
Abstract
Functional genomic screening with CRISPR has provided a powerful and precise new way to interrogate the phenotypic consequences of gene manipulation in high-throughput, unbiased analyses. However, some experimental paradigms prove especially challenging and require carefully and appropriately adapted screening approaches. In particular, negative selection (or sensitivity) screening, often the most experimentally desirable modality of screening, has remained a challenge in drug discovery. Here we assess whether our new, modular genome-wide pooled CRISPR library can improve negative selection CRISPR screening and add utility throughout the drug development pipeline. Our pooled library is split into three parts, allowing it to be scaled to accommodate the experimental challenges encountered during drug development, such as target identification using unlimited cell numbers compared with target identification studies for cell populations where cell numbers are limiting. To test our new library, we chose to look for drug–gene interactions using a well-described small molecule inhibitor targeting poly(ADP-ribose) polymerase 1 (PARP1), and in particular to identify genes which sensitise cells to this drug. We simulate hit identification and performance using each library partition and support these findings through orthogonal drug combination cell panel screening. We also compare our data with a recently published CRISPR sensitivity dataset obtained using the same PARP1 inhibitor. Overall, our data indicate that generating a comprehensive CRISPR knockout screening library where the number of guides can be scaled to suit the biological question being addressed allows a library to have multiple uses throughout the drug development pipeline, and that initial validation of hits can be achieved through high-throughput cell panels screens where clinical grade chemical or biological matter exist.
Introduction
Pooled CRISPR knock-out (CRISPRko) screens have successfully identified genome-wide collections of essential genes,1–4 as well as mechanisms of drug resistance and associated biomarkers.5–7 However, this technology is also a valid tool for the identification of biomarkers of drug sensitivity where perturbations increase the toxicity of a drug or another insult. Sensitivity screening, also known as negative selection screening, requires the identification of the loss of a key genotype or phenotype from the experimental population. This is a more challenging screen modality, as the experiment is looking to find the rarer event (i.e., loss of certain cell populations), which is contingent upon high penetrance of gene disruption and typically results in more complex datasets where the signal-to-noise ratio is greater. 8 Consequently, relatively fewer negative selection drug–gene interaction screens have been published,9–12 in contrast to many positive selection screens.13–15 While the rapid development of pooled lentivirus and deep-sequencing-led approaches have allowed us and others to exploit the positive selection approach in target identification (ID), drug mechanism of action analysis, and patient stratification focused screens, adapted tools are required to adequately capture the reciprocal interest in sensitivity screening.
Dual-direction and combined screening approaches have been of great value in improving screen datasets for negative selection screens6,16 These screens require an additional arm that provides gain-of-function information, and whilst powerful, they are experimentally complex and might not provide high-sensitivity loss-of-function genetic disruptions needed for rapid drug development. Adapting pooled phenotypic approaches 17 and simple haploid genetics18,19 has also increased screen power in some cases, but these specific examples required that the cell death pathways can be identified in a time-resolved way or use a restrictive cell line background, respectively, and therefore have limited generalized applicability. Much progress has been made in improving both the penetrance and the precision of CRISPR-based tools for screening, including the development of new algorithms to better predict guide sequences and modifications to the molecular infrastructure to increase editing rates,14,20,21 but robust screen outcomes for negative selection remain challenging. A common feature of existing algorithms for guide target site identification is the use of gene essentiality datasets to train them. However, the reductionist tendency of these algorithms often yields smaller, low complexity libraries.
Recognizing that screen success rarely uses a one-size-fits-all approach, here we have built a flexible pooled screening CRISPRko library that is partitioned into three parts to allow library design and complexity to meet the experimental challenges of a given individual screen. For instance, resistance screening could be viewed as a relatively low-risk endeavor in most cases, and screens can be successfully conducted using a lower complexity library with four guides targeting each gene. By contrast, negative selection screens require greater experimental power for robust hit ID, so a library with six or eight guides per gene could be deployed to accommodate the need for greater experimental power. We tested our library design of four guides per gene, six guides per gene, or eight guides per gene in a negative selection screen to identify genes that interact and specifically sensitize cells to the poly(ADP-ribose) polymerase 1 (PARP1) inhibitor olaparib. Some of the hits identified in the four-, six- and eight-guide library components are validated by high-throughput cell panel screening.
Materials and Methods
Cell lines
HT-29 cells (American Type Cell Culture) were cultured in McCoy's supplemented with 10% fetal bovine serum (all supplied by Gibco UK). Cells were routinely checked for mycoplasma and identity verified by short tandem repeat (STR) analysis.
Library generation
We generated three pooled, whole-genome guide RNA libraries using the guide RNA design algorithms from the Edit-R tool (Horizon Discovery). 22 Each single guide RNA (sgRNA) was synthesized (CustomArray, Inc.) using a modified trans-activating CRISPR RNA sequence, as described in Chen et al. 23 and Cross et al. 18 (5′-GTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTT-3′). An all-in-one lentivirus plasmid vector was used comprising a selection marker (puromycin resistance), the expression cassette for Cas9 and the sgRNA sequence. Each pooled sgRNA library was cloned into the vector backbone using a Gibson Assembly Master Mix kit (New England BioLabs) in accordance with the manufacturer's instructions. Library plasmids were purified using a Qiagen Plasmid Plus purification system in accordance with the manufacturer's instructions.
Lentivirus production
Lentivirus was produced as described in Le Sage et al. 6
Cell transduction and screening protocol
Cell culture and screening was conducted as described in Le Sage et al. 6 Deviations from the previously described protocol were the use of HT-29 cell line in the current study. A total of 1.5 × 108 cells were transduced; 48 h after library transduction, transduced and nontransduced cells were treated with puromycin at a final concentration of 0.6 μg/mL. Once selection was complete, puromycin was removed and cell populations were divided for treatments and replicates and maintained in multiple five-layer flasks (Falcon), counted, and reseeded at 5.5 × 107 cells per treatment every 3–4 days with appropriate treatments. Final pellets were harvested after 12 doublings of cells treated with DMSO. Genomic DNA was extracted using QIAamp DNA Blood Maxi kit (Qiagen). DNA concentration was determined using a Nanodrop spectrophotometer and at least 230 μg of genomic DNA for each sample was then amplified by PCR to generate amplicons of the sgRNA cassette using a forward primer, TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGU–[variable]–TGTGGAAAGGACGAAACACC; and a reverse primer, GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGATCAATTGCCGACCCCTCC. These amplicon samples were purified using Agencourt beads (Beckman) and deep sequenced on an Illumina NextSeq platform (Microsynth AG).
Data analysis
Raw next-generation sequencing libraries were evaluated for quality using FASTQC version 0.11.5. (Babraham Institute; Cambridge, UK). 24 Guide counts were obtained using an in-house customized version of k-mer counting, which took into account guide staggering from the experimental protocol. Briefly, guide-length fragments were trimmed and hashed before mapping with exact string counts from each file to provide raw counts for each guide found in the library. Guide counts were then normalized within each group (median based) and Log2 fold change was calculated to determine the change in abundance of each guide in each sample. DrugZ was used as the drug-interaction scoring tool, as described previously.25,26 Network analysis was conducted using EsyN and HumanMine with physical interaction. 27
Cell panel screening
Cell lines were grown in vendor recommended culture medium and STR-typed to verify identity. Cells were seeded into 384-well assay plates and compounds added after 24 h using a nine-point titration plus untreated control for the single agent assays or a 6 × 6 optimized dose matrix for the combinations. After a further 6 days of incubation, cell viability was assessed using CellTiter-Glo 2.0 (Promega). A baseline T0 measurement (at time of drug of addition) was also taken to enable calculation of growth inhibition, which enables the differentiation between cytostasis and cytotoxicity. Single agent responses were analyzed by calculating the area under the dose–response curve (response area) and combinations were analyzed using synergy score which was calculated using the Loewe additivity model using Horizon's proprietary Chalice Analyzer software. (For detailed methods see Kondo et al. 2002. 28 ) Pharmacogenomic analysis of response was performed using the mRNA expression and CRISPRko gene dependency data obtained from the Cancer Dependency Map (Depmap) portal release 2019q3. Correlation between drug response and PARP1 expression was analyzed by Pearson correlation. Cells were ranked according to drug sensitivity using Response Area and the top and bottom quartiles were assigned as responders or nonresponders, respectively. Significant differences in gene dependencies between these two populations were then identified using Fisher's exact test. Network analysis of the top 250 genes that sensitive cell lines were more dependent on was performed using EsyN and BioGRID with physical interaction, 27 and functional annotation and clustering was performed using the DAVID Bioinformatics Tool v6.8. 29
Results
We partitioned the guides in our library for synthesis across three independent arrays: one containing four guides (LibA), and a further two arrays containing two guides per gene each (LibB and LibC). When these library subsets are combined at the plasmid level in, they generate a library in which a total of eight guides targets each gene (Fig. 1). This approach facilitates screening using a library consisting of four, six, or eight guides, in accordance with the desired complexity. For the proof of concept screen, each library plasmid was combined in equimolar concentrations and packaged as a single lentiviral pool with eight guides per target gene (LibABC).
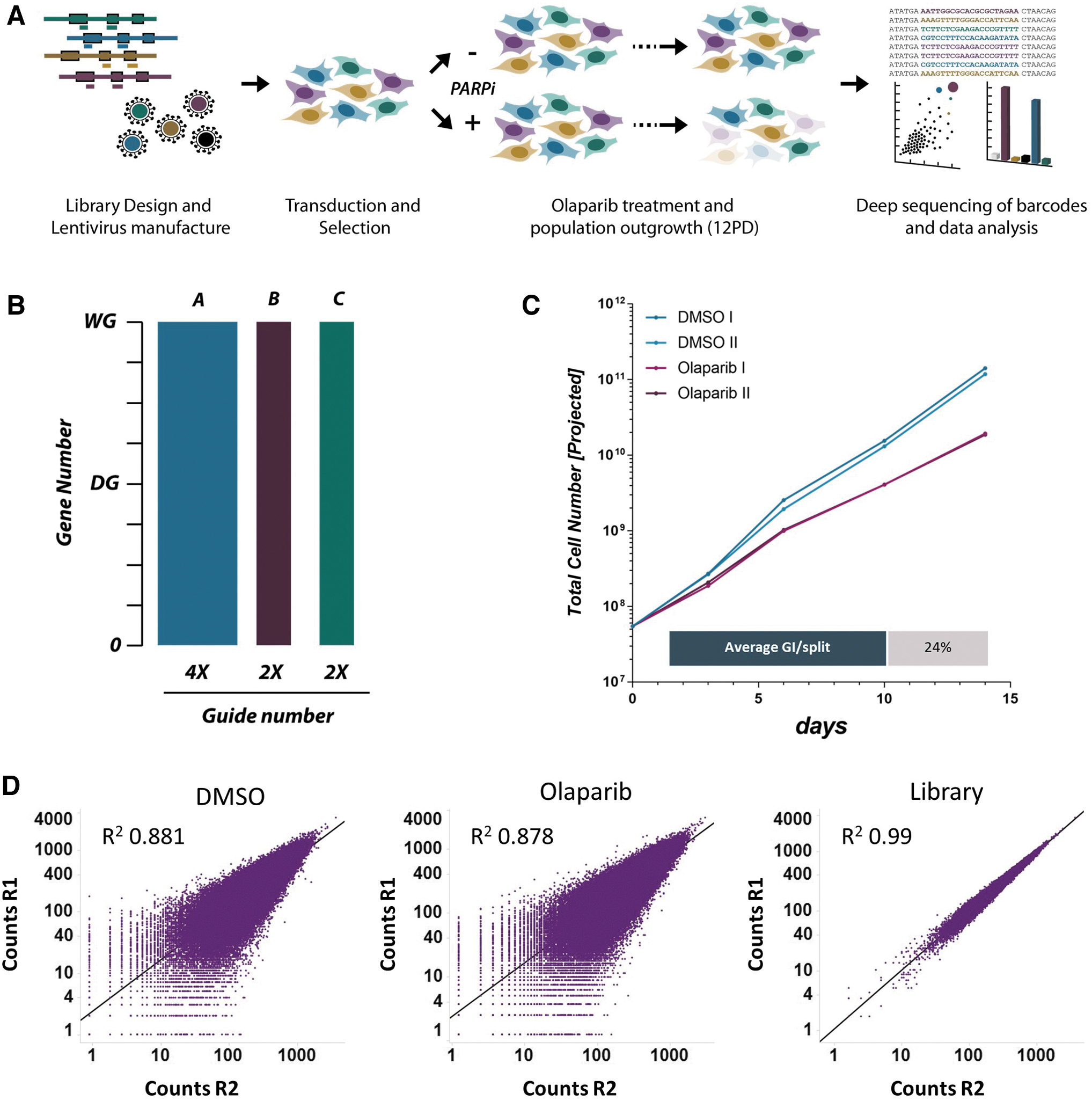
We tested this ABC pooled library in a drug–gene interaction screen using the PARP1 inhibitor olaparib (Fig. 1A). PARP1 inhibitors are well studied, and cells that have compromised BRCA1 or BRCA2 function are known to be sensitive to these inhibitors. However, other studies indicate the existence of BRCAness, where increased sensitivity to PARP1 inhibitors is evident in the absence of BRCA1 or BRCA2 mutations. 30 Moreover, mechanisms of response to PARP1 inhibition in tumors is etiologically dynamic and can result from diverse effects, including increasing DNA lesions resulting from loss-of-function of PARP1 and trapping of PARP at the loci of DNA damage. 31 Thus, our screen should be able to identify known sensitivity mechanisms to olaparib to check that our library is effective, and to potentially identify additional mechanisms of sensitivity.
HT-29 cells are a common-use colorectal carcinoma laboratory screening model, and these cells were infected with the ABC library at a low multiplicity of infection followed by antibiotic selection to eliminate untransduced cells. Following selection, replicate populations of cells were split off and treated with either DMSO (vehicle control) or a dose of 10 μM olaparib. Importantly, after each treatment phase (one passage), cells in each replicate were harvested and counted to monitor treatment response and maintain optimal dose effect. This was deemed to be a response rate of 20%–30% growth inhibition compared with control and was rigorously optimized in pilot experiments (data not shown; Fig. 1C). This growth inhibition provides both confidence of target engagement (which results in loss of cell viability) and a large assay window into which increments of effect resulting from CRISPR perturbation can be measured, allowing a suitable negative selection screening output.
Following the completion of the screen treatment phase, which was terminated at 12 population doublings of the vehicle control, samples were harvested and genomic DNA was extracted and subjected to amplicon-based sequencing to quantitatively identify genotype abundance in each sample. Replicates were analyzed side by side and showed high levels of concordance (Fig. 1D). Genes were annotated for known control groups, and the vehicle control sample was compared to baseline sequencing files derived from the plasmid samples ahead of lentivirus production. As we included nontargeting guides, positive controls (essential genes) and negative controls (nonessential genes) in our library, this comparison with plasmid samples allows a thorough qualification of expected effects on cell survival and fitness resulting from gene perturbation. Analysis of these control groups at the population level showed the expected behavior where essential genes were robustly lost from the population over time (Fig. 2A). Gene-level analysis showed drop out of essential targets up to 64-fold in some cases (Log2 −6); likewise, guide-level analysis showed low representation of all guides targeting essential genes at the screen endpoint. As expected, LibABC showed modest overall enrichment for non-targeting controls. 18 In silico comparison between each of the sub libraries was conducted to evaluate the performance of each module directly. This was determined by independent analysis of each of the library subsets and combinations thereof, to simulate the performance of a screen using LibA, LibAB and LibABC. The greatest overall drop-out and lowest variance of control essential genes was observed in the smallest library, LibA, as expected, since this library contains the highest-ranking guides (Fig. 2B). Interestingly, discovery of gene essentiality as measured by a receiver operating curve analysis was found to be robust with only two guides, but increasing guide numbers clearly improved this outcome (Fig. 2C). 32
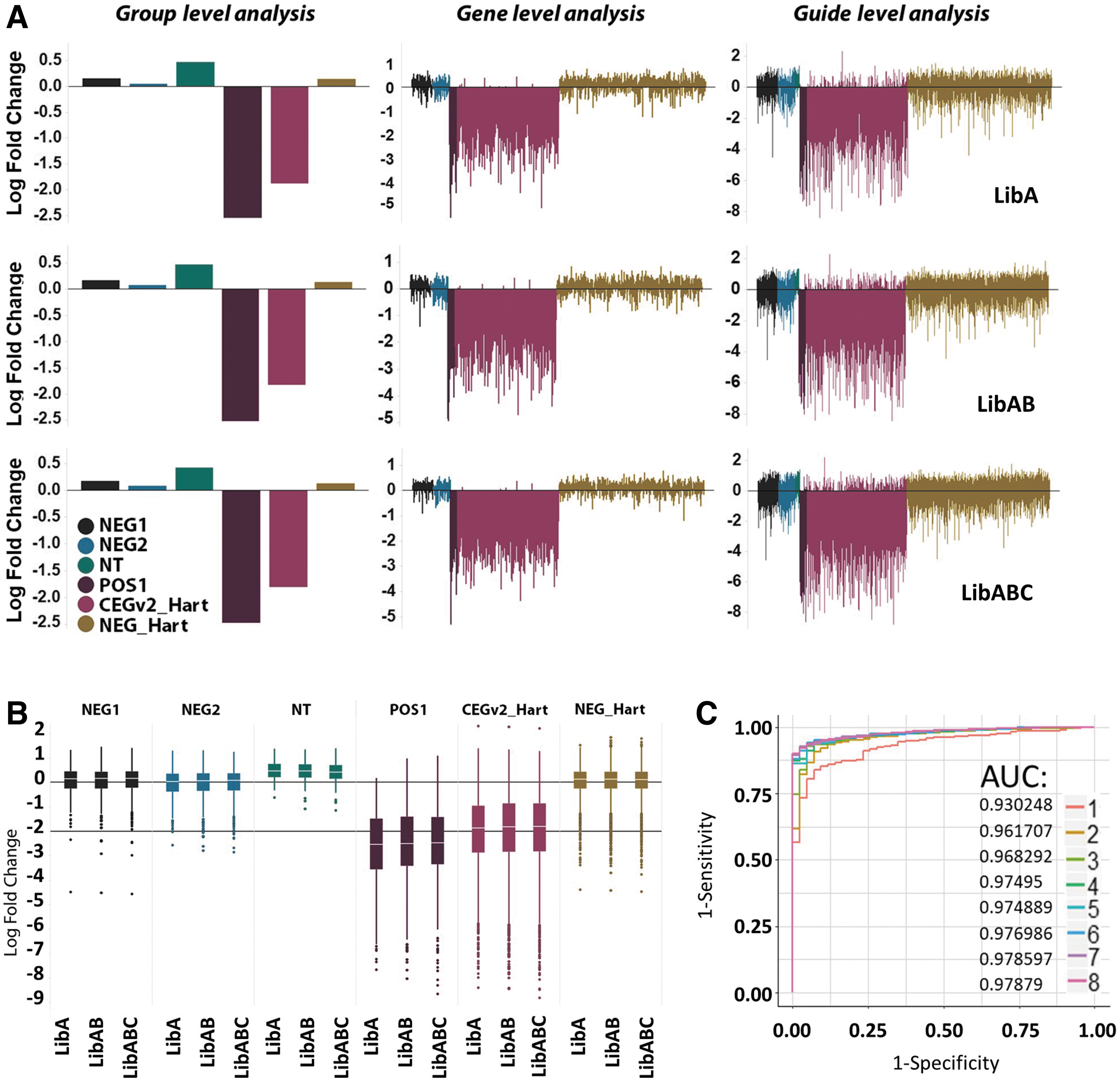
This result is reassuring for the quality control of the library. However, the identification of pan essential genes by CRISPRko is among the more straightforward of analytical tasks, since a defined list of expected essential genes exists. In order to explore the performance of our modular library in drug–gene interaction, we used the DrugZ analytical tool, 25 which for this data proved more sensitive than other methods, such as the more frequently used robust ranking aggregation tools (data not shown). 33 This analysis was conducted for each of the library modules. Discovery of sensitizing hits was surprisingly robust even with the low complexity LibA, which has four guides (Fig. 3A). We identified 39 hits with a false discovery rate <0.05 (Fig. 3B), including ATM and a large part of the Fanconi anemia (FA) pathway. Several other key agonists of DNA damage response (DDR) pathways such as MUS81, XRCC1, and POLB were also identified, which indicates a robust on-target effect in response to treatment with olaparib. All three components of ribonuclease H2 were found to be significantly depleted in the treated sample. More detailed guide-level analysis indicates a reproducible response for these top hits (Fig. 3C). These genes were also found as part of a previously published study that is similar to ours. 9 Comparing our data in HT-29 cells with the published data set 9 shows good concordance between the two studies (Fig. 3D).
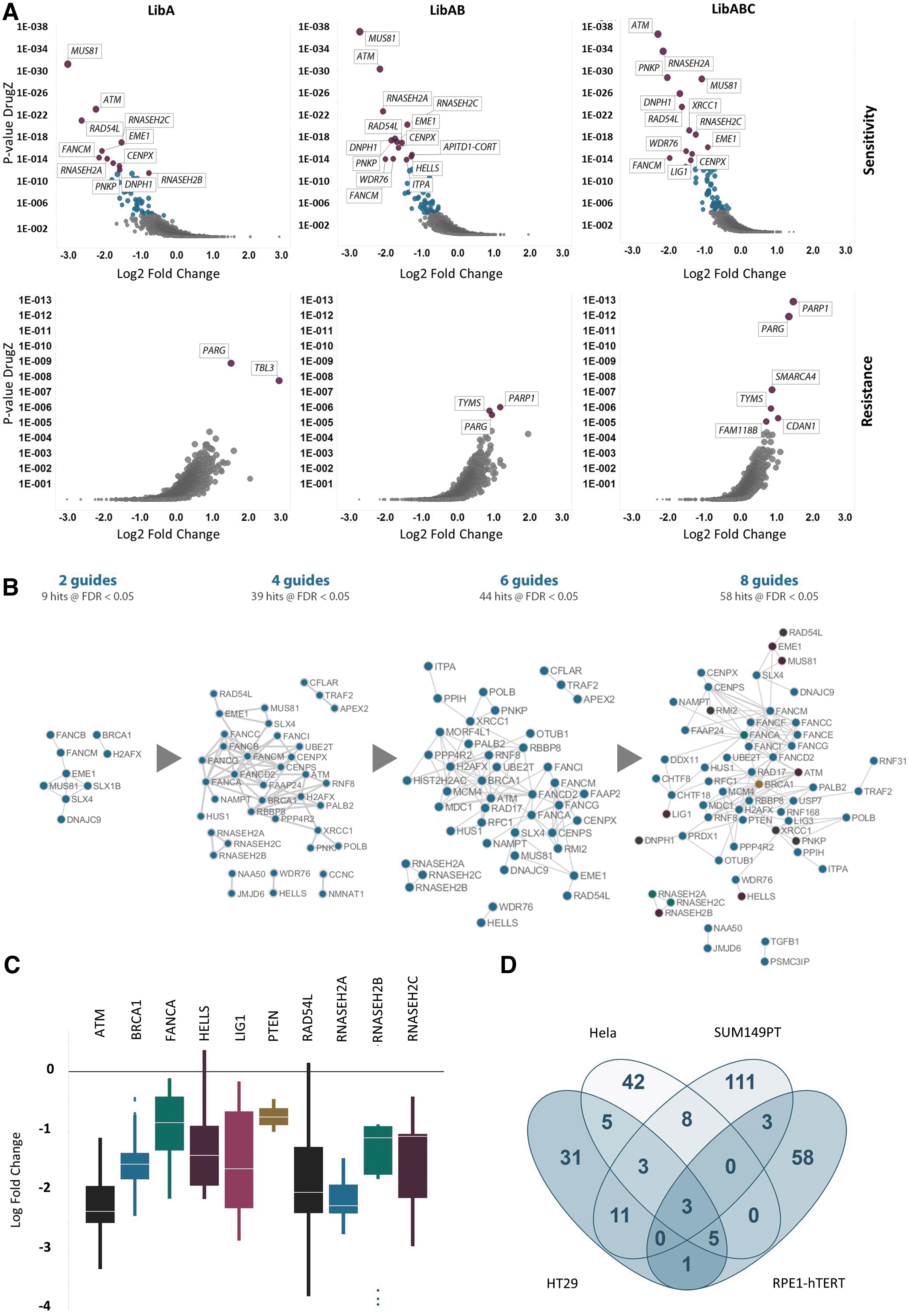
Although hit ID was found to be robust even with the simulated lower complexity library, a substantial increase of hits was observed with increasing numbers of guides. Up to 58 hits were found with the eight-guide library. Importantly, whilst BRCA1 was identified robustly in each library analysis, PTEN was found only when analyzing the screen with the eight-guide library (LibABC). Previous publications have indicated that cells lacking functional PTEN are more sensitive to PARP1 inhibitors, 34 and our data further supports these findings.
In addition to the analysis of genes that when lost sensitize to PARP inhibitors, we also used our data to look for resistance hits. Loss of PARG was a consistent hit found with all three simulated versions of the screen, with the highest representation of hits being evident with LibABC, which was able to identify PARP1 with substantially improved confidence (Fig. 3A). Overall, these observations illustrate the value of additional library complexity when looking for robust and maximum hit ID.
As our screen used a combined LibABC analyzed independently informatically, we sought additional validation for hits identified with each version of the component libraries. To do this, we used an orthogonal pharmacological cell panel screening approach. We ran a single agent screen with a viability readout following six days of drug treatment using a diverse panel of 326 cancer cell lines treated with olaparib and a second clinical-stage PARP inhibitor, talazoparib (BMN 673). The pattern of response across different tissue lineages was broadly similar for both compounds, and as previously reported, talazoparib was more potent than olaparib (Fig. 4A). 35 We used the publicly available genomic profile and genome-wide loss-of-function screening data for these cell lines in the DepMap portal to facilitate pharmacogenomic analysis of PARP inhibitor response. As expected, this revealed a significant positive correlation between PARP1 expression and inhibitor response (Fig. 4B), which is consistent with our finding that CRISPR-mediated knock out of PARP1 drives resistance to olaparib.
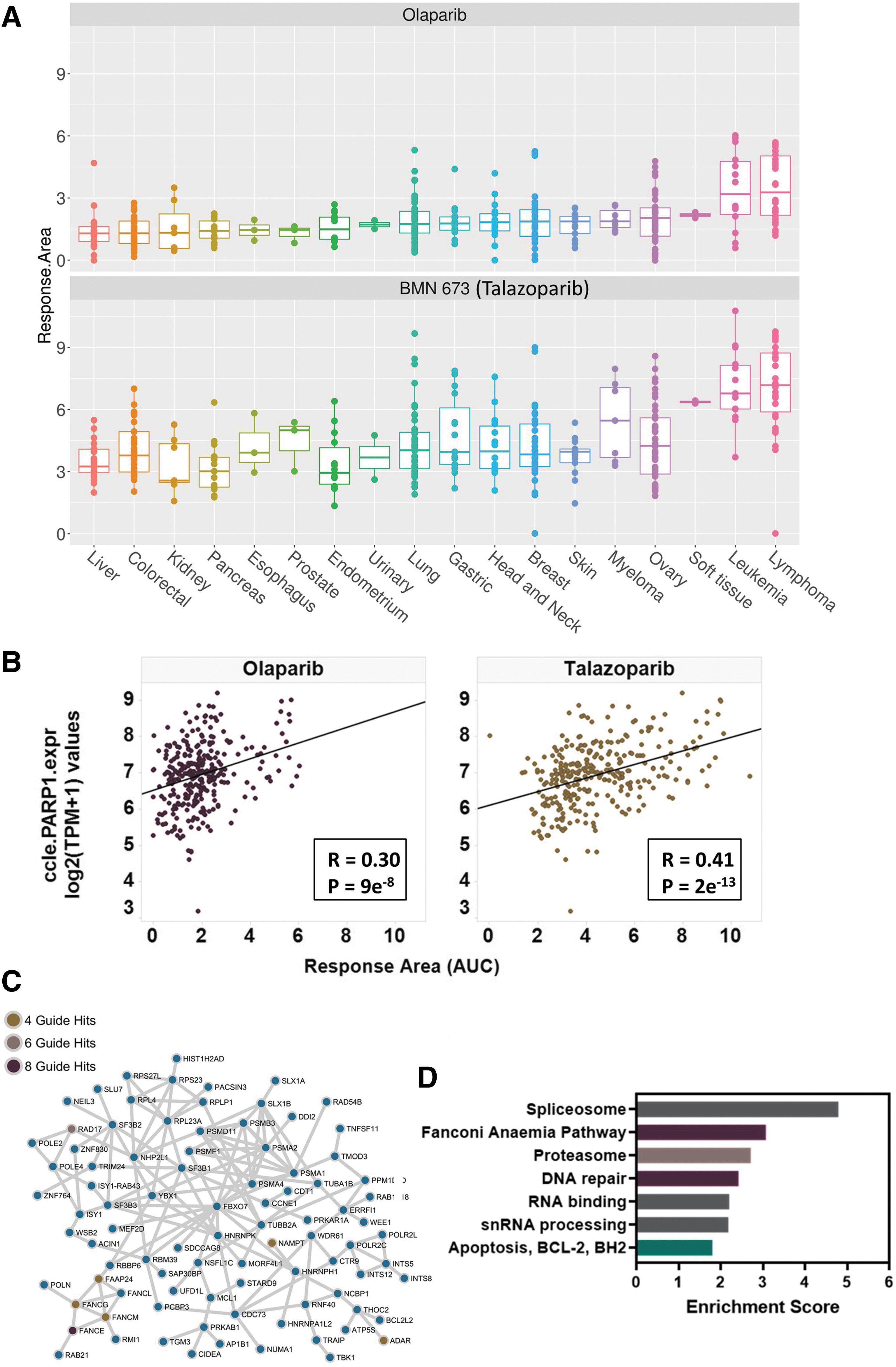
Pharmacogenomic analysis of PARP inhibitor single agent responses across a cell panel.
The integration of data obtained from drug sensitivity and loss-of-function screens is a valuable approach for identifying the underlying genes and pathways that govern drug responses.2,36 Therefore, to elucidate genes and pathways that could be important in modulating the cellular response to olaparib, we further used the DepMap CRISPR-mediated whole-genome loss-of-function data to explore associations between our cell panel drug sensitivity dataset and gene dependencies. This was achieved by categorizing cell lines in the cell panel screen as responders or nonresponders and then systematically searching for significant differences in gene dependencies between the sensitive and resistant populations. Strikingly, this revealed that olaparib sensitivity correlated with a greater dependence on genes or pathways that we and others identified using CRISPR sensitivity screens, including DNA damage and repair pathways and the spliceosome pathway (Fig. 4C and D). 9 Notably, the genes identified via the comparison of sensitive versus resistant cells included hits from the FA pathway that were identified using our lower complexity four-guide (FANCG, FANCM and FAAP24) and higher complexity eight-guide (FANCE) CRISPR libraries.
The identification of ATM (all libraries), BRD2 (LibAB and LibABC), and BCL2L1 (BCL-XL; LibABC only) as hits implicated the DDR pathway and BET- and BCL2-family members as potential therapeutic targets in combination with PARP1 inhibitors. To further validate these findings, we tested the ability of clinical-stage small molecule inhibitors against these targets to see whether there was synergy when used in combination with PARP1 inhibitors. We analyzed growth suppression in a panel of 10 cell lines that encompassed relevant cancer indications where PARP1 inhibitors are approved or are being evaluated in the clinic and two colorectal cancer cell lines, including HT-29, to compare with the data from our CRISPR screen (Fig. 5). The combinations of PARP inhibitors with ATM and other DDR pathway antagonists, such as inhibitors of ATR, CHK1, CHK2 and WEE1, displayed strong synergy independent of which pathway node was inhibited (Fig. 5). The inclusion of multiple inhibitors against different parts of the pathway and the use of different inhibitors against the same target confirms that the observed synergy was pathway- and target-specific, rather than owing to any compound-specific off-target effect. The combination of PARP inhibitors with the BET and BCL-2 inhibitors also exhibited robust and widespread synergy, although remarkably, synergy was only evident for the pan-BCL-XL/2 inhibitor ABT-263 with relatively little being observed for the more BCL-2-selective compound ABT-199 (Fig. 5). This could indicate that synergy is primarily driven through inhibition of BCL-XL either alone or in combination with BCL-2. Although PTEN, a negative regulator of the PI3K pathway, was identified as a sensitizing hit in the CRISPR screen, no well-characterized small molecule antagonists for PTEN exist. We therefore evaluated the effects of PI3K pathway modulation by combining PARP antagonists with a PI3K pathway inhibitor. This combination was generally not synergistic and is consistent with the hypothesis that upregulation (via PTEN loss) rather than downregulation of the PI3K pathway might be a greater driver of sensitization to PARP inhibitors.
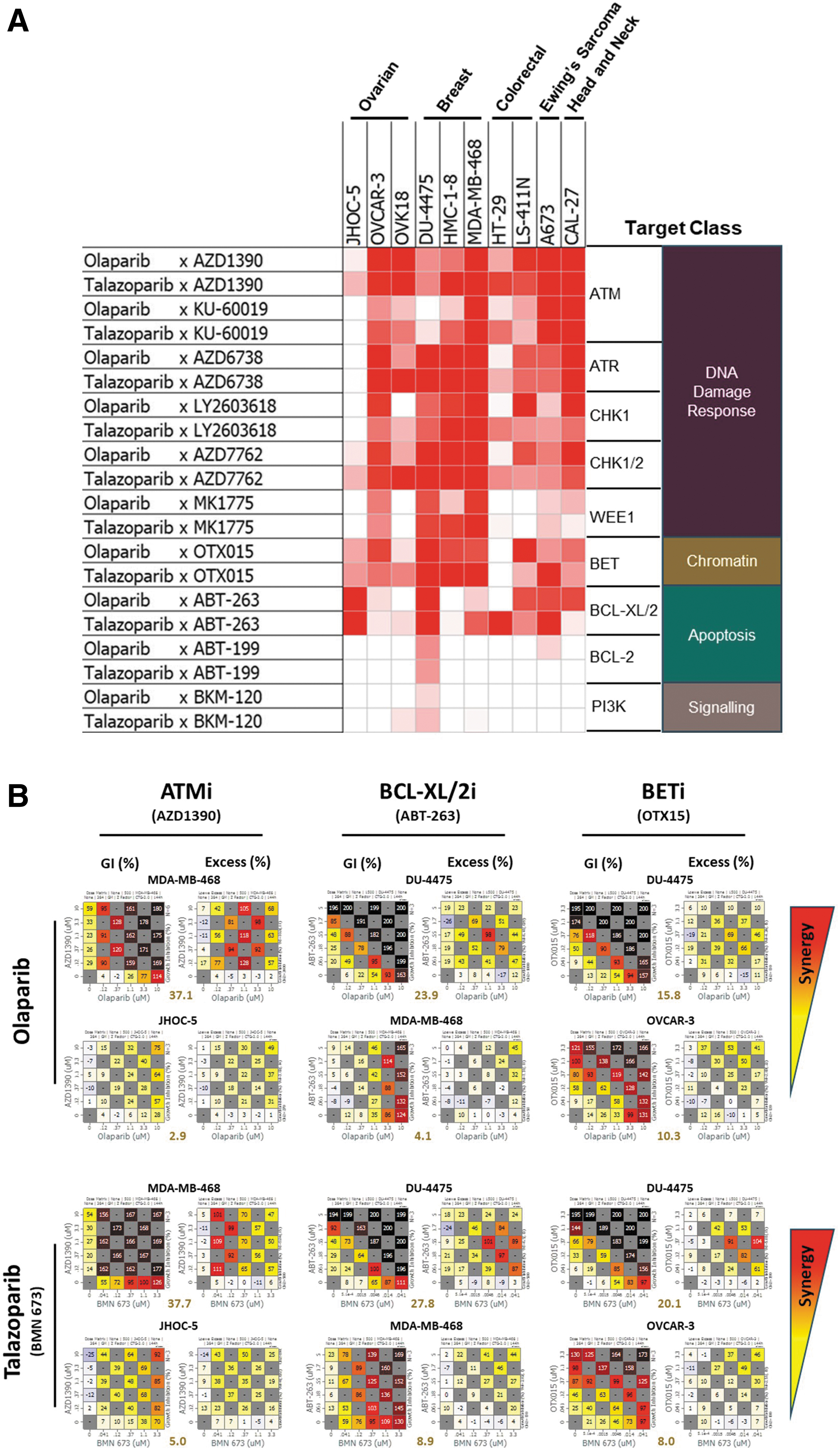
Validation of CRISPR screening hits using combinations.
Overall, the data from the cell panel screens support the sensitization and resistance hits identified with our component ABC CRISPR library.
Discussion
We have developed a flexible and modular pooled CRISPRko screening library that can be used to tailor the power of each screen to the experimental or biological complexity required. To support the data generated using our new library we used high throughput combinatorial cell panel screens as an orthogonal validation tool. Our modular libraries use either four, six, or eight guides to decrease or increase the resolution of each screen, as appropriate. As a first demonstration of this approach, we explored a known challenging paradigm, negative selection screening. In contrast to positive selection or enrichment screening, this often more valuable approach is typically subject to greater noise resulting in poorer hit discovery. Our results indicate that the higher complexity library of eight guides per gene can be used for negative selection screening or where an experimental paradigm is anticipated to yield more noise. Examples of this would be pooled phenotypic screening with flow cytometry sorting endpoints or CRISPR screens conducted in complex cell systems, such as primary or induced pluripotent stem cell (iPSC)–derived tissues. Conversely, in experiments where cell numbers need to be kept low because numbers are limiting, our data indicate that using LibA with four guides per gene will be sufficient to identify targets in a drug–gene sensitivity screen.
Our simulated analyses indicate that our component libraries are all able to find essential genes or controls with high confidence, and in fact only two guides were required to successfully complete this exercise. Indeed, robust identification of essential genes using a low complexity library can be expected, and is already established in multiple publications.2,3,37,38 Surprisingly, increasing the guide number in the case of essential gene discovery resulted in a minor increase in variance, most likely owing to the necessary but modest decrease in the ranking performance of guides included in each of the increasingly complex libraries.
The analysis of drop-out genes indicative of sensitization to olaparib provided a surprisingly rich dataset and showed excellent overall concordance with a previous publication. 9 Zimmermann and colleagues 9 exploited an alternative approach for enriching their dataset by simultaneous screening in several cell lines. We identify the RNaseH2 complex as a key hit in our screens, an observation consistent with the results from Zimmermann et al., 9 where the authors directly validated these hits. The substantial overlap of hits identified in our single CRISPR screen with previously published olaparib screens is further compelling evidence that our new component library is working effectively.
We validated some of the hits identified in our pooled CRISPRko screen by combinatorial drug screens in cell line panels with two clinical PARP inhibitors, olaparib and talazoparib. This orthogonal approach delivers rapid initial validation of selective hits in high throughput. Initial validation through this approach enables targets to be considered in terms of their tractability in the clinic. These data also illustrate how well CRISPR screens and high throughput cell panel screens can be used to rapidly validate new, clinically relevant genetic interactions.
Interestingly, the overlap between the hits in distinct cell models, both from this screen and those published previously, 9 indicate that while broad agreement is found between some of the major hits (as discussed above), there remains a high degree of difference between hits found in each cell line background. Although overall concordance between screens is broadly substantially great than that observed in RNA interference screens, for example,39–41 this observation is a valuable indication that robust conclusions around drug-gene interactions are best made from screens conducted across diverse genetic and lineage backgrounds, to more completely describe the effects. This can be achieved with our fully integrated pooled CRISPRko and high throughput cell panel screening platform.
Overall, our analysis of screening with a new flexible library and hit validation by combinatorial drug screening provides a powerful new approach, particularly well suited to the variable experimental demands of each screening campaign with potentially rapid clinical translation. Hit identification was broadly improved with higher complexity libraries, providing increased confidence and interrogation depth. The lower complexity screens also performed well, indicating that these tools will be poised to support robust hit discovery in future positive selection screening campaigns.
Footnotes
Acknowledgments
The authors thank all members of the screening group for their excellent technical support and Nicola McCarthy for help with reviewing and editing the manuscript.
M.B., D.A.S., and B.C.S.C. conceived the experiments; M.B., M.B.B-Z, G.R.A.M., C.l.S., H.N.P., G.S.T., and S.L. carried out the experiments; S.L. and J.E.M. ran the bioinformatics; and M.B., D.A.S., and B.C.S.C. wrote the paper. All authors reviewed the final version.
Author Disclosure Statement
At the time the experiments were carried out, all authors were employees of Horizon Discovery, Ltd.
Funding Information
No funding was received for this article.