
Abstract
Since its introduction, non-negative matrix factorization (NMF) has been a popular tool for extracting interpretable, low-dimensional representations of high-dimensional data. However, several recent studies have proposed replacing NMF with autoencoders. The increasing popularity of autoencoders warrants an investigation on whether this replacement is in general valid and reasonable. Moreover, the exact relationship between non-negative autoencoders and NMF has not been thoroughly explored. Thus, a main aim of this study is to investigate in detail the relationship between autoencoders and NMF. We define a non-negative linear autoencoder, AE-NMF, which is mathematically equivalent with convex NMF, a constrained version of NMF. The performance of NMF and the non-negative linear autoencoder is compared within the context of mutational signature extraction from simulated and real-world cancer genomics data. We find that the reconstructions based on NMF are more accurate compared with AE-NMF, while the signatures extracted using both methods exhibit comparable consistency and performance when externally validated. These findings suggest that AE-NMF, the linear non-negative autoencoders investigated in this article, do not provide an improvement of NMF in the field of mutational signature extraction. Our study serves as a foundation for understanding the theoretical implication of replacing NMF with non-negative autoencoders.
INTRODUCTION
Non-negative matrix factorization (NMF) is a popular tool for unsupervised learning (Lee and Seung, 1999). In NMF, a non-negative data matrix is factorized into a product of two non-negative matrices of lower dimension: a basis matrix consisting of basis vectors and a weight matrix consisting of the basis vector’s weights for each observation in the data matrix. NMF has gained a strong footing in various scientific fields due to its high interpretability (Alexandrov et al., 2013; Fang et al., 2018; Ozer et al., 2022). Specifically, NMF has proven to be a useful tool to derive mutational signatures from cancer genomics data.
In mutational signature analysis, it is typically assumed that all somatic mutations in a cancer genome are caused by mutagenic processes that leave a characteristic pattern of mutations in the genome. These patterns are denoted mutational signatures. Several signatures have been identified and linked to different mutagenic processes such as ultraviolet light exposure and tobacco smoking (Nik-Zainal et al., 2015). Alexandrov et al. (2013) proposed using NMF on mutational count data from cancer genomes to decipher the mutational signatures of the processes the patients have been exposed to throughout the development of the disease. NMF has since then been the dominating model for mutational signature extraction (Alexandrov et al., 2020; Blokzijl et al., 2018; Islam et al., 2022). When extracting mutational signatures with NMF, the data matrix consisting of a number of patients’ mutational profiles is decomposed into a matrix representing the signatures of mutagenic processes (basis vectors) and an exposure matrix dictating the number of mutations that can be attributed to each specific process in the mutational profiles of each patient (weight matrix).
In this study, we analyze the frequency distribution of the 96 possible trinucleotides, which represent all single-base substitutions (SBS) flanked by the nucleotides immediately to their left and right, commonly referred to as the trinucleotide context.
Recently, several studies have proposed substituting NMF with non-negative autoencoders, which are increasingly popular for dimensionality reduction (Hosseini-Asl et al., 2016; Khatib et al., 2018; Lemme et al., 2012; Özer et al., 2022; Smaragdis and Venkataramani, 2017). This is also the case in mutational signature extraction (Pancotti et al., 2024; Pei et al., 2020). Pei et al. (2020) suggested using a sparse autoencoder to identify mutational signatures from cancer genomics data and generated estimates that were not only in concordance with existing literature but also correlated meaningfully with observed exogenous exposures. Pancotti et al. (2024) suggested a hybrid architecture with a deep encoding and shallow decoding to relax the assumption of linearity NMF imposes on mutational signature extraction. This trend of using non-negative autoencoders as an alternative to NMF with promising results, especially within mutational signatures, prompts the questions: What is the mathematical relation between autoencoders and NMF? And how do they compare in mutational signature extraction?
This study compares the performance of linear non-negative autoencoders to NMF in the field of mutational signatures. In particular, we compare the in- and out-of-sample reconstruction error, as well as the consistency of the extracted signatures from the ovary, prostate, and uterus tumors from the Genomics England 100,000 Genomes Project (GEL) (Turnbull, 2018; Turro et al., 2020). Moreover, we show theoretically that autoencoders can be constructed as convex NMF, a special case of NMF where the basis vectors are restricted to be convex combinations of columns in the data matrix (Ding et al., 2010). Based on this, we deduce how it impacts interpretation of the estimates generated by linear non-negative one-hidden-layer autoencoders in general and especially within the field of mutational signatures.
In Section 2, we show under what constraints autoencoders are equivalent to convex NMF and use this to establish a meaningful comparison between non-negative linear autoencoders and NMF. Section 3 compares NMF and non-negative linear autoencoders’ performance on simulated and cancer genomics data. Lastly, we discuss and conclude on the results in Sections 4 and 5.
METHODS
Consider a non-negative data matrix
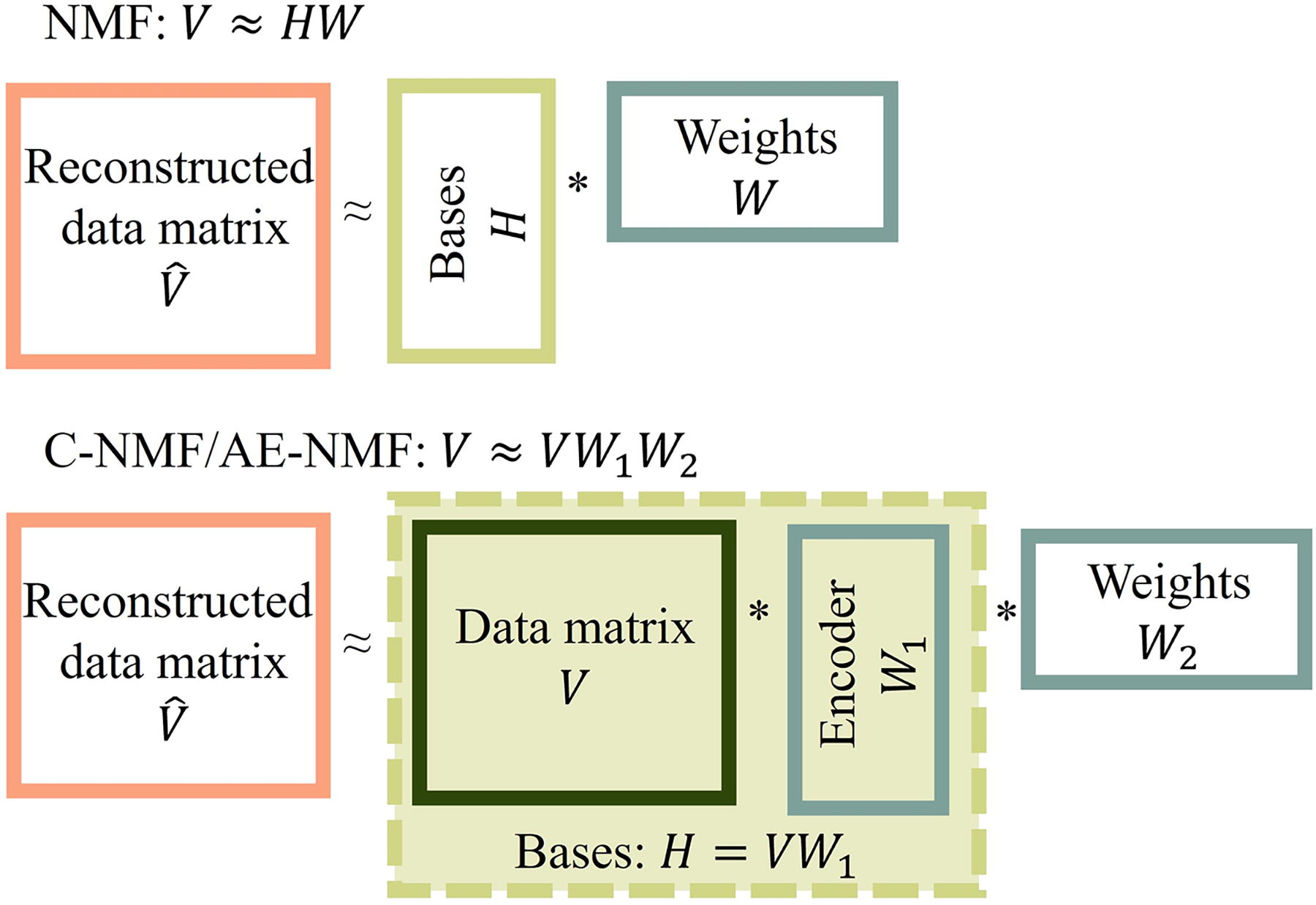
Schematic representation of the composition of basis vectors and weights in NMF (top) and C-NMF and AE-NMF (bottom). NMF, non-negative matrix factorization.
NMF decomposes a matrix with non-negative entries into a matrix product of two factor matrices with non-negative entries, one containing a set of basis vectors and one containing a set of weights. The shared dimension, K, of the factor matrices, is typically chosen to be much smaller than the dimensions of the input matrix, making NMF a dimensionality reduction technique.
Standard K-dimensional NMF was introduced by Lee and Seung (1999) and aims to make a reconstruction,
Convex NMF, as introduced by Ding et al. (2010), is a special case of NMF, where the basis vectors are constrained to be spanned by the columns of the data matrix,
Autoencoders consist of an encoder applied to the input to create a latent representation and a decoder that maps the latent representation to a reconstruction of the input (Kramer, 1991). Choosing the dimension of the latent representation to be lower than the dimension of the input makes the autoencoder a dimensionality reduction technique. A single hidden layer and fully connected autoencoder’s reconstruction,
In this study, we define linear autoencoders as single-hidden-layer autoencoders with identity activation functions.
There are several methods to enforce non-negativity in autoencoders as defined in Equation (3). An overview of those considered in this article is provided in Supplementary Table S1 and elaborated on in Sections 1.3 and 2.3 in Supplementary Data. Here, non-negativity is enforced by taking the absolute value of the encoding and decoding weight matrices in the forward pass.
Mathematical equivalence and interpretation
Setting
The core architecture of this autoencoder is analogous to that of Pei et al. (2020), but differs by fixing the bias terms to zero instead of estimating them through training and choosing the identity function as activation functions instead of the rectified linear unit (ReLU) for the encoding layer and softmax for the decoding layer. The proposed autoencoder differs from the autoencoder proposed by Pancotti et al., (2024) in the orientation of the input matrix and in the depth of the encoding network. The latent representation in this study corresponds to the exposure matrix rather than the signature matrix, and the latent representation in this article is derived from a shallow encoding instead of a deep, non-linear encoding. An overview of the architectural differences of autoencoders proposed for mutational signature extraction can be seen in Table 1. Although all models described in Table 1 differ at various assumptions, they do share the common trait that decoding is performed in a single layer.
Overview of Architectural Differences and Similarities of Autoencoders Suggested for Mutational Signature Extraction
Overview of Architectural Differences and Similarities of Autoencoders Suggested for Mutational Signature Extraction
We will use “C-NMF” to refer to convex NMF optimized with the multiplicative updating steps derived by Ding et al. (2010) and use “AE-NMF” for the class of autoencoders constructed equivalently to C-NMF.
The encoded data matrix in AE-NMF and C-NMF,
Though the interpretation of AE-NMF, C-NMF, and NMF is similar, there are still considerable differences between C-NMF, AE-NMF, and standard NMF. In particular, NMF estimates
AE-NMF and C-NMF will estimate a larger number of parameters in the factor matrices than NMF when the number of observations N surpasses the number of features M, which is often the case within mutational signature extraction. Moreover, the computational complexity for training NMF with respect to the Frobenius norm is of order
For each cohort in both simulated and real cancer data, patients were divided into 30 training/test splits with the ratio of 80/20. De novo extractions by NMF and AE-NMF were performed on the training sets optimized with respect to both the Frobenius norm (mean squared error [MSE]) and the Kullback-Leibler divergence (KLD) according to the updates defined in Section 1.1 in Supplementary Data. The results generated from optimizing with respect to the MSE are reported in this section, but corresponding results for the KLD can be found in Section 2 in Supplementary Data. Refits on the test sets were performed according to Section 1.1.5 in Supplementary Data, yielding the test errors. All training was performed with a relative tolerance of
To emulate the terminology used in the cancer data analysis, the matrix of basis vectors will be denoted as signatures, and the weight matrix will be denoted as exposures.
Simulated data
The dataset analyzed in this section is “Scenario 8” from Islam et al. (2022), which consists of 1000 trinucleotide mutational profiles simulated to emulate a mixture of renal cell carcinomas and ovarian adenocarcinomas, alongside the corresponding true signature matrix and exposure matrix. There are three ground-truth signatures resembling the catalogue of somatic mutations in cancer (COSMIC) signatures SBS3, SBS5, and SBS40, which are all considered to be relatively flat and featureless signatures (Tate et al., 2019). In all analyses of this dataset, the true number of signatures, K = 3, is used.
The reconstruction loss in the training and test settings, ACS with the ground truth signatures, and the SC of all models can be seen in Table 2. These analyses reveal that while all models perform equally well in recovering the ground truth signatures with equal signature consistencies, NMF consistently performs most accurately in reconstruction of the input data. This is both the case for training and test data and for both loss functions.
Average Training and Test Error Between the Input and Reconstructed Data for Each Method Across the 30 Splits of the Scenario 8 Cohort (Islam et al., 2022)
Average Training and Test Error Between the Input and Reconstructed Data for Each Method Across the 30 Splits of the Scenario 8 Cohort (Islam et al., 2022)
The lowest reconstruction error in each cohort is highlighted in bold.
ACS, average cosine similarity with true signatures; MSE, mean squared error; KLD, Kullback-Leibler divergence; SC, signature consistency.
Extraction of mutational signatures is performed on the trinucleotide representations of the tumor-normal whole genome sequences of the 713 ovary, 311 prostate, and 523 uterus tumors from GEL (Turnbull, 2018; Turro et al., 2020).
The number of signatures for each cohort was determined with the method described in Section 1.2 of Supplementary Data using 10 bootstrap samples. The test errors as a function of
Extraction performance
As shown in Section 2.4, C-NMF and AE-NMF are mathematically equivalent. Furthermore, since the resulting factor matrices of C-NMF and AE-NMF overlapped in the proof of concept in Section 2.1 in Supplementary Data and C-NMF and AE-NMF performed similarly in both the simulated data and in the cancer data analysis with respect to reconstruction error (Table 2, Supplementary Figs. S2 and Figs. S5) and consistency (Supplementary Fig. S7), we consider C-NMF and AE-NMF as practically equivalent. Thus, the following analyses will be performed comparing only NMF and AE-NMF. Boxplots of the training and test errors for each method and cohort are shown in Figure 2 and Supplementary Figures S5 and Figure S6. Table 3 reports the average training and test error across the 30 splits for each method, cohort, and loss function as well as the average ratio between the NMF reconstruction loss and the AE-NMF reconstruction loss.
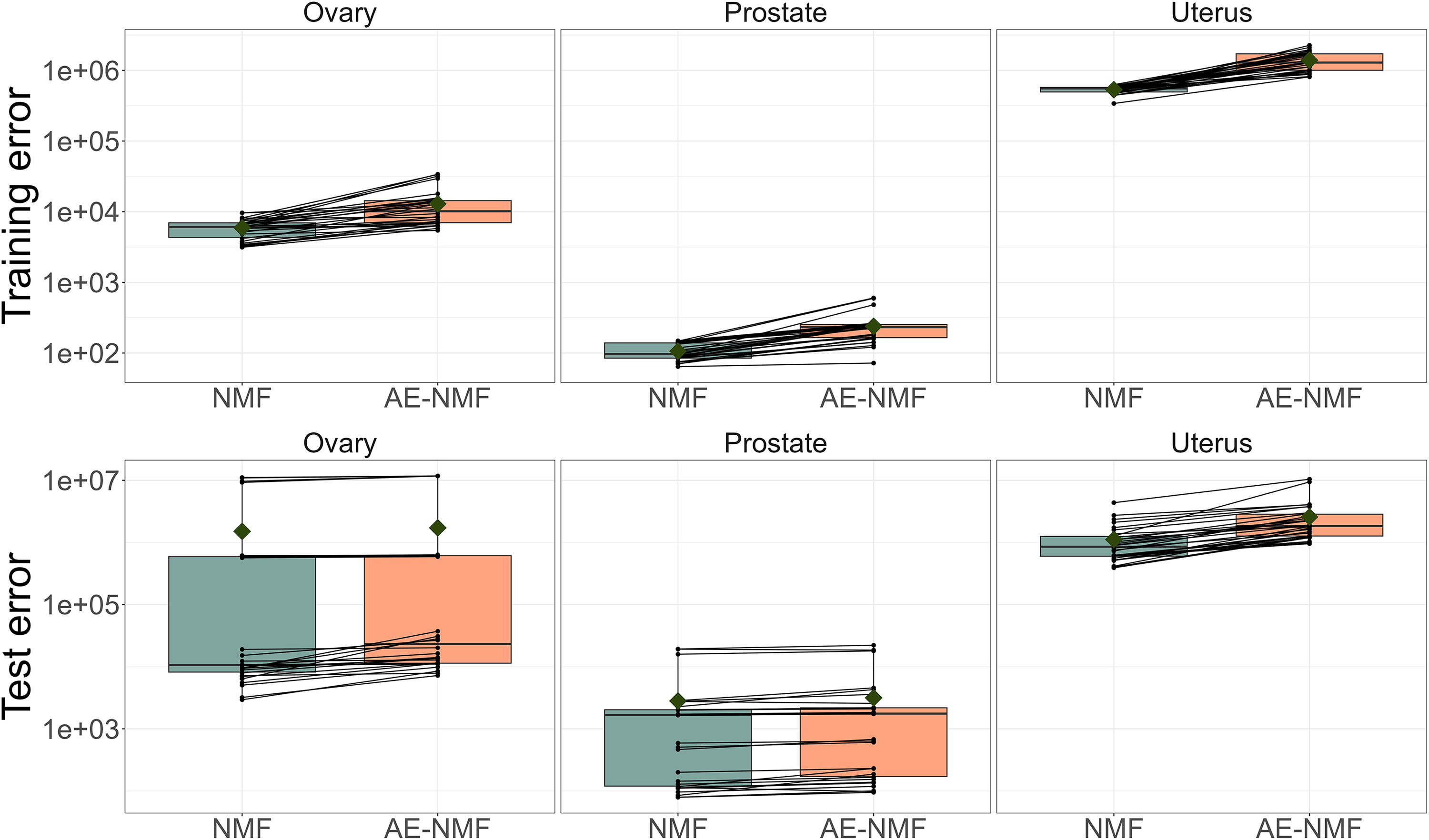
Boxplots of the training and test MSE of 30 train/test splits of the ovary, prostate and uterus cohorts. NMF and AE-NMF errors resulting from the same splits are connected by a black line. The boxes are colored corresponding to the method used. Note that the y-axis is on log scale. MSE, mean squared error.
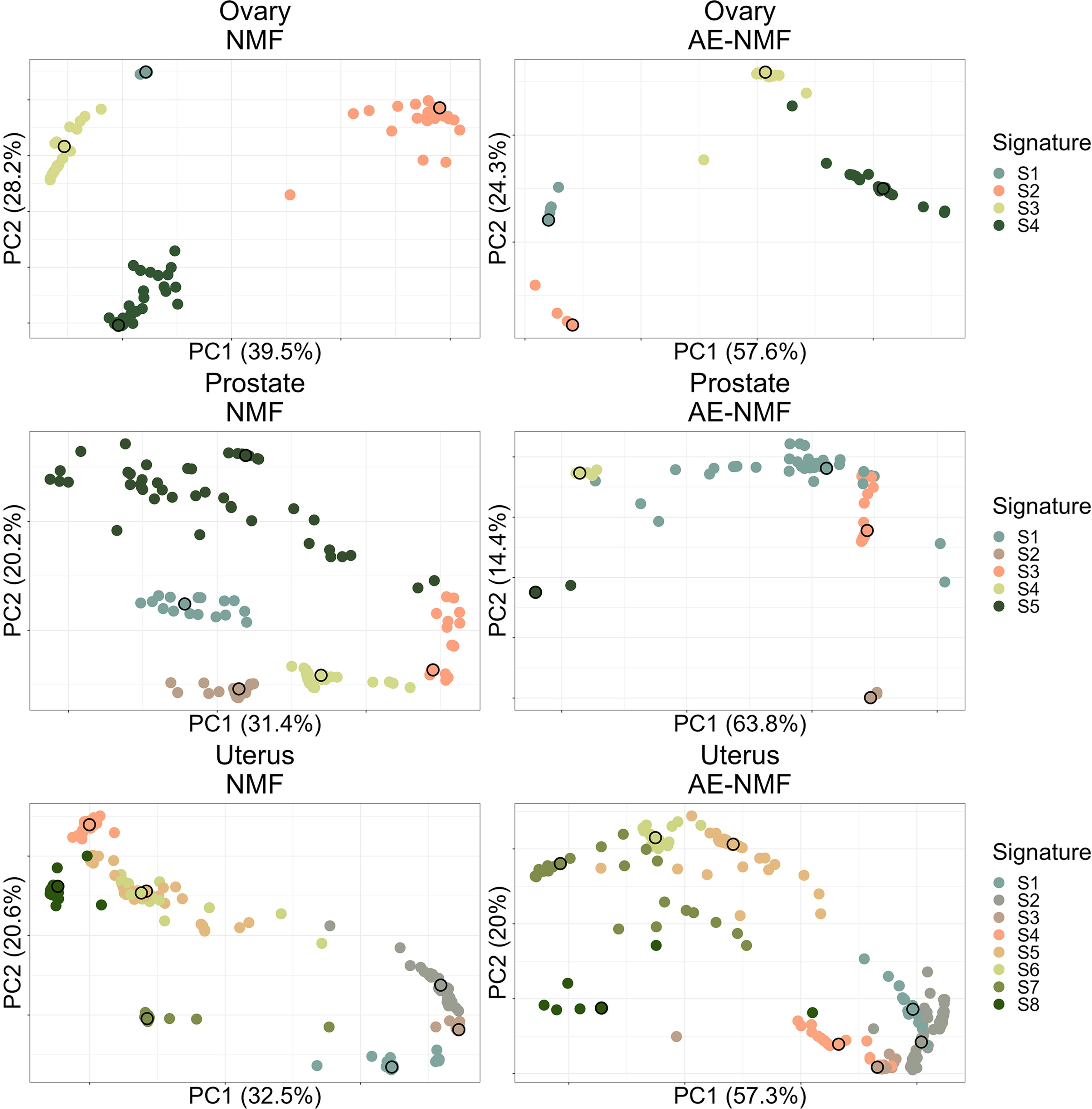
First and second principal component of the de novo extracted signatures from the 30 train/test splits for MSE optimized AE-NMF and NMF (columns) and each cancer type (rows). Points are colored by the PAM clustering assignment, and the cluster mediods are highlighted with a black outline. PAM, partitioning around medoids.
Average Training and Test Error Between the Input and Reconstructed Data for Each Method Across the 30 Splits of the Ovary, Prostate, and Uterus Cohort and the Average Ratio between the Errors
The lowest reconstruction error in each cohort is highlighted in bold.
NMF consistently performs better than AE-NMF in terms of reconstructing the input data for both training and test data across all cohorts and for both loss functions. The ratios in Table 3 reveal that the difference is more expressed in the training splits than the test splits.
For each method, cohort, and loss function, the analyses yielded 30 signature sets, one from each training set. Plots of the first and second principal component of the signatures from the 30 training sets are depicted in Figure 3 for MSE and in Supplementary Figure S9 for the KLD. The consistency of the estimated signatures extracted within each method was thus based on a total of
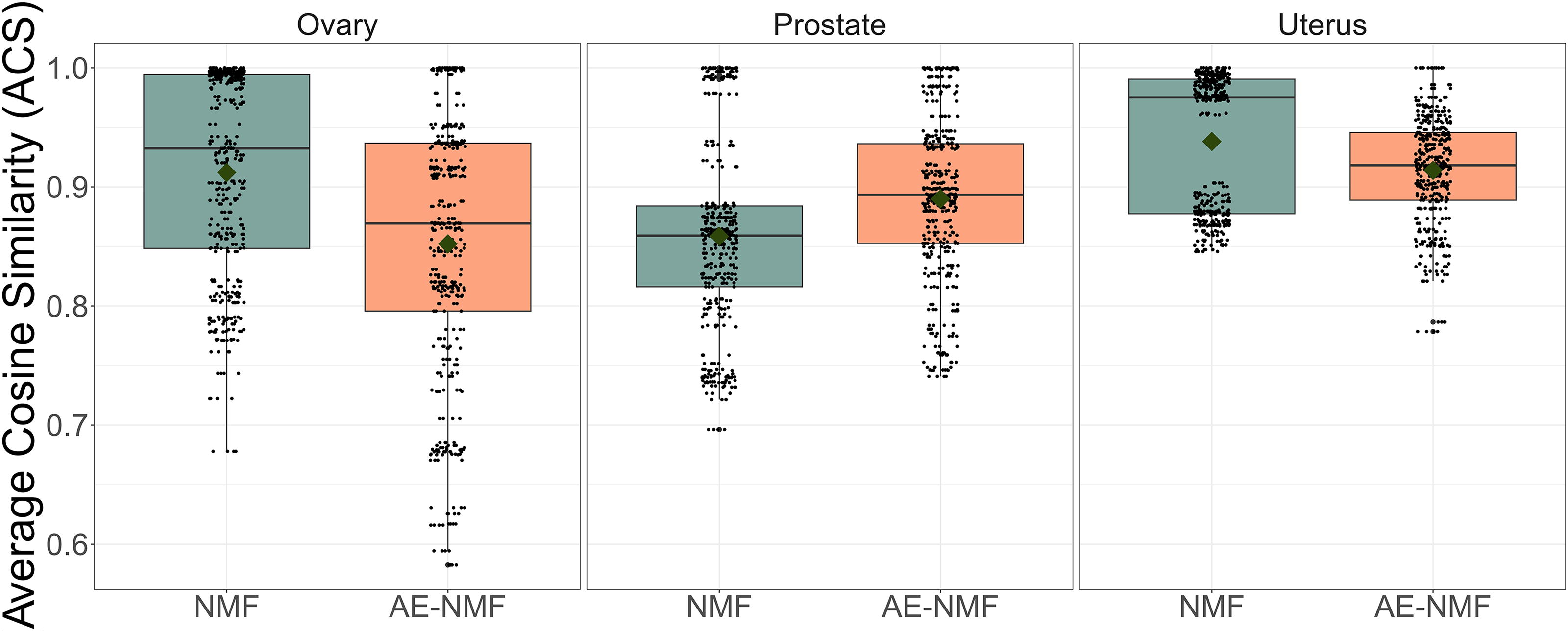
Boxplots of the ACS between each combination of AE-NMF and NMF signatures extracted using the MSE across the 30 splits of the data matrix for the ovary, prostate, and uterus cohort. ACS, average cosine similarity.
To compare the estimated signatures with known libraries, the signatures were clustered to form sets of consensus signatures using the partitioning around medoids (PAM) algorithm (Kaufman and Rousseeuw, 1990) with the number of clusters equal to the number of signatures used in the initial extractions. The clustering is depicted in Figure 3 and Supplementary Figure S9, where the first and second principal components of all de novo signatures are depicted and the points are colored by their assigned PAM clustering. The consensus signatures were subsequently matched to the COSMIC v. 3.4 signatures (Tate et al., 2019), as well as the Signal library of mutational signatures, which is based on the publication from Degasperi et al. (2022) where mutational signatures were extracted based on the GEL cohorts, as in this study. Comparisons with COSMIC signatures were made with the full library and comparisons with the signal signatures were made with the related organ-specific signatures extracted solely on the GEL cohort. The matched signatures along with their cosine similarity can be seen in Table 4.
The COSMIC and Signal Signatures Matched to the Consensus Signatures Estimated with Respect to the MSE and the Corresponding Cosine Similarity
The COSMIC and Signal Signatures Matched to the Consensus Signatures Estimated with Respect to the MSE and the Corresponding Cosine Similarity
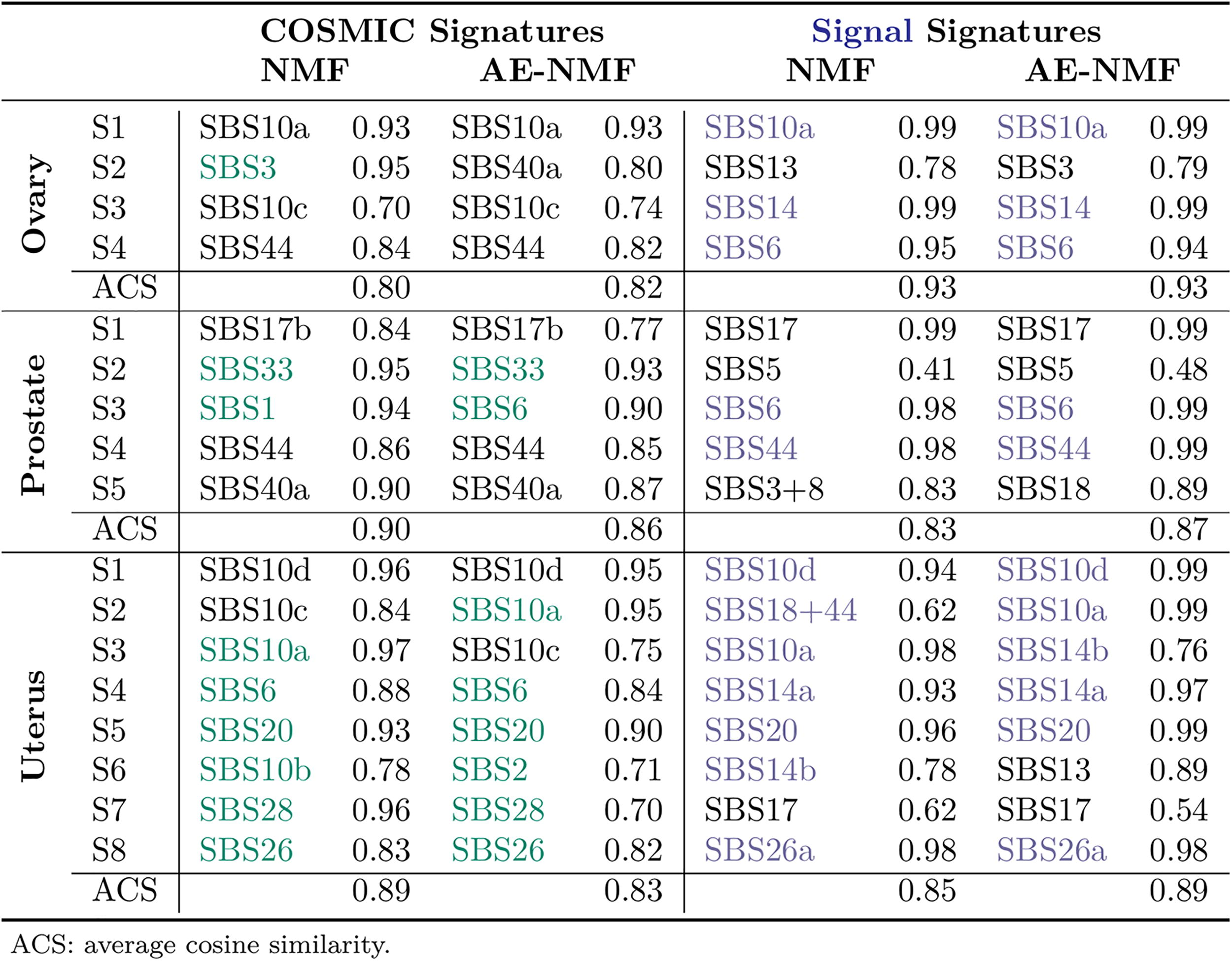
The MSE-estimated NMF and AE-NMF signatures displayed similar (with a slight advantage to NMF) average cosine similarities to the COSMIC library (0.80, 0.90, and 0.89 for NMF against 0.82, 0.86, and 0.83 for AE-NMF for the ovary, prostate, and uterus signatures, respectively), the average cosine similarities with the signal signatures display an advantage to the AE-NMF signatures (0.93, 0.83, and 0.85 for NMF against 0.93, 0.87, and 0.89 for AE-NMF for the ovary, prostate, and uterus signatures, respectively). Among the four, five, and eight consensus signatures generated for the ovarian, prostate, and uterine cancer cohorts, the matched COSMIC signatures had previously been observed in the corresponding cancer types in 1/4 of NMF and 0/4 of AE-NMF signatures in the ovarian cohort, 2/5 of both NMF and AE-NMF signatures in the prostate cohort, and 6/8 of both NMF and AE-NMF signatures in the uterus cohort. Overall both NMF and AE-NMF show a high degree of conformity with both COSMIC and signal SBS signatures, and the extracted signatures are relevant to the diagnoses in which they have been identified. In SC, conformity with COSMIC, and choosing relevant signatures, NMF and AE-NMF perform similarly, perhaps with a slight advantage to NMF. The matched COSMIC signatures for all splits before clustering for NMF and AE-NMF can be seen in Supplementary Figures S10–S15.
In this study, we compare NMF and AE-NMF by their ability to extract valid and consistent basis vectors and creating accurate reconstructions of the input data. We assert that such comparisons are theoretically meaningful since we demonstrate that AE-NMF and C-NMF, a constrained version of NMF, are mathematically equivalent.
The study focuses on extracting mutational signatures in the ovary, prostate, and uterus cancer genomes of Genomics England’s 100,000 Genomes cohort, optimizing both with respect to the MSE and the KLD. Across all cancer types and both loss functions NMF consistently outperformed AE-NMF in terms of reconstruction error; the differences being more expressed in the training data than in the test data. While AE-NMF constrains parameters to convex combinations of patients’ profiles, NMF can freely assume non-negative values, giving it an advantage in training set reconstruction. When reconstructing the test set, the signature matrix is fixed, and the task is, thus, identical for NMF and AE-NMF. One could expect the constrained nature of AE-NMF to regularize the signatures such that they would reconstruct the test splits better compared with NMF, but as this was not the case in this study, AE-NMF signatures may be generally less informative than the corresponding NMF signatures. The validation with existing mutational signature libraries revealed that both models recovered relevant signatures with high cosine similarity, which is interesting considering that the majority of signatures in these libraries are extracted using NMF-based methods (Alexandrov et al., 2020).
By establishing the mathematical equivalence between C-NMF and AE-NMF, we underline that it is meaningful to interpret the parameters in AE-NMF similarly to NMF. We deem this equivalence is necessary for proper comparison of NMF and non-negative autoencoders, which has been missing in previous attempts to replace NMF with autoencoders while using the same interpretation. Squires et al. (2019) also came to the conclusion that an autoencoder with the architecture of AE-NMF yields a hidden layer consisting of convex combinations of the data points but did not make the connection to convex NMF.
The architecture of AE-NMF is atypical for autoencoders by its shallow and linear nature and by transposing the input data matrix. By orienting the input matrix,
This article defines under what exact conditions the non-negative autoencoder yields parameters that can be interpreted equivalently to those resulting from NMF but also highlights a disadvantage of the linear non-negative autoencoder used in this study in regards to reconstruction accuracy. In contrast, the autoencoder proposed by Pancotti et al., (2024) utilizes deeper extraction and conventional input orientation, which jeopardizes the link to NMF and the exact interpretation of parameters, but the increased complexity holds a potential for improved extraction performance. On the contrary, AE-NMF has an advantage over especially C-NMF when considering the computational complexity and runtime of the estimation. Section 2.4 shows that C-NMF scales quadratically in both the number of patients (N) and the number of signatures (K), NMF scales quadratically in the number of signatures (K) and linearly in the number of patients, and AE-NMF scales linearly in the number of signatures (K) and patients (N). These theoretical complexities are consistent with the runtimes observed in the bootstrap analysis for determining the number of signatures, where we observe C-NMF veering from NMF and AE-NMF from early values of K, while NMF diverges from AE-NMF at higher K’s dependent on the number of patients N.
In this study, a relative tolerance convergence criteria of
CONCLUSION
This study compares NMF with linear non-negative autoencoders in mutational signature extraction, arguing that such comparisons are, in fact, theoretically relevant as this study shows that linear non-negative autoencoders and convex NMF are mathematically equivalent. This bridges a gap in the comparison of NMF and linear non-negative autoencoders by offering insights into parameter interpretation that were previously lacking. The choice between convex NMF and its autoencoder equivalent is a question of choosing between a multiplicative or gradient descent-based optimizing algorithm to solve the same optimization problem and the linear non-negative autoencoder described in this study can, thus, be used as a faster alternative to convex NMF. In the comparison with NMF, linear non-negative autoencoders exhibit higher reconstruction errors and similar consistencies when validated in external signature libraries, therefore the linear non-negative autoencoder investigated in this study is not a suitable alternative to NMF in mutational signature extraction. This study underscores the significance of methodological considerations when replacing NMF with non-negative autoencoders. On the contrary, autoencoders hold promise for modeling non-linearity, a capability absent in NMF, but, as this article asserts, such advancements are made at the cost of exact parameter interpretation.
Footnotes
ACKNOWLEDGMENT
The authors are grateful to the two anonymous reviewers for constructive and helpful comments on this article.
AUTHORS’ CONTRIBUTIONS
I.E.: Conceptualization (equal), formal analysis (lead), methodology (equal), software (lead), visualization (lead), writing—original draft (lead), writing—review and editing (equal). R.F.B.: Conceptualization (equal), formal analysis (supporting), supervision (equal), writing—original draft (supporting), writing—review and editing (equal). M.P.: Conceptualization (equal), formal analysis (supporting), writing—review and editing (equal). A.H.: Conceptualization (equal), formal analysis (supporting), software (supporting), writing—review and editing (equal). M.B.: Conceptualization (equal), formal analysis (supporting), supervision (equal), writing—original draft (supporting), writing—review and editing (equal).
CODE AND DATA AVAILABILITY
Code used for this study can be found at Github (https://github.com/CLINDA-AAU/AE-NMF) and the Genomics England WGS data used in this study was provided by Turro et al. (2020) on Zendo (![]() ).
).
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.