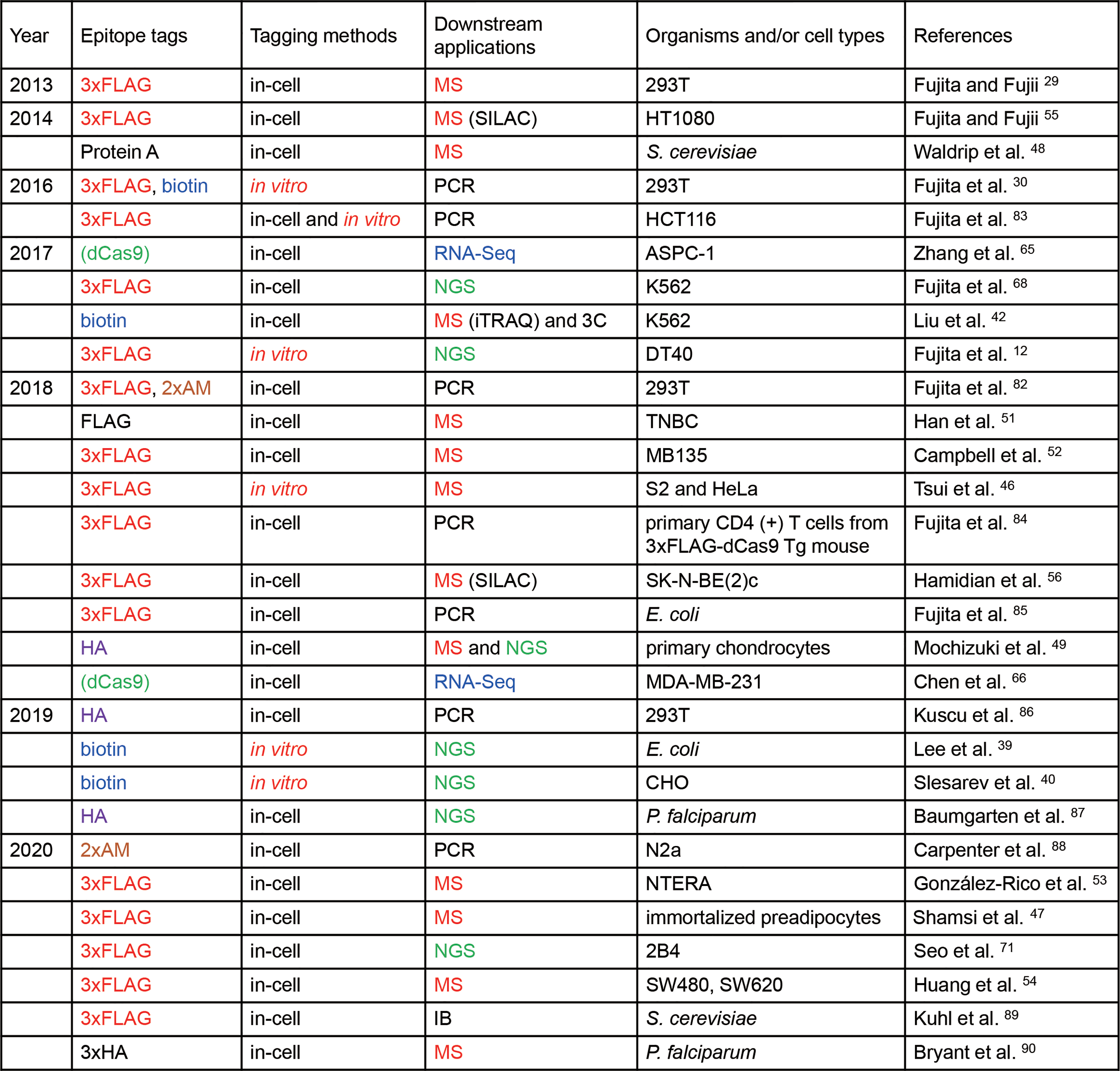
Publications using CRISPR-based purification of specific DNA species. Tags for affinity purification used in two or more publications are shown in different colors. In vitro tagging is highlighted in red. In the “Downstream applications” column, MS, RNA-Seq, and NGS are shown in red, blue, and green, respectively. All studies employed Sp-dCas9. One study used Sa-dCas9 in addition to Sp-dCas9. 82 Publications are listed in order of publication date. IB, immunoblot analysis. Color images are available online.
In this study, we aim to outline the recent advances of the locus-specific CRISPR purification method and its applications. This review will first introduce the strategies for affinity purification of a specific genomic locus. Subsequently, we describe the downstream applications to identify the interacting molecules and compare this method with others.
The Principles of Locus-Specific CRISPR-Based Purification
The CRISPR-based locus-specific DNA purification is based on the DNA-binding ability of catalytically inactive CRISPR and chromatin immunoprecipitation (ChIP) with or without the use of antibodies (Abs). The target DNA region can be tagged with the CRISPR complex inside of the cell or in vitro.29,30
In-cell tagging
The in-cell tagging approach labels a target locus with the CRISPR complex in the cell of interest by expression of the CRISPR complex (Fig. 2). If necessary, the chromatin-CRISPR complex can be cross-linked with formaldehyde or other cross-linkers to preserve the chromatin structure during purification. After chromatin fractionation, the chromatin DNA is fragmented by sonication or enzymatic digestion. In either case, shearing conditions of chromatin DNA should be optimized so that average length of chromatin DNA is between 1 and 2 kbp. The chromatin-CRISPR complex is affinity purified with antibodies (Abs) as well as other molecules recognizing either the tags fused to dCas9 or guide RNA (gRNA), or the anti-Cas9 Ab. If applicable, the cross-linking is reversed, and either mass spectrometry (MS) is performed for associated proteins, or next-generation sequencing (NGS) or microarray analysis for RNAs and DNAs. The detailed technical variations, considerations, and downstream applications are discussed as follows.
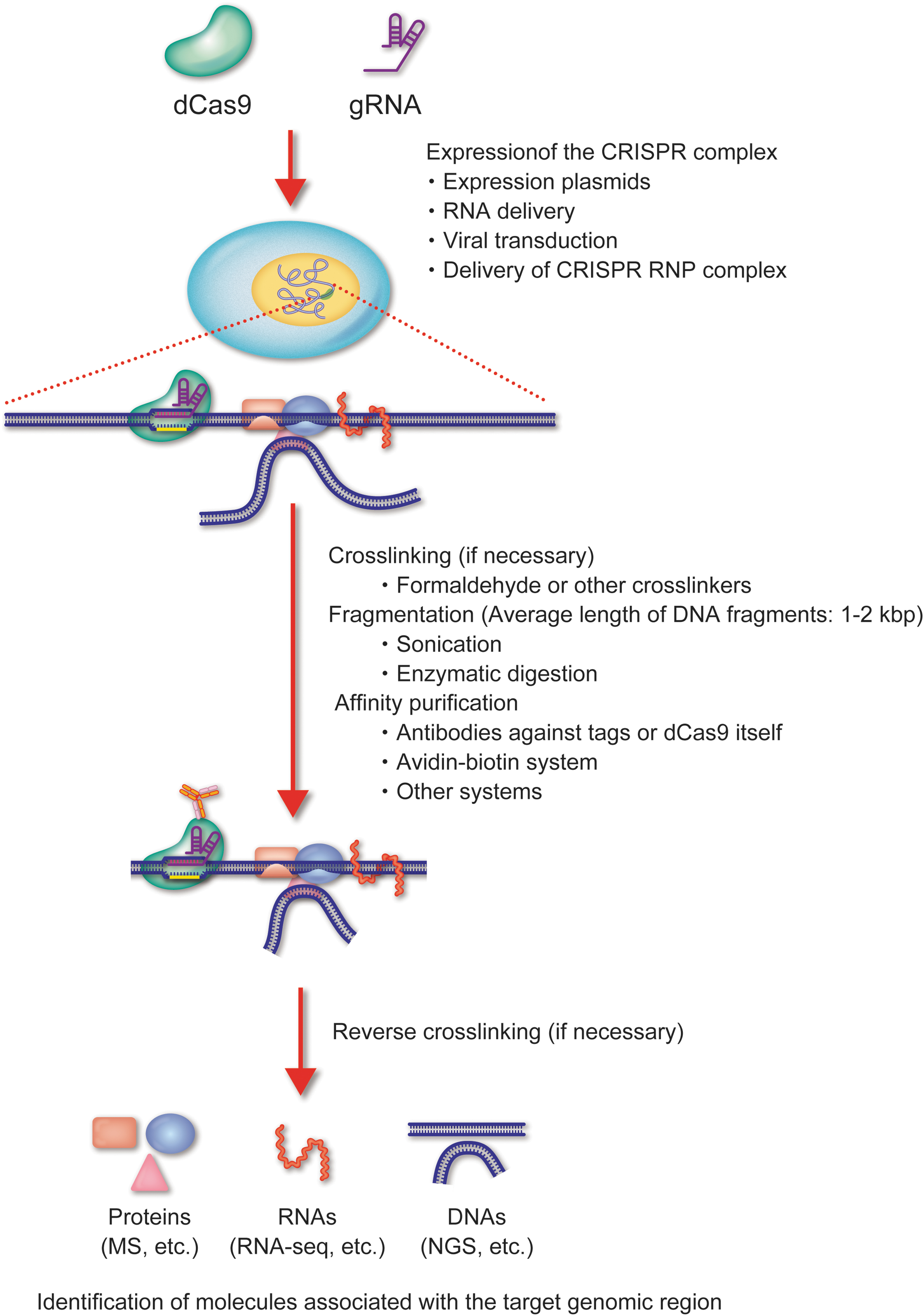
In-cell tagging of CRISPR-based DNA purification of a specific genomic region. Schematic of the in-cell tagging approach. A target locus is tagged in a cell with the CRISPR complex, and the chromatin-CRISPR complex is affinity purified. The molecules associated with the purified genomic region are identified by downstream analyses. Color images are available online.
The CRISPR machinery can be expressed transiently or stably. The transient expression of the CRISPR complex from transfected DNA or RNA, or transduction of CRISPR ribonucleoprotein (RNP) complexes is convenient for easy-to-transfect or easy-to-transduce cells. However, some cell lineages have poor expression levels or low transfection/transduction efficiency resulting in low yields of affinity purification. In such cases, the cells stably expressing the CRISPR complex should be generated using a retrovirus-, a lentivirus-, or an adeno-associated virus-derived expression system. Nowadays, researchers can easily obtain plasmids for expression in bacteria, budding yeast, and mammalian cells, including retroviral/lentiviral expression vectors for CRISPR-based DNA purification from Addgene.* In addition, transgenic mouse lines carrying dCas9 are options to purify the target locus from primary cells and tissues by expression of a gRNA(s) (RIKEN BioResource Center; No. RBRC09976, 10188-90).
In vitro tagging
The in vitro tagging approach captures the fragmented chromatin using a CRISPR-dCas9 complex, which is prepared in a test tube by mixing a synthesized gRNA and a recombinant dCas9 protein (Fig. 3). The advantage of in vitro tagging is that it does not require the expression of the CRISPR complex in the cell of interest. This is beneficial for the analysis of CRISPR-based DNA purification for several reasons (Fig. 4). First, the direct tagging of chromatin without the need to produce cells expressing the CRISPR complex makes it more time efficient and cost-effective. This is important in situations where expression of the CRISPR complex is difficult or impossible, such as in clinical specimens, pathogens, or specific lineages of primary cells and tissues. In addition to nuclear DNA, organelle DNA, including mitochondrial DNA (mtDNA) and chloroplast DNA, is a potential target of in vitro tagging approach. Because of the insufficient delivery of gRNA into organelles, in-cell tagging and purification of a specific region of intracellular mtDNA by the CRISPR system might be difficult.31,32 In this regard, the in vitro tagging could be a practical approach for the isolation of a region of organelle DNA, as it does not require the intracellular delivery of the CRISPR complex. Second, in vitro tagging avoids inherent risks of in-cell tagging, such as interfering with the genomic functions or altering chromatin accessibility.33,34 Moreover, the presence of the CRISPR complex at the locus might hinder the interaction of endogenous DNA-binding factors. The in vitro capture of the target locus after the fixation of chromatin eliminates these risks of in-cell tagging. However, compared with in-cell tagging, the in vitro method has a lower efficiency of affinity purification because the CRISPR complex needs to access the target locus, especially after fixation, whereby the chromatin is cross-linked. Thus, more cells might be required for identification of interacting molecules than with the in-cell tagging method.
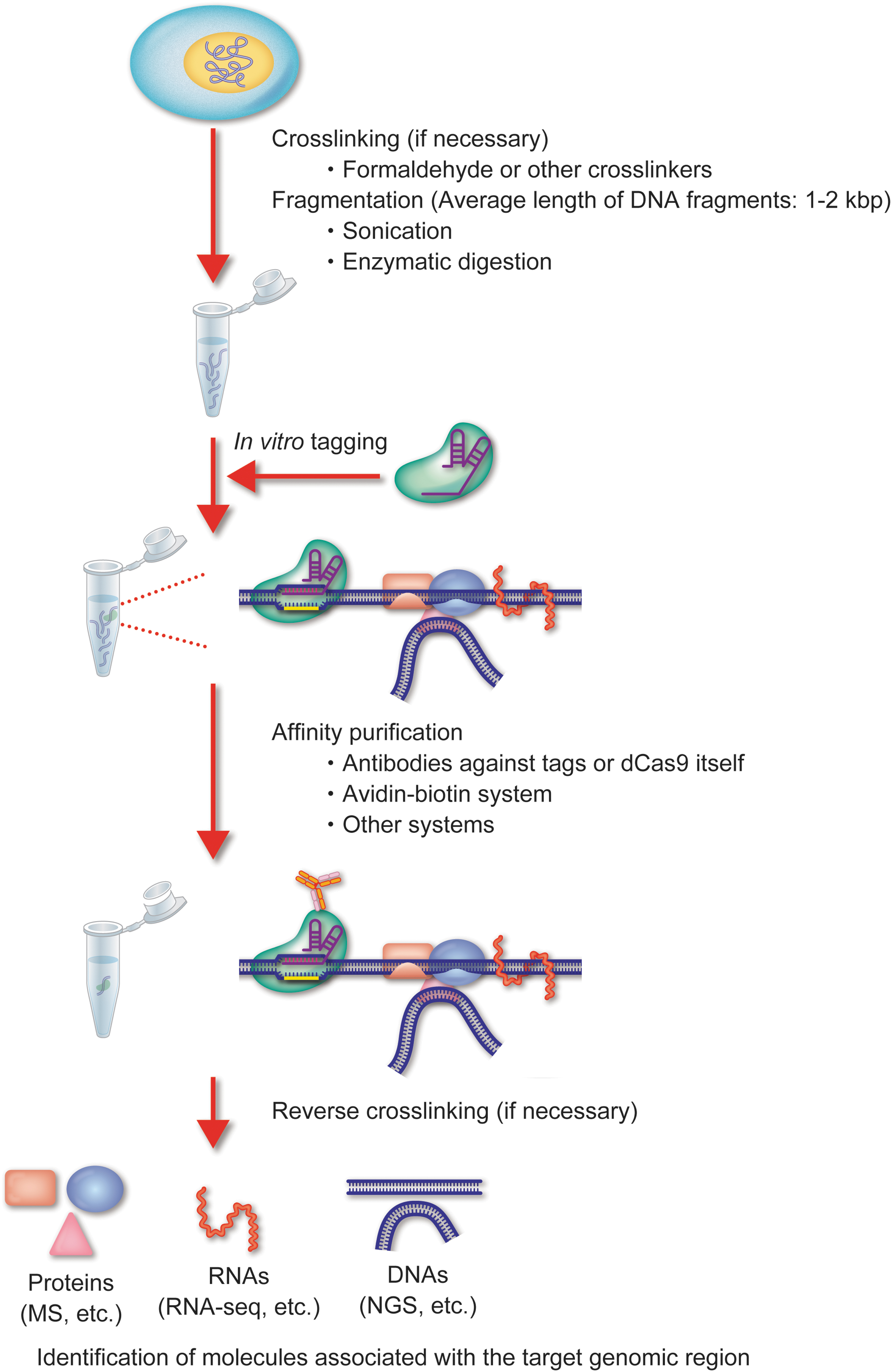
In vitro tagging of CRISPR-based DNA purification of a specific genomic region. Schematic of the in vitro tagging approach. A target DNA is captured by the CRISPR complex consisting of recombinant dCas9 and synthetic gRNA, lysed, DNA fragmented, and then purified. Color images are available online.
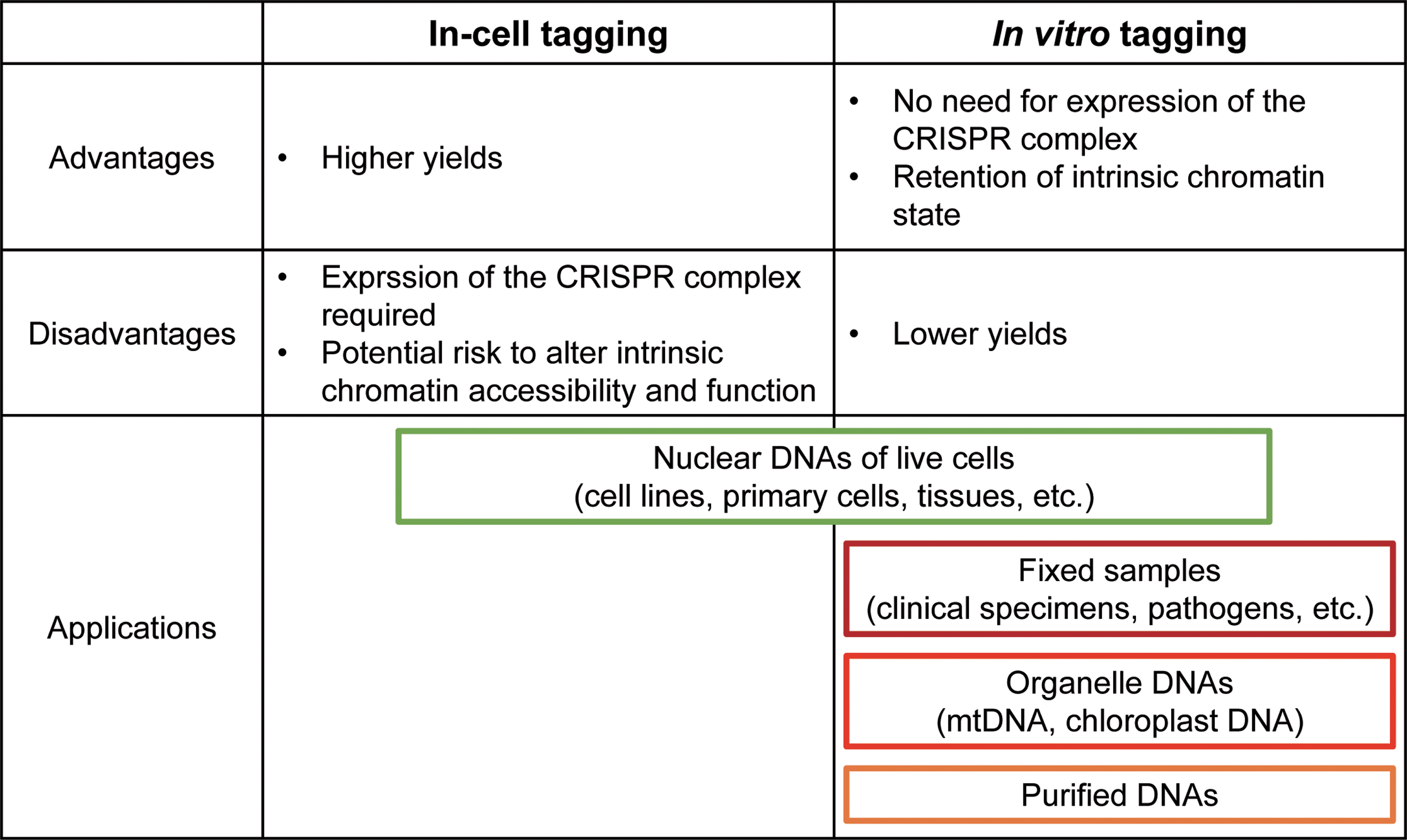
Comparison of in-cell and in vitro tagging systems. Summary of the advantages/disadvantages and application examples of in-cell and in vitro tagging methods. Color images are available online.
The in vitro tagging approach can also be used for enrichment of specific DNA species from a heterogenous population of purified DNA molecules, such as fragmented whole genomic and complementary DNAs. Sequencing the enriched species of interest is more time and cost-effective than a nonselective analysis method, such as whole-genome sequencing. Various target-enrichment methods have been widely used, such as multiplexed polymerase chain reaction (PCR), hybridization capture using complementary strands, and selective circularization using molecular inversion probes.35–38 The combination of CRISPR-based DNA enrichment and NGS can be used to detect the rare variants and to quantify the gene copy number.30,39,40 Because the CRISPR-based DNA-enrichment approach does not require a PCR amplification step before sequencing, this is one of the few enrichment techniques applicable to long-read sequencing. Long-read sequencing is a state-of-the-art technology for improved mapping accuracy, throughput, and identification of structural variants. 41 The combination of CRISPR-based DNA purification with long-read sequencing is, therefore, not only useful for biomedical research, but also for clinical diagnostics in the future.
Tag systems of CRISPR complex for affinity purification
CRISPR-based DNA purification of a specific locus is compatible with virtually any affinity purification systems by tagging of the CRISPR complex with 3 × FLAG, protein A, biotin, 2 × AM, HA (1 × and 3 × ) as well as using endogenous epitopes of the CRISPR complex (Fig. 1). It has been reported that the biotin tag system using biotinylation enzymes expressed in the target cells improves the specificity and efficiency of CRISPR-based DNA purification compared with the 1 × FLAG tag and anti-Cas9 Ab. 42 However, this needs to be investigated in comparison with other high-affinity tag systems, such as 3 × FLAG. Indeed, the 3 × FLAG tag was more specific than the biotin tag with the in vitro tagging system. 30 Because the competitive elution of 3 × FLAG fusion proteins using 3 × FLAG peptides is milder than the dissociation or denature condition of biotin-avidin bonds, the 3 × FLAG tag may reduce contamination derived from nonspecific binding of affinity purification beads, for example. At least, the 3 × FLAG and biotin-tagged CRISPR systems have been shown to achieve high yields of isolated interacting molecules.
Technical Considerations
Design of gRNAs
As mentioned earlier, the CRISPR complex can interfere with genomic functions and change chromatin accessibility when expressed in living cells. In addition, off-target binding of the CRISPR complexes can lead to nonspecific isolation of genomic regions. The design of target-specific gRNAs can overcome these issues and various design tools are available.43,44 Researchers can choose the appropriate gRNAs from the candidates listed from these design tools based on the following guidelines. First, to target promoter regions, gRNAs should be designed to bind several hundred base pairs upstream of the transcription start site to avoid interference with the recruitment of transcription factors and RNA polymerases by the CRISPR complex. In contrast, to target regulatory regions such as enhancers and silencers, the gRNA binding site should be located proximal to the regulatory sequence because they often have more distinct boundaries and binding of the CRISPR complex to the outside of those regulatory regions is less likely to interfere with their functions. Second, the target sites of gRNAs should not overlap with the binding sequences of endogenous DNA-binding molecules or sequences conserved among species, since these sequences may be functional regulatory regions. Third, on-target enrichment efficiency and specificity of gRNAs should be validated using CRISPR-based DNA purification combined with quantitative PCR (qPCR) and/or NGS. Finally, the genomic functions of target regions in the presence of the CRISPR complexes should be experimentally evaluated. If the gRNAs are not specific or interference with genomic functions is observed, the gRNAs or target sites should be reconsidered.
Experimental controls
One major concern of CRISPR-based DNA purification is the off-target binding of the CRISPR complex. To cancel out effects of the off-target binding and contamination of nonspecific molecules, proper control experiments should be conducted in parallel during CRISPR-based DNA purification. Experiments using cells lacking the gRNA target sequence provide a good reference for off-target binding and nonspecific interacting molecules. However, the deletion of the target genomic sequence is not always practical because it may require significant time and labor. Alternatively, the following comparison sets would be sufficient to exclude nonspecific binding molecules.
First, interacting molecules consistently detected with several different gRNAs for the same target genomic region are likely true positives because the off-target binding would differ among each gRNA. In parallel with this comparison set, the molecular profile obtained from cells expressing dCas9 alone or dCas9 plus an irrelevant gRNA would be helpful to exclude the contaminations of nonspecific binding molecules. Second, affinity purification using the same gRNA under different experimental conditions, for example, the presence or absence of stimulation and between different cell types, can identify the candidate molecules regulating the function of a genomic locus in a stimulation-dependent or cell type-specific manner. In the quantitative analysis, it is necessary to pay attention to the difference in efficiency of target enrichment between each condition. The difference in enrichment efficiency among different conditions is caused by various factors, such as chromatin accessibility and the expression levels of the CRISPR complex, leading to artifactual changes in the relative amounts of identified molecules. To avoid this problem, the observed quantitative changes can be normalized to the yield of a target region under each condition. In any case, however, additional independent analyses by other methods are required to unambiguously demonstrate the interaction of the candidate molecules with the target genomic region (details are discussed in the next section).
In the case of systems using enzymes biotinylating dCas9, nonspecific biotinylation of other proteins inside of cells may increase backgrounds. 42 Therefore, additional controls such as samples expressing only the biotinylation enzyme must be required.
Downstream Analyses of CRISPR-Based Purification of a Specific Locus
Identification of proteins associated with a specific locus
MS-based proteomics has been combined with CRISPR-based locus purification, enabling the comprehensive identification of interacting proteins. Many studies have applied the label-free “shotgun” MS approach to identify novel binding proteins in addition to known regulators of target regions.29,45–54 Quantitative proteomics using MS is more informative for characterizing the function of locus-specific binding proteins and their associated genomic regions. The relative amounts of associated proteins changing in response to extracellular stimulation can be detected by comparing the protein profile using stable isotope labeling by amino acids in cell culture (SILAC).55,56 It is postulated that the proteins that change in a stimulus-dependent manner may play essential roles in regulating gene function, that is, are candidates of regulator proteins. Compared with SILAC, the isobaric labeling strategies, such as tandem mass tags and isobaric tags for relative and absolute quantitation (iTRAQ), enable the simultaneous analysis of more samples, thereby increasing its throughput. The multiplex capacity of iTRAQ allows the sorting of locus-specific, off-target, and nonspecific binding proteins from a pool of proteins identified by CRISPR-based locus purification. This was achieved by comparison with multiple experimental controls, including CRISPR-based locus purification from cells lacking a gRNA target sequence, or cells expressing either dCas9 alone, or dCas9 with a gRNA targeting a different site. 42
In addition to the detection of molecules associated with a specific locus, a potential application of CRISPR-based locus purification is the detection of novel post-translational modifications (PTMs) of proteins, such as histones, transcription factors, and others. A vast diversity of PTMs and its combination on proteins contributes to gene transcription, DNA replication, and DNA repair.57,58 MS of cell extracts can identify and quantify the relative abundance of single and combinatorial PTMs in a large-scale manner. 59 However, this analysis cannot detect the relatively low-abundant PTMs restricted in a specific genomic region and define its distribution. Enrichment of proteins at a specific genomic region by CRISPR-based locus purification can allow the detection of rare and locus-specific PTM combinations. Furthermore, their location information would help link the PTMs of proteins with genomic roles.
After detecting candidate proteins interacting with the locus of interest by CRISPR-based locus purification, further analysis is necessary to confirm the interaction and reveal functional relationships. In this respect, conventional ChIP is a powerful technique to detect the DNA–protein interactions in cells. Modern ChIP-based methods profile the genome-wide distribution of given DNA-binding proteins by combining with NGS or DNA microarray analysis. However, since the ChIP assay depends on the affinity and specificity of Ab in use, its application is restricted to the interaction with a known target protein; therefore, a stoichiometric comparison between different proteins is limited. Thus, the CRISPR-based locus purification and ChIP methods are complementary approaches to reveal unknown mechanisms underlying genomic events.
Identification of RNAs associated with a specific locus
Accumulating evidence suggests that various types of RNA play essential roles in regulating genomic DNA functions such as gene transcription and genomic imprinting.60,61 Chromatin isolation by RNA purification (ChIRP) and capture hybridization analysis of RNA targets (CHART) are biochemical methods to identify chromatin-binding regions of an RNA of interest, similar to ChIP for proteins.62,63 Both methods are based on a hybridization-based strategy using biotin-tagged oligonucleotides to purify the target RNA and interacting chromatins. Similar to ChIP, these methods are based on the assumption that an RNA of interest interacts with the target genomic regions to be detected by PCR or other methods, unless nonbiased methods such as NGS are used.
Previously, a locus-specific DNA purification method using TAL has been combined with reverse transcription-qPCR (RT-qPCR) or RNA sequencing (RNA-Seq) to identify RNAs associated with a specific genomic region.6,64 More recently, the CRISPR-based locus purification method combined with RNA-Seq has been used to detect various types of RNAs, including a circular RNA interacting with the FLI1 promoter and a micro-RNA interacting with the IGF2 promoter.65,66 These methods are practical tools to reveal unknown RNA regulators of genomic functions.
Identification of DNAs associated with a specific locus
Genomic DNA, compacted into nucleosomes, is further compartmentalized to organize the nuclear territories. This 3D genome architecture plays important roles in genomic functions. 67 In particular, inter- or intrachromosomal regulatory elements, such as enhancers and silencers, interact with target genes by forming long-range DNA loops, resulting in distal gene regulation. The combined CRISPR-based locus purification and NGS analysis enables the identification of genomic regions associated with a specific locus without the need for a ligation step. In this approach, a target locus is tagged in-cell or in vitro with a CRISPR complex, affinity purified, followed by subsequent NGS analysis to identify the interacting regions.12,68 This analysis results in the detection of many interacting genomic regions, which are candidates of regulatory elements of a target gene. Additional experimental controls and analyses are required to narrow down these candidates. These include negative control cells to compare the interacting regions, characterization of epigenetic markers, analysis of open chromatin structures detected with formaldehyde-associated isolation of regulatory elements (FAIRE) or assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-Seq), and deletion of candidate genomic regions.69,70 For example, the combined approach of CRISPR-based locus purification and NGS analysis identified enhancer regions overlapping with open chromatin regions and epigenetic markers of H3K4me1 and H3K27Ac. Moreover, the deletion of these accessible regions resulted in the downregulation of target genes.12,49,71 Together, these studies have demonstrated the capability of the CRISPR-based approach for identification of locus-specific interactions between a promoter and an enhancer(s).
The chromosome conformation capture (3C), and its derivative methods, have advanced the understanding of genome interaction and chromatin architecture.72–76 The basic concept behind the 3C-based methods is that the frequency of ligation correlates with the physical DNA–DNA interactions. However, because this is an indirect detection approach, there is a potential risk of detecting not only physical interactions but also accessible genomic regions, thereby occasionally leading to discrepancies with other techniques.77,78 Several methods have been developed to detect the physical genome interactions in a ligation-free manner, including genome architecture mapping (GAM), split-pool recognition of interactions by tag extension (SPRITE), and chromatin interaction analysis via droplet-based and barcode-linked sequencing (ChIA-Drop).79–81 GAM measures the statistical proximities of chromosomes by sequencing the DNA extracted from ultrathin cryosectioned nuclear slices. SPRITE includes repeated rounds of splitting and barcoding of individual chromatin complexes, and subsequent identification of the interacting genomic regions by matching the terminal barcodes. In the case of ChIA-Drop, amplicons arising from gel-bead-in-emulsion (GEM) droplets of each chromatin complex are tracked with a barcode sequence. These methods allow the genome-wide mapping of simultaneous contacts of chromatin; in contrast, the CRISPR-based locus purification approach detects multiple genomic regions interacting with one target locus. Advantageously, it does not require specialized equipment nor training to perform because it is a similar procedure to ChIP-Seq after tagging the target locus with the CRISPR complex. In summary, the combination of CRISPR-based locus purification and NGS is a practical and cost-effective approach to identify regulatory elements interacting with a genomic region of interest.
Concluding Remarks
There is a rising consensus that macromolecules consisting of numerous proteins, RNAs, and DNAs orchestrate the series of nuclear events such as the chromatin organization and gene transcription. To help define the regulatory principles, we need to consider both the role of single molecule and the global control of genomic functions. Purification of a specific genomic locus was previously hindered by technological limitations; however, the advances of engineered DNA-binding molecules have provided a feasible and flexible approach. In this study, we described the recent advances of CRISPR-based DNA purification tools. The CRISPR-based DNA purification system, coupled with a high-throughput identification method, can allow a comprehensive identification of molecules interacting with a specific locus to provide snapshots of the genomic process. This approach will provide new insights into how the function of a specific locus is regulated. Therefore, the CRISPR-based DNA purification approach will facilitate the unbiased discovery of chromosome biology.
Footnotes
Authors' Contributions
All authors participated in the writing and editing of the article.
Author Disclosure Statement
H. Fujii and T.F. are inventors of granted patents owned by Osaka University for the technology of purification and subsequent analysis of specific DNA including genomic DNA with chromatin structure using insertion of a recognition sequence of an exogenous DNA-binding molecule such as LexA into the target DNA and affinity purification of the exogenous DNA-binding molecule bound to its recognition sequence (Patent name: “Method for isolating specific genomic regions”, Patent numbers: US 8,415,098, Japan 5,413,924). Osaka University licensed the patents to Epigeneron, Inc., H. Fujii and T.F. are also inventors of granted patents and a patent pending owned by Osaka University for the technology of purification and subsequent analysis of specific DNA including genomic DNA with chromatin structure using an engineered DNA-binding molecule including the CRISPR complex binding to the target DNA (Patent name: “Method for isolating specific genomic region using molecule binding specifically to endogenous DNA sequence,” Patent numbers: Japan 5,954,808, EP 2,963,113; Patent application number: WO2014/125668). Osaka University licensed the patents to Active Motif, Inc., and Epigeneron, Inc., H. Fujii and T.F. are co-founders of Epigeneron, Inc. H. Fujii and T.F. are a director and an advisor of Epigeneron, Inc., respectively. H. Fujii is a member of the advisory board of Addgene. H. Fujita has no conflicts of interest with the contents of this study.
Funding Information
This study was supported by the Karoji Memorial Fund for Medical Research (H. Fujii), Grant-in-Aid for Scientific Research (C) (No. 18K06176) (T.F.), and Grant-in-Aid for Scientific Research (B) (No. 15H04329) (T.F., H. Fujii), “Transcription Cycle” (No. 15H01354) (H. Fujii) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.