Most sequence sketching methods work by selecting specific k-mers from sequences so that the similarity between two sequences can be estimated using only the sketches. Because estimating sequence similarity is much faster using sketches than using sequence alignment, sketching methods are used to reduce the computational requirements of computational biology software. Applications using sketches often rely on properties of the k-mer selection procedure to ensure that using a sketch does not degrade the quality of the results compared with using sequence alignment. Two important examples of such properties are locality and window guarantees, the latter of which ensures that no long region of the sequence goes unrepresented in the sketch. A sketching method with a window guarantee, implicitly or explicitly, corresponds to a decycling set of the de Bruijn graph, which is a set of unavoidable k-mers. Any long enough sequence, by definition, must contain a k-mer from any decycling set (hence, the unavoidable property). Conversely, a decycling set also defines a sketching method by choosing the k-mers from the set as representatives. Although current methods use one of a small number of sketching method families, the space of decycling sets is much larger and largely unexplored. Finding decycling sets with desirable characteristics (e.g., small remaining path length) is a promising approach to discovering new sketching methods with improved performance (e.g., with small window guarantee). The Minimum Decycling Sets (MDSs) are of particular interest because of their minimum size. Only two algorithms, by Mykkeltveit and Champarnaud, are previously known to generate two particular MDSs, although there are typically a vast number of alternative MDSs. We provide a simple method to enumerate MDSs. This method allows one to explore the space of MDSs and to find MDSs optimized for desirable properties. We give evidence that the Mykkeltveit sets are close to optimal regarding one particular property, the remaining path length. A number of conjectures and computational and theoretical evidence to support them are presented. Code available at https://github.com/Kingsford-Group/mdsscope
INTRODUCTION
Sketching methods, such as minimizers (Roberts et al., 2004a) or open-syncmers (Edgar, 2021), distill a long sequence into a smaller “sketch,” a set of k-mers and their positions in the sequence. By comparing these sketches, it is possible to quickly estimate whether two sequences are similar and may have a good quality alignment between them or not. Because sketching methods greatly reduce the computational needs in many genomics algorithms with usually little impact on the quality of the result, they are used in many computational biology software packages [see Zheng et al. (2023) for a review].
For our purposes, a k-mer sketching method is modeled by a function φ that takes a context as an input (a substring of the input sequence of fixed length c) and outputs a set of positions within the context of the selected k-mers. The output of φ can be the empty set, meaning that nothing is selected in this context. The sketch for a sequence S is the union of all selected positions over all the contexts of S (see Section 2). This sketch contains a subset of all the k-mers in S as the function φ might not pick any k-mer in a context or adjacent contexts may pick the same locations.
The two properties of sketching methods that downstream applications rely on to prove correctness are as follows:
Locality The property that similar sequences (i.e., that have reasonably long identical subsequences) will have common elements in their sketches, and hence, long enough matches will be detected using the sketches. This is naturally satisfied because the selection is done using a deterministic function (φ); therefore, two sequences that share an exact substring of length at least c will select the same k-mers in that context.
Window guarantee The maximum distance w between two selected k-mers is the window size or guarantee. A small window size guarantees that no large part of a sequence is ignored. Equivalently, the window guarantee means that k-mers are selected at approximately regular intervals.
Sketching methods are usually optimized for two metrics, density (Schleimer et al., 2003) and conservation (Edgar, 2021). The density is the relative size of the sketch, formally defined as . A lower density is desirable as a smaller sketch usually implies less computation and lower memory requirements. The conservation is the proportion of elements that are common between a sketch of S and a sketch of a slightly mutated sequence , where the common elements are either k-mers or subsequences covered by these k-mers. Higher conservation is desirable because it usually correlates to higher sensitivity to detect sequence similarities in the face of mutations and errors. For a fixed k, a smaller context size leads to higher conservation, as the presence of a k-mer in the sketch of the mutated may be affected by mutations in the entire context (Shaw and Yu, 2022).
Not all sketching methods satisfy the window guarantee property (i.e., for some sketching methods, there are infinitely long sequences S with an empty sketch; see Section 3). However, sketching methods that do not satisfy the window property are problematic in two ways. First, most algorithms using a sketching method do not have a proof of correctness in cases without the window property (e.g., an aligner may miss arbitrarily long, good quality alignments, preventing claims of sensitivity).
Second, the sketch optimization problem is ill-formed without the window property. The empty selection function that returns the empty set for any input sequence satisfies vacuously the locality property, it has perfect conservation, and it has the lowest possible density. But of course, no information is preserved in an empty sketch and this trivial solution is not useful. The existence of trivial solutions is not a purely theoretical concern. When optimizing sketching methods using machine learning, almost empty (and not practically useful) solutions are found if no window constraint is used in the loss function (Hoang et al., 2022a).
A set of k-mers M is unavoidable if any infinitely long sequence must have k-mers from M. Because any sequence uniquely corresponds to a path in the de Bruijn graph Dk of order k, an equivalent point of view is the decycling sets (DSs): M is an unavoidable set of k-mers (and a decycling set) if and only if , the de Bruijn graph Dk with the k-mers from M removed, is a directed acyclic graph (DAG).
There is a strong two-way connection between such decycling sets and sketching methods with a window guarantee. Consider the set of possibly selected k-mers (the union of all k-mers selected over every possible context) for sketching method φ. If the sketching method has a window guarantee, then is a decycling set. Moreover, the window size of φ is equal to the remaining path length of , that is, the length of the longest path in the DAG .
The function φ of a sketching method with the smallest possible context (c = k, aka context-free methods, such as syncmers) must return an empty set for some input contexts, otherwise it would select every k-mer and would be equivalent to no sketching. Consequently, in the context-free case, φ is equivalent to the indicator function of its set : as the input context contains only one k-mer, the output of φ is not empty exactly when the input k-mer is in . A sketching method with a larger context may not select every occurrence of k-mers in from S. For example, a context may contain multiple k-mers from but the function φ only selects one of them (DeBlasio et al., 2019). In other words, given two sketching methods, one context-free and one with a context, having the same set of possibly selected k-mers, the method with a context can lower its density at the expense of having a lower conservation. Conversely, given a decycling set M, the indicator function of M defines a context-free sketching method with a window guarantee.
This connection between decycling sets and sketching methods suggests, first, that the properties of the decycling sets ultimately define the properties of the associated sketching method. In other words, by studying the space of decycling sets we gain insights into the design space of sketching methods. Second, the space of decycling sets is much larger than the decycling sets generated by the few families of sketching methods currently used. Rather than creating ad hoc sketching methods, a promising strategy is to find a decycling set with desirable properties and use the sketching method associated with this set.
In this study, we focus on minimum decycling sets (MDSs), i.e., deycling sets of minimum size. MDSs provide a logical starting point for the study of decycling sets. First, the MDSs are by definition as small as possible, therefore reducing as much as possible the cost of storing and querying such a set. Second, for context-free case, a small set corresponds, in expectation, to smaller sketches (aka low-density method). Finally, these sets are likely to have short remaining path lengths (say polynomial in k), corresponding to sketching methods with small window guarantee.
The connection between MDSs and sketching methods was already explored (Orenstein et al., 2017, 2016; Ekim et al., 2020; Pellow et al., 2023), but mostly using one particular MDS construction by Mykkeltveit (1972). In this study, we give new methods to explore the space of all MDSs as a way to define new sketching methods with desirable properties.
After describing the window guarantee of common sketching methods, we describe the structure of the de Bruijn graph and of its cycles. We then give two simple graph operations that can be used to enumerate MDSs. Provided Conjecture 1 is true (for which we provide ample theoretical and experimental evidence); all MDSs can be reached with these operations. Using these operations, we design an optimization procedure to find MDSs with short remaining path lengths. This optimization procedure gives further insight on the range of possible window guarantees for sketching methods and of the well-known Mykkeltveit set.
The conjectures and optimization methods proposed here are the basis to further the understanding of MDSs and the design space of the sketching methods that are central to computational biology algorithms, in particular sketching methods with a small context and a strong window guarantee.
PRELIMINARIES AND NOTATIONS
An alphabet is a set Σ of size . Although the results generalize to any alphabet size, we consider the binary alphabet and the DNA alphabet of size 4. A sequence S is an element of , and sequences are indexed starting at 1. represents the subsequence starting at position a of length k, that is, the ath k-mer of S. is the set of integers .
We assume that . A sketching scheme is defined by its selection function , where denotes the power set. A context is a subsequence of length c of S: with . The sketch of S is the set of the positions of the selected k-mers in S: . The set of all possibly selected k-mers for the sketching method φ is .
The de Bruijn graph of order k is the directed graph , where each k-mer is a node and the edges represent the suffix–prefix relationship . The de Bruijn graph is σ-regular, Eulerian, and Hamiltonian. For convenience, short strings, such as k-mers, are commonly represented as base-σ numbers.
WINDOW GUARANTEE OF EXISTING SKETCHING SCHEMES
We review sketching methods commonly used in computational biology and evaluate their window guarantee.
Hash-based methods. Hash methods use a hash function h and select the k-mers m that satisfy, for example, or h(m) < t for some predefined constants p, t (Karp and Rabin, 1987; Ekim et al., 2021). Effectively, the hash function randomizes the k-mers, and the criteria selects a subset of the k-mers. Other approaches apply a sketching method like minimizers or syncmers and further down-sample the sketch using a hash function (Rouzé et al., 2023; Edgar, 2021).
In general, these methods do not have a window guarantee and, historically, this was one of the motivations for Schleimer et al. (2003) to introduce the winnowing scheme (which is equivalent to minimizers). Although hash-based schemes can have low density and have a short context (c = k), it is achieved at the cost of having no window guarantee. For example, by choosing low values of the threshold t, the density can be made arbitrarily low, but the number of distinct cyclic sequences not covered by the scheme increases dramatically.
Window-based methods. These methods always pick at least one k-mer in each context and therefore the context and the window guarantee are closely linked.
The minimizer scheme has three parameters and in each window of w consecutive k-mers (i.e., the context is a substring of length ), the selection function returns the position of the smallest k-mer according to the order (Roberts et al., 2004a,b). There are many ways to select the order (Zheng et al., 2021, 2020b; Hoang et al., 2022b; Jain et al., 2020), for example to improve the density, but because the selection function never returns the empty set, all these methods have a window guarantee of w, independent of the choice of .
The density of minimizers schemes is usually between and (Marçais et al., 2017, 2018), and the context length is . Density can be lowered by increasing w, although this increases the context length (hence weakens the locality and lowers the conservation). Having a coupling between the window guarantee and the context length constrains the parameter choices for minimizer schemes.
Compared to minimizers, the minmers scheme (Kille et al., 2023) adds a fourth parameter d: in each window of dw consecutive k-mers the selection function returns the position of the d smallest k-mers according to . Minmers achieve a density closer to while having a significantly longer context of .
Positional minimums. Under this generic name are methods such as open-syncmers (Edgar, 2021), masked minimizers (Hoang et al., 2022a), and parameterized syncmers (Dutta et al., 2022).
Parameterized syncmers schemes have four parameters where and m is a nonempty bit-mask of length k. A context of length c = k is selected if the smallest s-mer in the context (choose left-most to break ties) is at position i and bit i is set in the mask m. This is a generalization of the syncmers schemes: the mask of syncmers has exactly one bit set to 1.
Masked minimizers have a two-step process as follows: the first step selects an element similarly to parameterized syncmers, and, second, a reporting function returns the final selection (which can be, e.g., the smallest s-mer or the k-mer containing it). This two-step approach unifies syncmers and minimizers like schemes.
Whether these schemes have a window guarantee depends on whether the first bit of the mask m is set. If the first bit is set and a k-mer is not selected, then this implies that an s-mer at position i > 1 is strictly smaller than the s-mer at position 1, forming a decreasing list of s-mers. As the k-mers are shifted along the sequence, this decreasing list of s-mers must eventually come to an end, hence there is a window guarantee. This window guarantee is weak as the window can be as long as as seen in the following construction.
Assume and create an order on the s-mer using a de Bruijn sequence D of order s (D contains all the s-mers once and only once) and by definition s1 < s2 if and only if the s-mer s1 appears after s2 in D. The sequence D is a decreasing sequence of s-mers of length . With , we created a sequence of length without a selected k-mer.
If the first bit is not set, because of the left-most tie breaking rule, there is no window guarantee. Hence, these methods have a short context and a weak or missing window guarantee.
CYCLE STRUCTURE OF THE DE BRUIJN GRAPH
There exist two methods to generate decycling sets of minimum size by Mykkeltveit (1972) and Champarnaud et al. (2004). These algorithms are of great theoretical importance as they settled a conjecture of Golomb (1967) on the size of an MDS. They are also practical algorithms as membership in these MDSs is testable in time and memory polynomial in k (i.e., the entire set does not need to be precomputed and stored). But, as we shall see, the space of all MDSs is much larger than these two MDSs.
We provide a method that uses only two simple graph operations—called F-move and I-move—that transform an MDS into another MDS. Furthermore, we conjecture that these two operations are sufficient to enumerate all MDSs. In other words, given a graph where the nodes are all the MDSs and the edges represent these operations, Conjecture 1 states that this graph is strongly connected. We give theoretical and computational evidence to support this conjecture.
This section describes the structure of the cycles in the de Bruijn and how through these two operations MDSs interact with the cycles. Although these two operations are similar in nature and together they might enumerate all MDSs, we describe them separately as they have qualitatively distinct effects on the MDSs (see Proposition 2 and Conjecture 2).
A pure cycling register (PCR), aka a conjugacy class, is a cycle in the de Bruijn graph made of the circular permutation of a k-mer. For example, the PCR of the 4-mer 1011 over the binary alphabet is . The PCRs form a partition of the k-mers, and therefore, any MDS must contain at least one k-mer from each PCR. We call a k-mer set with exactly one k-mer in each PCR a PCR set. The theorems of Mykkeltveit (1972) and Champarnaud et al. (2004) show that every MDS is a PCR set. In contrast, not every PCR set is an MDS.
F-moves
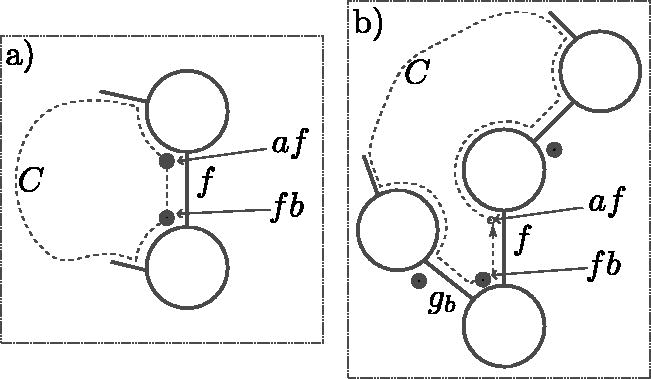
The left-companions (respectively right-companions) is the set of k-mers that have the same suffix (respectively prefix). Given , then are the left companions sharing the suffix f and are the right companions. See Figure 1 for examples. If , then the k-mers af and fa are equal (homopolymer ak), and this k-mer is in both the left- and right-companion sets for f. The homopolymers are the only such k-mers. Every other k-mer is a left companion for exactly one suffix and a right companion for a different prefix.
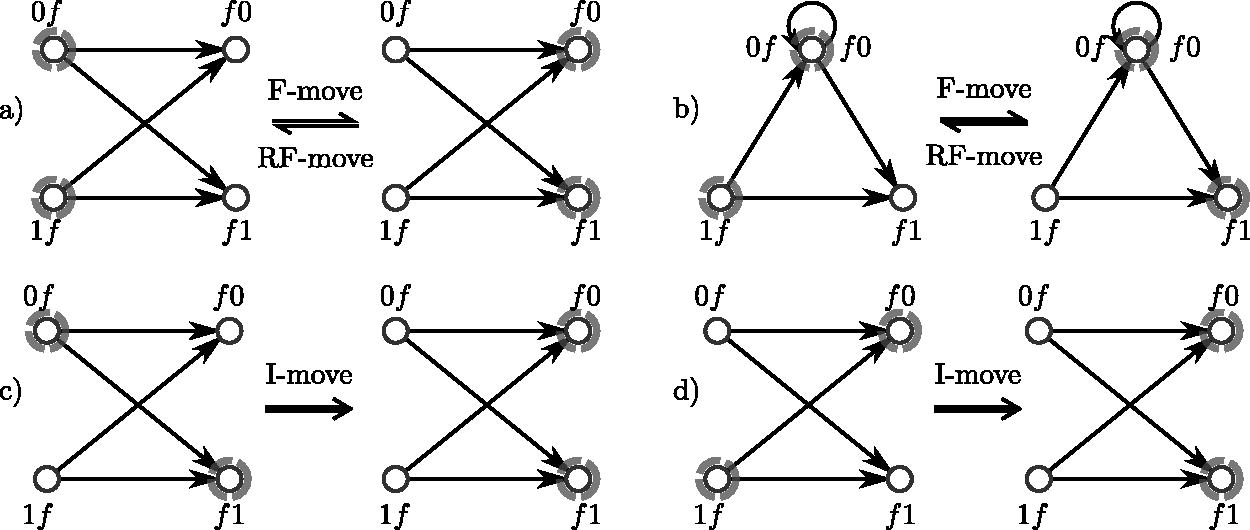
For , the left-companions (k-mers 0f and 1f for the binary alphabet) and right-companions (f0 and f1) induce a directed complete bipartite . (a) When the left-companions are in the set (left subgraph, highlighted in gray), an F-move replaces these nodes with the right-companions (right subgraph). An RF-move is the reverse operation, replacing the right-companions with the left-companions. (b) When one k-mer is a homopolymer (shown here with , so ), the induced subgraph is slightly different, but the F-moves and RF-moves are defined analogously. (c) One of the possible I-moves, , where a mixture of left- and right-companions is in the set. (d) The other possible I-move, . For any there are 1F-move, 1 RF-move, and I-moves possible, unless f is a homopolymer.
Proposition 1. (Existence of F-moves). In any MDS M, there existssuch that M contains the left companions of f and the right companions of.
Proof. By contradiction, assume that there is no such . Color all the nodes of the graph blue and do a random walk in the graph, starting from any node not in M, avoiding the nodes in M. Color in red the nodes traversed. Any k-mer m is the left companion of a suffix, say fm, and every outgoing edge from m is an incoming edge to a right companion of fm (see Fig. 1). Because no right-companion set is in M, it is always possible to continue the walk avoiding M from any m. Given that the graph is finite, the red nodes will eventually create a cycle, contradicting M being a decycling set. The same reasoning applies for the existence of f traversing edges in the reverse direction.☐
An F-move [named after Fredricksen (1992)] in M for is the operation of exchanging the set of left companions of f for the set of right companions, as shown in Figure 1. We use the functional notation fM to designate the set obtained by the valid F-move f from M: . This is a valid operation only when M contains lc(f). As a consequence of Proposition 1 there always exists a valid F-move in an MDS. The RF-move (reverse F-move) is the inverse operation, valid when M contains rc(f), , satisfying .
Proposition 2. (F-moves preserve decycling sets). Let M be an MDS such that, then fM is also an MDS.
Proof. If there is a cycle that avoids fM, then it must use one of the nodes in lc(f), otherwise it was already a cycle avoiding M. Any cycle using a node in lc(f) then must use a node in .☐
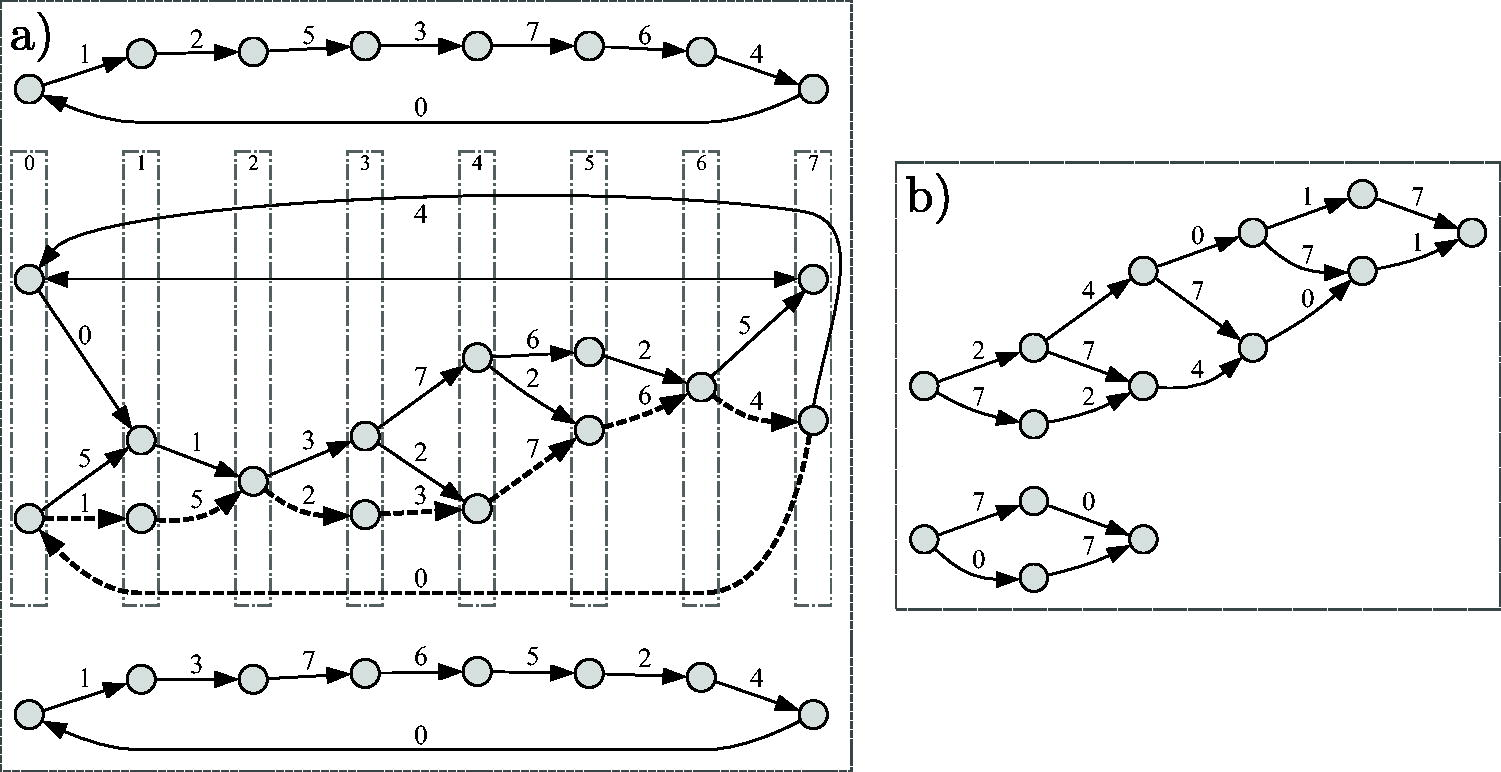
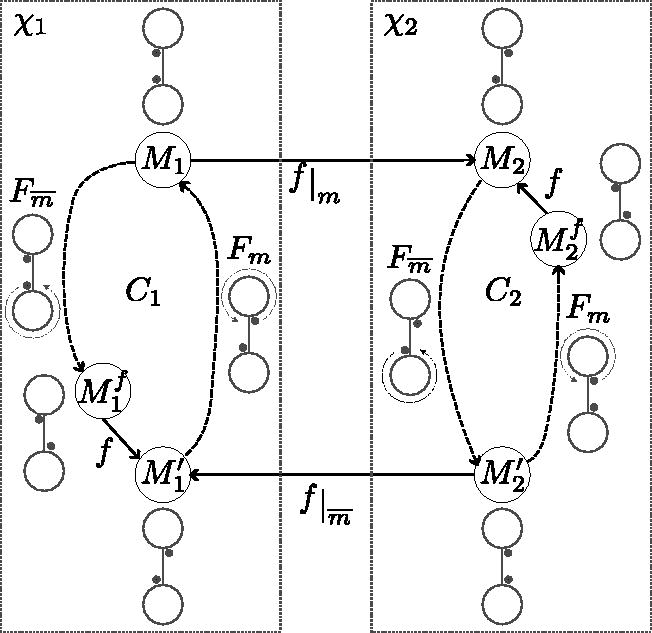
An analogous statement holds for RF-moves. F-moves give a procedure to enumerate some MDSs, starting, for example, from either the Mykkeltveit or Champarnaud set and repeatedly applying a (guaranteed-to-exist by Prop. 1) F-move. Unfortunately, not all MDSs are reachable using only F-moves. The MDS graph has all the MDSs as nodes and edges that represent F-moves operations between MDSs. is not connected, as seen in Figure 2, but its components have a well characterized structure (proof in Section 8.1).
(a) MDS graph with edge labels as numbers in representing the F-moves. There are 3 components. Each component is strongly connected and can be partitioned into layers with edges only from one layer to the next. The gray vertical boxes in the middle component highlight the layers, numbered from 0 to . Each layer in the middle component has size 1 or 2. An example of a cycle of length 8 with every F-move done exactly once is highlighted with dashed edges. (b) Example of 2 components of nondecycling PCR sets. The components are DAGs with a longest path less than 8 edges.
Proposition 3. ( component structure). For any σ and k, the components ofsatisfy the following:
Every component is strongly connected.
The length of every cycle is a multiple of, that is, for every cycle C, there existssuch that C is of length.
In a cycle of length, every possible F-moveoccurs exactly α times.
Every node is in a cycle of length(hence the girth is).
Each component is a-partite directed graph.
I-moves
An I-move, as in an “incomplete F-move,” is valid when M contains a mixture of left- and right-companions: for some and , either af or fa is in M. See Figure 1 for an example. For a given , there are distinct I-moves as follows: one for each possible choice of left-companion nodes in M, excluding the F-move [all of lc(f)] and the RF-move [none of lc(f)]. There is one exception as follows: when is a homopolymer, af = fa is both in lc(f) and rc(f), and the number of possible I-moves for f is .
An I-move is denoted by where is interpreted as a bit mask giving the nodes from lc(f) (i.e., the ath bit ma = 1 if and mb = 0 if ). With this notation, the F-move f would be equivalent to , whereas the RF-move would be . By definition the notation implicitly implies that it is a potential I-move and that m is neither the empty nor the full mask ( and ). An identical argument as for Proposition 2 shows that applying a valid I-move to an MDS also gives an MDS.
Although F-moves and I-moves seem like similar operations and both preserve MDSs, they have distinct effect on MDSs. First, empirically we observe that I-moves, unlike F-moves, are not always possible. MDSs always have a valid F-move (Proposition 1), whereas an MDS may not have any valid I-move. All of the F-moves are represented by an edge in every component of the MDS graph. By contrast, out of the potential I-moves, only a subset of those are valid operations. σ = 2 and k = 5 is an extreme case where no MDS has any valid I-move. Moreover, some I-moves can be a valid operation in one component and not in another.
Second, F-moves not only preserve the decycling property of MDSs, but they also preserve the “coverage” of every cycle by an MDS. To make this notion precise, define the hitting number of a cycle C of Dk by the MDS M as the size of their intersection: . Because M is a decycling set, necessarily . PCRs, for example, have a hitting number of 1, whereas any Hamiltonian cycle has a hitting number equal to .
Furthermore, the cycle signature of MDS M is the vector of all hitting numbers for all possible cycles: . Per the following proposition, F-moves preserve hitting numbers and signatures, whereas I-moves do not.
Proposition 4.
Let M be an MDS and f a valid F-move in M, then for any cycle C,.
For every valid I-movein MDS M, there exists a cycle C of Dk such that.
For any MDS M1, M2from the same component of.
For any MDS M1, M2from different components of.
Proof. Let f be a valid F-move in MDS M and C be a cycle of Dk. Because every outgoing edge of a node in lc(f) is an incoming edge to a node in rc(f), C must contain as many nodes from lc(f) as from rc(f) (which can be 0). Before the F-move, all the nodes from lc(f) and none from rc(f) are in M, whereas the opposite is true for fM. Hence the hitting number is unaffected by the F-move, proving 1.
Let be a valid I-move in M, , such that ma = 1 and mb = 0 (i.e., both af and fb are in M). Because Dk is -vertex connected (Sridhar, 1988), there exists a path P from fb to af that avoids . Path P followed by edge form a cycle C such that (af is in M but not in fM). By the same construction, there exists a “complementary” cycle using bf and fa such that . This proves 2.
As a component of GMDS is strongly connected by F-moves, statement 3 is a direct consequence of 1. A proof for 4 is given in Section 8.2.☐
As a consequence of this proposition, the hitting number and signature are constant over a component of the MDS graph, and the hitting number and the signature are well defined for a component χ. Because an I-move changes the signature, every I-move links MDSs from different components. Consider now the component graph with one node for each component of GMDS and a directed edge from component if there is an I-move from an MDS to . In fact, as stated in the following Proposition, Gcomp is an undirected graph (proof in Section 8.3).
Proposition 5. ( is undirected). Letbe a valid I-move from MDS M1 in component χ1 to M2 in χ2. Then there existsin, respectively, such that(whereis the bit-complement of m) is a valid I-move fromto.
Enumerating All MDSs
We make the following two conjectures regarding the use of I-moves to enumerate all MDSs.
Conjecture 1 (Connectivity by I-moves). The Gcomp graph is connected. Equivalently, every MDS is reachable from the Mykkeltveit MDS using a sequence of F-moves and I-moves.
This conjecture is supported by the previous theoretical results, in particular that all the components have a different signature and that an I-move always changes the signatures. For reasonable values of k (σ = 2, ), it is computationally feasible to enumerate all PCR sets and check which of them are also decycling sets. Using this brute force method we can confirm that is connected up to k = 7.
The following conjecture is computationally also verified up to k = 7 and exposes another fundamental difference between F-moves and I-moves. Every F-move is always valid in every component, whereas the valid I-moves identify a component (similar to the cycle signature). For a component χ, let the list of I-moves be .
Conjecture 2 (I-move signature). Every component inhas a distinct list of valid I-moves.
The validity of this second conjecture is likely related to the previous one. To prove Conjecture 1, one needs to show that for any two components there is a path of I-moves to go from χ1 to χ2. Conjecture 2 can be used as a guide to find that path: because , then there exists a valid I-move in either or (note that it is possible to have, for example, ). Do that I-move and repeat with the new components. Although in our testing Conjecture 2 is useful to find a path from χ1 to χ2, it is not sufficient as it does not guarantee that the size of the difference between the I-move lists is decreasing.
To create Table 1 we use both conjectures as follows: one to traverse the graph and the other to avoid enumerating a component more than once. The results in this table empirically show that, independent of the validity of the two preceding conjectures, the space of MDSs reachable using F-moves and I-moves is very large. The ability to traverse that previously unexplored space of MDSs allows us to create optimizing methods to create new sketching methods.
Gcomp and GMDS Properties for σ = 2
Method
Exhaustive
I-moves
k
2
3
4
5
6
7
8
9
10
# components
1
1
3
1
273
4
194,133
4,318,173
195,740,496
# MDSs
2
4
30
28
68,288
18,432
—
Layer range
1–1
1–1
1–2
1–2
1–48
28–153
—
“Layer range” gives, when possible, the range of the number of Minimum Decycling Sets (MDSs) in each layer of GMDS. The numbers for are exact, computed from the exhaustive list of MDSs. For columns , the number of components is correct provided the conjectures are correct, otherwise the numbers provided are under-estimations. For k = 8, the layer size and number of MDSs are estimated by sampling 100 random components. For k = 9, the numbers are likely severe under-estimations. For k = 10, computation is too expansive. For the DNA alphabet σ = 4, these numbers would grow even more quickly
Nondecycling PCR Sets
Nondecycling PCR sets may also have valid F-moves and I-moves, but there are significant differences with MDSs. Unlike MDSs (see Proposition 1), a nondecycling set is not guaranteed to contain sets of left- and right-companions. Even more, the analog graph to GMDS with nondecycling PCR sets as nodes and F-moves for edges is a nonconnected graph where each component is a DAG (see Fig. 2 and Section 8.4). There cannot be any F-moves between an MDS and a nondecycling set. In contrast, there can be an I-move from a nondecycling set to an MDS (but not the other way around).
REMAINING PATH LENGTH AND WINDOW GUARANTEE
By traversing the component graphs and the MDS graph, one can search for MDSs with desirable properties. Unfortunately, as seen in Table 1, every aspect of these graphs (i.e., number of MDS, number of components, layer size, and so on) seems to have super-exponential growth. Enumerating all MDSs for with the binary alphabet is likely not reasonable, and for the DNA alphabet it is even more difficult. In this section, we provide some methods to explore the space of MDSs more efficiently and study the window guarantee of MDSs.
Efficiently Traversing the Component Graph
As is seen in Table 1, the number of MDSs and components is increasing quickly with k, although an actual estimate of the growth as a function of k is not known. The memory used to traverse a component can be reduced by noticing that each component is partitioned into layers with edges only from one layer to the next (see Fig. 2). Therefore, it is only necessary to keep in memory the MDSs of the current and next layer to exhaustively enumerate every MDS in the component.
As each component contains at least one cycle of length , the number of MDSs grows by at least a factor of faster compared with components. In fact, it grows much faster as each of the layers has a size that grows fast with k as well (see Table 1). While the number of MDSs and the size of the layers varies significantly between components, in general it is not efficient to traverse an entire component to find all the valid I-moves. Using the following proposition, it is possible to find all the valid I-moves in a component by considering only one MDS.
Given an MDS M, any cycle C satisfies . The cycles with a hitting number of exactly 1, called constrained cycles, play an important role in the existence or not of a valid I-move: an I-move is only valid if there is no constrained cycle using edges of the I-move.
Proposition 6.Let, and let χ be a component of. Thenis not a valid I-move in any MDS of χ if and only ifsuch thatand there exists a constrained cycle using the edge.
This proposition, proved in Section 8.5, shows that to find the list of valid I-moves in the entire component it is sufficient to find the edges not covered by a constrained cycle in just one of the MDSs of the component. This holds, as by Proposition 1, that the list of constrained cycles is constant across the MDSs of a component. Moreover, tagging the edges covered by constrained cycle can be done with one depth first search for each k-mer in the MDS. The main advantage of this method is that its run time is independent of the number of MDSs in the component.
Remaining Path Length
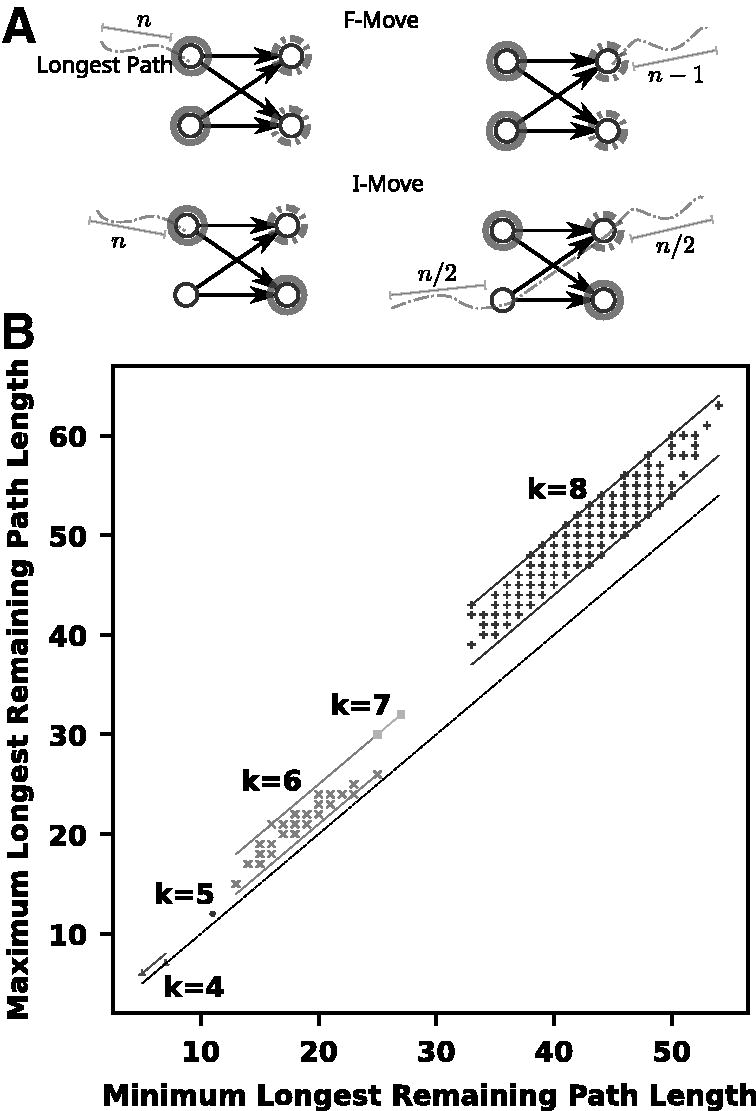
The remaining path length of an MDS, M, is the length of the longest path in the DAG obtained by removing the k-mers of M from Dk. Given a selection scheme that selects in a sequence the k-mers from M, the remaining path length is precisely the window guarantee of the scheme. The following proposition gives bounds on the effect of an F-move or I-move on the remaining path length (see Fig. 3).
Left: If a longest path does not start at a valid F-move f, i.e., one of the left-companion of f in solid gray is missing, then it could be extended to the left, contradicting maximality. Doing F-move f (changing solid gray for dashed nodes) can shorten the longest path by 1 node. Also, after doing F-move f, a path now ending in one of the solid gray nodes could be the longest and was extended by 1 node. If the path goes through an I-move , then doing the I-move cuts the path in two possibly equal parts. Right: Comparison of the minimum and maximum remaining longest path for components of for . Each point represents one connected component of the graph. The minimum and maximum remaining path lengths are computed over all the MDSs of a component. Therefore, the vertical distance of a point from the diagonal y = x (in yellow) shows the variation of remaining path length within a component. For k = 8, a subsample of 500 components was examined, as the total number of components is exceedingly large. The lines are drawn to depict the bounds of the increase between components. In all cases seen, the difference between the minimum and maximum remaining length within a component is in some range for an alpha that is less than k.
Proposition 7.An F-move or RF-move can increase or decrease the remaining path length by at most 1. An I-move can increase the remaining path length by at most 1 or decrease it by at most half.
Proof. First, notice that the longest path in must start at a valid F-move and end at a valid RF-move. Let be a longest path. The k-mer m1 is the right companion of some suffix f. Suppose there exists such that , then the path avoids M and is longer than P, contradicting its maximality. Therefore and f is a valid F-move in M. The proof is symmetrical for mn as the left-companion of some prefix with .
Because , the path P is shortened by 1 by the F-move f, which may shorten the longest path if there were no other paths of that length. In addition, (i.e., f is a valid RF-move in fM but it was not in M); hence, there might be maximal path ending at a left-companion of f with . Because the F-move only moved nodes forward by one edge, and the longest path may have increased by 1. The same argument applies to an RF-move.
For a valid I-move in M, the same reasoning applies for increasing by 1. In contrast, a longest path may have used an edge where . That is, . After the I-move, , and the path is now broken in up to two parts as follows: and . Therefore, the remaining path length could be halved if . ☐
Based on this, we implemented a simulated annealing algorithm to find the smallest and largest remaining path lengths among MDSs. The longest path for the MDS M is computed using a modified topological sort of the DAG . Suppose that we are computing the smallest remaining path length. Starting from a component of the MDS graph, the program performs a fixed number of random F-moves (2k by default) and computes the remaining path length for each MDS and keeps the minimum. Then, it finds all the valid I-moves in the current component as explained in Section 5.1, and it picks one at random.
After performing the I-move, in the new component, the remaining path length is computed for 2k MDSs reachable by F-moves and a new minimum is computed. If this new minimum is lower than the previous minimum, then the new component becomes the current component. Otherwise, it becomes the current component only with some small probability. Then the process is repeated from the current component for a fixed number of iterations. As is traditional with simulated annealing, the probability to jump to “worse” components decreases over time.
Table 2 shows the remaining path length for the two previously known algorithms to generate MDSs and the range of remaining path length. These ranges are either exact when an exhaustive list of MDSs is computable and approximated using simulated annealing otherwise. Based on the pattern that the Mykkeltveit set is always at or close to the minimum remaining path length, we conjecture that it holds for all parameters k and σ.
The Remaining Path Length for the Mykkeltveit and Champarnaud Sets Compared With the Range of Remaining Path Length
σ
Algorithm
k
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
2
Mykkeltveit
5
11
21
27
39
55
74
89
119
143
194
219
253
299
408
437
539
Champarnaud
7
11
21
27
47
57
94
112
190
209
367
415
683
756
1343
1393
2560
SA Min
5
11
13
25
32
48
70
89
119
143
194
SA Max
7
12
26
32
55
80
116
158
257
288
387
4
Mykkeltveit
21
41
77
111
145
231
330
403
616
Champarnaud
27
39
119
141
429
520
1601
1765
6180
SA Min
20
41
77
111
145
SA Max
34
66
149
270
530
For σ = 2 and , the range of remaining path length is computed exactly from the exhaustive list of MDSs. The other values in the simulated annealing (SA) Min and SA Max rows are estimated using an SA algorithm and are underlined.
Conjecture 3.For a given σ, let, respectively, be the smallest, largest, and Mykkeltveit set remaining path lengths. Thenasymptotically in k.
Per-Component Remaining Path Length
Proposition 7 gives a bound on the change in the remaining path length as the MDS graph is traversed using F-moves and I-moves. Within one component, given that every MDS is in a cycle of length σk, the remaining path length along this cycle could change by up to . In other words, this proposition only gives an exponential bound on the range of remaining path length within a component.
The graph in Figure 3 has a point for each component at the coordinate where is the minimum of the remaining path length over all the MDSs of the component χ, and is the maximum. The vertical distance from the diagonal y = x represents the range of remaining path lengths within a component. We observe for on the binary alphabet that the range is bounded by O(k).
Conjecture 4.Within a component of GMDS, the range of remaining path length is O(k).
There are plausible reasons for having such a small range. Consider the following two extremes: (1) there are many F-moves and RF-moves valid at the same time in an MDS M, (2) there is only 1 F-move and 1 RF-move valid in M. In the first case, doing one of these F-moves or RF-moves affects the maximal paths that start or end at these moves. Consequently, many of these moves change the length of paths that are not the longest. In other words, these moves have no effect on the remaining path length. In the second case, it is possible to show that doing the 1 valid F-move does not change the remaining path length (the longest path is truncated by its first node and extending by one node, hence not changing in length). This type of situation is likely to happen when there are few F-moves and RF-moves possible. In both cases, most F-moves do not affect the remaining path length.
This conjecture partially justifies only exploring O(k) MDSs within one component in the simulated annealing algorithm in Section 5.2.
DISCUSSION
Proportion of MDSs. A simple algorithm to generate a random MDS, sampling the space of MDSs uniformly, is to select at random k-mer from each PCR and check whether it is decycling, and to resample if not. Even though the space of MDSs is (maybe surprisingly) large, it is nonetheless only a tiny fraction of the PCR sets. The number of PCR sets is easily computable (Fredricksen and Kessler, 1986), and asymptotically, there are PCR sets. There is no formula for the number of MDSs, but based on the numbers from Table 1, for k = 8, of the PCR sets the proportion that are MDSs is only . For k = 9 that proportion is essentially 0. Thus, the random sampling method is not of any practical use.
In that sense Conjecture 1, provided it is true, is an efficient method to enumerate all MDSs as only MDSs are ever considered without the need to filter out an overwhelming number of nondecycling sets. Even if this conjecture is eventually proven wrong, the F-moves and I-moves allow us to explore a large subspace of MDSs and, using simulated annealing or more advanced machine learning methods, to find MDSs with desirable properties.
Moreover, on the theoretical side, providing evidence for this conjecture lead us to a deeper understanding of the space of MDSs and to formulate the other conjectures.
Mykkeltveit set and short windows. It is surprising (or lucky) that the first algorithm for constructing MDSs by Mykkeltveit (1972) gives a set with close to the shortest remaining path length. This fact may explain retrospectively the success of previous methods using this set as the starting point to design minimizers schemes (Orenstein et al., 2016, 2017; Ekim et al., 2020; Pellow et al., 2023). The growth of the remaining path length for the Mykkeltveit set is well characterized (Zheng et al., 2020a): it is and . Fitting the data from Table 2 we obtain an exponent of 3.12 ± 0.14, suggesting an actual growth of . Provided that Conjecture 3 holds, this would answer the question of the shortest window guarantee that is possible using an MDS. For comparison, fitting the Champarnaud data gives an exponent of 6.1 ± 0.59.
Longest remaining path length. Conjecture 4 only suggests a bound on the range of remaining path length within a component of GMDS. A legitimate question is what is the bound of the range in GMDS as a whole. Figure 3 could suggest that this range is polynomial in k, although the trend in this figure is much too short to elevate this statement to a conjecture. Given the known results bounding the longest remaining path of the Mykkeltveit set by , this would mean a polynomial bound on the remaining path length of MDSs.
This statement seems counterintuitive at first (and is, of course, not proven). We saw in Section 3 that syncmers have a window guarantee of ; hence, there exists DSs that are not of minimum size that have exponentially long remaining paths. How then can sets with fewer k-mers (MDSs) have a shorter remaining path length? The intuition is as follows. In the syncmers construction, we chose one exponentially long path (length ) through the graph, whereas every node not on this path is added to the DS M. The size of the DS is exponential as well: it takes many nodes, guiding that long path, to prevent cycles. In contrast, the size of an MDS is , which is . The average remaining path length is k and there are too few k-mers in an MDS to guide an exponentially long path to prevent it from creating cycles (i.e., to have back edges).
In practice, even a window guarantee may be too long, and an MDS may need to be extended to a decycling set with even shorter remaining path length [as done in Orenstein et al. (2017)]. Hence starting with an MDS with the shortest possible remaining path length is advantageous. Even if Conjecture 3 is true, it does not prevent the existence of decycling sets with smaller remaining path lengths than the Mykkeltveit set. Whether MDSs with remaining path length of exist is still an open question.
CONCLUSION
The window guarantee is an important requirement, theoretically and practically, to define and optimize sketching methods. As discussed, an underlying concept that can be extracted from the definition of this guarantee in any local sketching method is a set of nodes in the de Bruijn graph which are unavoidable (i.e., decycling). While many such sets exist, the minimum-sized sets have important properties that can be exploited and examined. In this work, we described some of the first theoretical findings on properties of these sets, as well as a method to traverse many (if perhaps not all) MDSs for a given k-mer length. We also showed that the choice of MDS, whether direct or as an implication of the design of the sketching method, does have an impact on the strength of the window guarantee. Although we provide our major results as conjectures, we present significant evidence to support these claims.
EXTENDED PROOFS
MDS Graph Structure
Lemma 1 (Commutative property). Let M be an MDS andbe two valid F-moves in M, then f1 is a valid F-move in, f2 is valid in, and.
Proof. The left companions of f1 and f2 are all in different PCRs. Hence, after doing the F-move f1 or f2, the other F-move is still valid. Moreover, regardless of the order in which the F-moves are performed, the resulting set is the same.
There is no equivalent statement to Lemma 1 for I-moves: if are two valid I-moves in M, then may not be valid in .
Lemma 1 applies to a sequence of F-moves . Suppose that this is a valid sequence of F-moves starting from MDS M and that both fi and are valid F-moves in , then and .
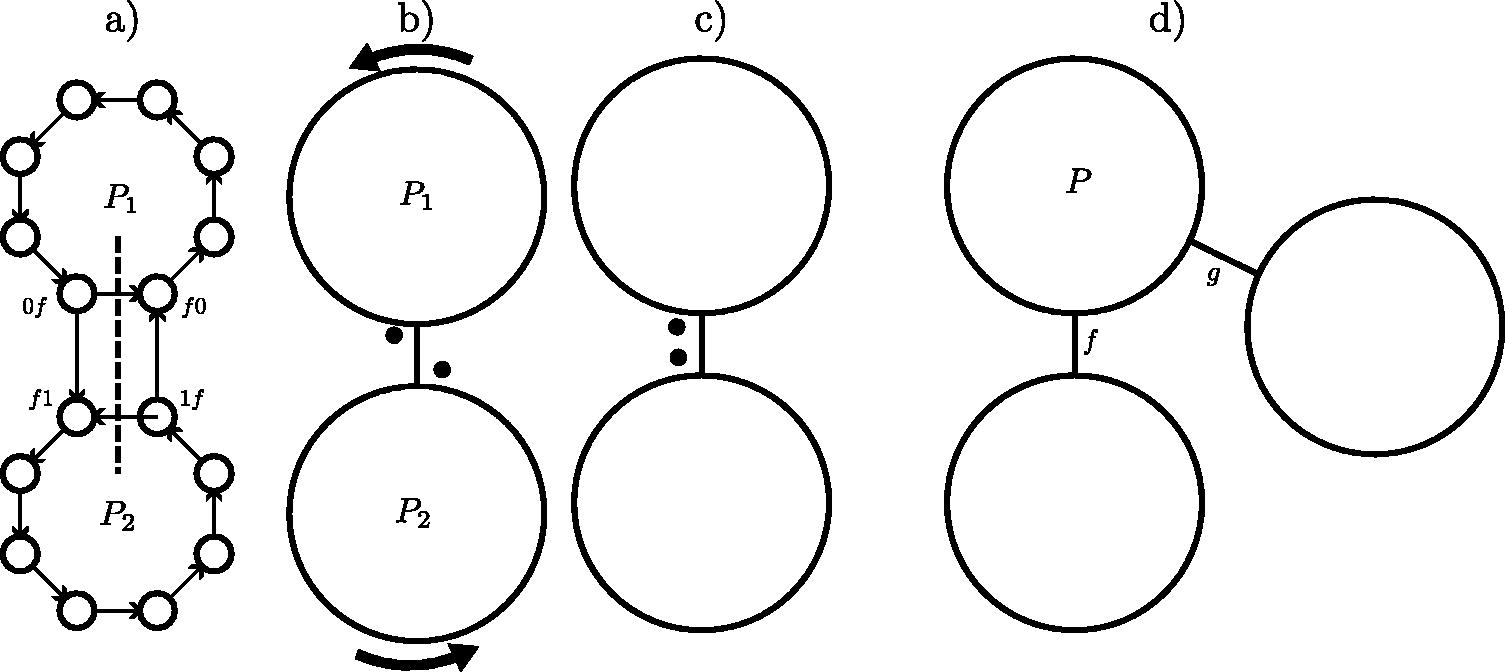
In the following proofs, we use the simplified representation for PCRs, F- and I-moves given in Figure 4. For simplicity, the figure shows an example with the binary alphabet. When , an F-move f represents a hyperedge between σ PCRs rather than a simple edge as shown.
Simplified representation of PCRs, F-moves, and I-moves when σ = 2. (a) shows two PCRs from the de Bruijn graph Dk. Every PCR is a circle, and they are all oriented counterclockwise (see PCR P1 and P2 here). Let f be an F-move that involves P1, P2. Here P1 has the edge , and P2 has : these are the PCR edges. The cross-PCR edges and form antiparallel edges between P1 and P2. (b) The simplified PCR/pebbles representation shows PCRs as large cycles without representing individual k-mers and only representing the F-move edges of interest. The elements from the MDS in each PCR (the pebbles) are small black circles that can travel only counterclockwise around the PCR. An F-move is an edge between P1 and P2 and acts as a semaphore: a pebble can move one step around the PCR and across the edge of f only when the other pebbles are present next to the edge in the other PCR [i.e., lc(f) is in the MDS], as shown in b), and all pebbles move across the edge at the same time. (c) The position of the pebbles for the I-move : bit 0 is set but not bit 1, so the pebbles are on 0f and f1 (left side of the edge of f). The top pebble can move across the edge, counterclockwise, whereas the lower one stays still. For I-move with bit 0 unset and bit 1 set, the pebbles would be on 1f and f0, on the right side of the edge of f. (d) If F-moves f and g have a PCR P in common, then, because F-moves act like semaphores, it is not possible to do the F-move f twice before g is done once. For the pebble to go around P to do f a second time, necessarily the F-move g was done as well. PCR, pure cycling register.
Proposition 3. ( component structure). For any σ and k, the components ofsatisfy the following:
Every component is strongly connected.
The length of every cycle is a multiple of , that is, for every cycle C, there exists such that C is of length .
In a cycle of length , every possible F-move occurs exactly α times.
Every node is in a cycle of length (hence the girth is ).
Each component is a -partite directed graph.
Proof of points 2 and 3, length of cycles. Every PCR is a cycle in Dk, and an MDS M is seen as pebbles sitting on the k-mers (see Fig. 4b). There is one pebble per PCR. An F-move involves σ distinct PCRs (edges are each in their own PCR). Hence, an F-move is a hyperedge connecting σ PCRs. An F-move is like moving the pebbles along σ PCRs at a time, from left-companions to right-companions, and this move is legal only if . In that sense, an F-move is like a semaphore: pebbles can move only if all their left-companions are present in the set.
First, because every MDS has a valid F-move and a component of GMDS is finite, a component must have a cycle. Let be a cycle of MDSs in GMDS, and equivalently is a list of F-moves such that (indices taken modulo n). After doing F-move f0, the pebble on at least one PCR, say P0, has moved. Because C is a cycle, by the time is done, all pebbles are back on their respective starting spot. Meaning the pebble on P0 went all the way around (possibly multiple times) P0. To move around P0 with F-moves, the pebbles in the PCRs adjacent to P0 must have moved as well. By the time is done, every pebble in the PCRs adjacent to P0 has moved around its respective PCR. By transitivity, and because the de Bruijn graph is strongly connected, every pebble on every PCR has gone around its PCR after is done. Because every pebble went around its PCR, this means that every one of the F-moves was done and .
Conversely, because the F-move/hyperedge act as semaphores, it is not possible for a pebble on a PCR to do more rotations around its own PCR than the pebbles on the adjacent (by hyperedge) PCRs. To see this, consider the starting position of the pebble on PCR P0. For this pebble to start a second turn around P0, all of its left-companions must be back on their starting spot and also start a second turn around their own PCRs. This holds for all PCRs by transitivity.
Hence, in a cycle of the MDS graph, the pebbles of all PCRs go around the same number of times, say α, and the number of F-moves in the cycle C is .☐
Proof of point 1, strongly connected. As in the previous proof, there exists a cycle in GMDS, and its edges are with .
We show that for any node Mi of this cycle and any neighbor M of Mi, reachable by an F-move or RF-move from Mi, M and Mi are in a cycle. If this holds, by transitivity of the relation “being in the same strongly-connected component”, any pair of nodes in the component are in a cycle and the component is strongly connected.
Without loss of generality, we prove this property for the neighbors of M0 (see Fig. 5). It is a consequence of the commutativity of the F-moves (Lemma 1). Let be a neighbor of M0 for some . Because in a cycle all F-moves occur, there exists a first such that (and ). f is valid in M0; hence, it is also valid in M1, and recursively in . Therefore, f commutes with , and the sequence of F-move is another path from M0 to that is going through M. This path followed by the remainder of C from back to M0 is a cycle that includes both M0 and M.☐
Example of a cycle in . The outer circle is , a cycle of length . is a neighbor of M0 not on C. Because f must occur in C, here , then f commutes with . Hence is also a cycle in , and it contains M0 and M.
Proof of point 4, cycle length. Let M be a MDS on a cycle C in GMDS. It is of length , with by point 2. Suppose that . Let be the sequence of F-moves representing that cycle. Every distinct F-move occurs exactly α times in that sequence. We show that the sequence can be reordered so that the different F-moves occur at the first positions of the sequence.
If it is not already the case that the first F-moves are distinct, there must be an F-move f that occurs twice in the list before an F-move g occurs for the first time. Let i < j be two indices which are the first two occurrences of f in the sequence (i.e., ) and such that j + 1 is the first occurrence of g (). If any of the PCRs involved in the F-move f are also involved in the F-move g, then it is not possible to use f twice in C before using g (see Fig. 4d). Therefore the PCRs involved in the F-moves f and g are distinct, and g must be a valid F-move just before the second use of f as well. In other words, fj and commute.
Repeated swapping of F-moves leads to the desired sequence of F-moves with all distinct F-moves in the first positions, which induces a cycle of length containing M.☐
Proof of point 5,-partite. Partition the nodes of a component of GMDS as follows. We create sets: . Let M0 be an arbitrary MDS of the component and assign it to the set . For every other MDS M, take a shortest path in GMDS. Assign M to the partition with index .
Because M0 is in a cycle of length , every set has at least one MDS assigned to it. Moreover, every MDS is assigned to exactly one set. Hence the sets form a partition of the MDSs in the component.
An edge between MDSs in sets and with would imply the existence of a cycle containing M0 of length , which is not possible.☐
Cycle Signature Is Unique per Component
An MDS M is called f-terminal if the only valid F-move in M is f.
Lemma 2.For anyand in any component of GMDS, there exists an f-terminal MDS.
Proof. From Proposition 3, in any component there exists an MDS where f is a valid F-move. If there exists other valid F-moves than f in , do them recursively. That is, we do every possible F-move in but refuse to do f. This creates a path P of MDSs in GMDS starting at that does not contain f as an edge.
Because every cycle in GMDS contains every possible F-move, P cannot induce a cycle, and it must terminate at an MDS M. By construction M is f-terminal.☐
An f-terminal MDS M has a useful property: every maximal path in Dk that avoids M (as created by a walk like in Proposition 1) must start at a k-mer . Equivalently, any walk in Dk that avoids M following edges backward ends at some .
Proposition 4.
Let M be an MDS and f a valid F-move in M, then for any cycle C, .
For every valid I-move in MDS M, there exists a cycle C of Dk such that .
For any MDS M1, M2 from the same component of .
For any MDS M1, M2 from different components of .
Points 1–3 were proven in Section 4.2.
Proof of point 4, different signatures. Fix. Because the signature is constant in a component by point 1, and because by Lemma 2 there always exists an f-terminal MDS in a component, it suffices to show that the signatures are different for two f-terminal MDSs M1, M2 from different components. We will construct a cycle C in Dk that has different hitting numbers between the components: .
M1 and M2 are in different components, so they are distinct MDSs and there exists a PCR R where the selected k-mer is different. That is, . Take a path P in Dk following edges backward from node 0f (which is in both M1 and M2 because they are f-terminal) to m1 that avoids nodes . Path P exists because Dk is -connected. Because , there must exist a first node which is in .
Let P1 be the restriction of the path P from 0f to m and, without loss of generality, assume that . By construction, .
Let P2 be a path created by a maximal random walk in Dk, following edges backward, starting from m and that avoids M2. Because M2 is f-terminal, the walk ends at a node . By construction, (P2 avoids nodes from M2 but may contain nodes from M1).
Two cases can happen. First case, there exists a first node . Then define the cycle C as the restriction of P1 from to m followed by the restriction of P2 from m to . Second case, and define the cycle C as the concatenation of P1, P2 and backward edge .
In both cases, C satisfies by construction .☐
Gcomp Is Undirected
Proposition 5. ( is undirected). Letbe a valid I-move from MDS M1 in component χ1 to M2 in χ2. Then there existsin, respectively, such that(whereis the bit-complement of m) is a valid I-move fromto.
Proof. See Figure 6. In component χ1, by Proposition 3, there is a cycle C1 of length that contains MDS M1, and this cycle has f that has an F-move. Hence, , where is the MDS where f is a valid I-move and . Equivalently, looking at the edges, , where are sequences of F-moves.
Simplified example for finding the complementary I-moves, when σ = 2. On the left box, component χ1 and component χ2 on the right of GMDS. The cycles C1, C2 are cycles in χ1 and χ2, respectively. The simplified PCR/pebble drawings represent the position of the pebbles on the PCRs of Pm (top PCR) and (bottom PCR). The edge between these PCRs represents f. The PCR/pebbles drawings next to the MDS nodes represent the state of the PCRs for these MDSs, whereas the drawings next to the dotted line (sequences of F-moves) represent the action of the sequence of F-moves on the pebbles. From the cycle C1 in χ1, we construct cycle C2 in χ2 by swapping the order of the F-moves: . These cycles go through the desired MDSs and that are linked by the complementary I-move .
By hypothesis is a valid I-move in M1, which means that if ma = 1, then and otherwise.
Let’s call Pm the set of PCRs that contain af when ma = 1 and the PCRs containing af when ma = 0 (Pm contains only the top PCR in Fig. 6 and the bottom PCR).
In , f is a valid F-move, which means that for all . In other words, the sequence of F-moves made by the pebbles in the PCRs in goes around from fa to af, whereas the pebbles in the PCRs in Pm did not move. (The only way for the pebbles in the PCRs in Pm to move is to do F-move f, which by construction is not in ).
Similarly, the sequence of F-moves Fm made by the pebbles in the PCRs in Pm goes around from fa to af, whereas the pebbles in the PCRs of did not move.
Now from M1 do the valid I-move . This advances the pebbles in the PCRs of Pm from af to fa (forward by 1 edge), to get to M2 in component χ2, where . The position of the pebbles in M1 and M2 agrees everywhere except on the PCRs of Pm. Because the F-moves in do not affect the PCRs of Pm, the sequence is a valid sequence of F-moves in M2 as well.
for all . Applying to M2 leads to MDS where if ma = 0 and otherwise. In other words, I-move is valid in . It is easy to check that doing the I-move gets back to .
For completion, one can check that the sequence of F-moves Fm applies to because and only differ on the pebbles on the PCRs of and Fm does not affect those PCRs. Applying Fm to get to where f is a valid F-move and .
Therefore, the cycle is a valid cycle in χ1 and contains M1 and , whereas is valid in χ2 and contains M2 and .☐
Nondecycling PCR Sets
Proposition 8.Let GPCR be the graph with nondecycling PCR sets as nodes and F-moves as edges. Then each component of G is a DAG.
Proof. Suppose there exists a cycle in GPCR, where . Because M1 is not decycling, then there exists a cycle C in . Because RF-moves preserve the hitting number, C is also a cycle in , and by induction a cycle in . From the proof Proposition 3, any cycle must do every F-move to return to the starting set, and the union of all the left-companions of the F-moves is the set of all k-mers. This is a contradiction.☐
I-moves and Constrained Cycles
Proposition 6.Let, and let χ be a component of. Thenis not a valid I-move in any MDS of χ if and only ifsuch that, and there exists a constrained cycle using the edge.
Proof. Let be a potential I-move with ma = 1 and mb = 0 ().
Suppose there exists a constrained cycle C in the de Bruijn graph Dk using the edge and . If is a valid I-move in an MDS , then by definition , hence . This contradicts that C is constrained (see Fig. 7a).
A) The with ma = 1 and mb = 0 is not possible because . When the I-move is valid, necessarily C’s hitting number must be at least 2. B) Suppose is never valid, then a backward walk creates a cycle with hitting number 1 using the edge .
Conversely, suppose that is not a valid I-move in any MDS of χ. Let be an MDS where f is a valid F-move and . Then . Define , that is, for all right-companion of f, .
From M recursively do all valid F-moves except for the F-moves gc where mc = 0 to obtain where the only valid F-moves are exactly those that we refused to do. There must exist such that ma = 1 and , otherwise is a valid I-move in (see Fig. 7b). From af do a walk that avoids using backward edges. This walk must end at one of the right-companions of the valid F-moves in , that is, there exists b such that walk ends at . By construction there is a backward edge . Then follow the backward edge to create a cycle C. By construction the only node from in cycle C is fb, hence and C uses the edge with ma = 1 and mb = 0.☐
Footnotes
ACKNOWLEDGMENT
This article was deposited as an arXiv preprint: doi:10.48550/arXiv.2311.03592.
AUTHORS’ CONTRIBUTION
G.M.: Conceptualization, Formal analysis, Software, Funding acquisition, and Writing—Original Draft. D.D.: Software, Validation, Visualization, and Writing—Review & Editing. C.K.: Conceptualization, Supervision, Funding acquisition, and Writing—Review & Editing.
AUTHOR DISCLOSURE STATEMENT
C.K. is a cofounder of Ocean Genomics, Inc; G.M. is VP of software engineering at Ocean Genomics, Inc. D.D. has no conflict of interest.
FUNDING INFORMATION
This work was supported, in part, by the US National Science Foundation [DBI-1937540, III-2232121], the US National Institutes of Health [R01HG012470], and by the generosity of Eric and Wendy Schmidt by recommendation of the Schmidt Futures program.
References
1.
ChamparnaudJ-M, HanselG, PerrinD. Unavoidable sets of constant length. Int J Algebra Comput, 2004; 14(02):241–251; doi: 10.1142/S0218196704001700
2.
DeBlasioD, GbosiboF, KingsfordC, et al.Practical universal k-mer sets for minimizer schemes. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, BCB ’19, 167-176, New York, NY, USA, 2019. ACM; doi: 10.1145/3307339.3342144
EdgarR. Syncmers are more sensitive than minimizers for selecting conserved k-mers in biological sequences. PeerJ, 2021; 9:e10805; doi: 10.7717/peerj.10805
5.
EkimB, BergerB, ChikhiR. Minimizer-space de Bruijn graphs: Whole-genome assembly of long reads in minutes on a personal computer. Cell Syst, 2021; 12(10):958–968.e6; doi: 10.1016/j.cels.2021.08.009
6.
EkimB, BergerB, OrensteinY. A randomized parallel algorithm for efficiently finding near-optimal universal hitting sets. In SchwartzR., editor, Research in Computational Molecular Biology, Lecture Notes in Computer Science, 37–53, Cham, 2020. Springer International Publishing. 10.1007/978-3-030-45257-5_3
7.
FredricksenH. A new look at the de Bruijn graph. Discrete Applied Mathematics, 1992; 37:193–203; doi: 10.1016/0166-218X(92)90133-U
8.
FredricksenH, KesslerJI. An algorithm for generating necklaces of beads in two colors. Discrete Mathematics, 1986; 61(2–3):181–188; doi: 10.1016/0012-365X(86)90089-0
HoangM, ZhengH, KingsfordC. DeepMinimizer: A differentiable framework for optimizing sequence-specific minimizer schemes. In Pe’erI., editor, Research in Computational Molecular Biology, 52-69, Cham, 2022b. Springer International Publishing; doi: 10.1007/978-3-031-04749-7_4
12.
JainC, RhieA, ZhangH, et al.Weighted minimizer sampling improves long read mapping. Bioinformatics, 2020; 36(Suppl_1):i111–i118; doi: 10.1093/bioinformatics/btaa435
13.
KarpRM, RabinMO. Efficient randomized pattern-matching algorithms. IBM J Res & Dev, 1987; 31(2):249–260; doi: 10.1147/rd.312.0249
14.
KilleB, GarrisonE, TreangenTJ, et al.Minmers are a generalization of minimizers that enable unbiased local Jaccard estimation. Bioinformatics, 2023; 39(9); doi: 10.1093/bioinformatics/btad512
MarçaisG, PellowD, BorkD, et al.Improving the performance of minimizers and winnowing schemes. Bioinformatics, 2017; 33(14):i110–i117; doi: 10.1093/bioinformatics/btx235
17.
MykkeltveitJ. A proof of Golomb’s conjecture for the de Bruijn graph. Journal of Combinatorial Theory, Series B, 1972; 13(1):40–45.
18.
OrensteinY, PellowD, MarçaisG, et al.Compact universal k-mer hitting sets. In Algorithms in Bioinformatics, Lecture Notes in Computer Science, 257–268. Springer, Cham, 2016; doi: 10.1007/978-3-319-43681-4_21
19.
OrensteinY, PellowD, MarçaisG, et al.Designing small universal k-mer hitting sets for improved analysis of high-throughput sequencing. PLoS Comput Biol, 2017; 13(10):e1005777; doi: 10.1371/journal.pcbi.1005777
20.
PellowD, PuL, EkimB, et al.Efficient minimizer orders for large values of k using minimum decycling sets. Genome Res, 2023; 33(7):1154–1161; doi: 10.1101/gr.277644.123
21.
RobertsM, HayesW, HuntBR, et al.Reducing storage requirements for biological sequence comparison. Bioinformatics, 2004a;20(18):3363–3369; doi: 10.1093/bioinformatics/bth408
22.
RobertsM, HuntBR, YorkeJA, et al.A preprocessor for shotgun assembly of large genomes. J Comput Biol, 2004b;11(4):734–752; doi: 10.1089/cmb.2004.11.734
23.
RouzéT, MartayanI, MarchetC, et al.Fractional hitting sets for efficient and lightweight genomic data sketching, 2023.
24.
SchleimerS, WilkersonDS, AikenA. Winnowing: Local algorithms for document fingerprinting. In Proceedings of the 2003 ACM SIGMOD International Conference on Management of Data, SIGMOD ’03, ACM. New York, NY, USA, 2003. 76–85; doi: 10.1145/872757.872770
25.
ShawJ, YuYW. Theory of local k-mer selection with applications to long-read alignment. Bioinformatics, 2022; 38(20):4659–4669; doi: 10.1093/bioinformatics/btab790
26.
SridharMA. On the connectivity of the De Bruijn graph. Information Processing Letters, 1988; 27(6):315–318; doi: 10.1016/0020-0190(88)90219-0
27.
ZhengH, KingsfordC, MarçaisG. Lower density selection schemes via small universal hitting sets with short remaining path length. In SchwartzR., editor, Research in Computational Molecular Biology, Lecture Notes in Computer Science, 202-217, Cham, 2020a. Springer International Publishing; doi: 10.1007/978-3-030-45257-5_13
28.
ZhengH, KingsfordC, MarçaisG. Improved design and analysis of practical minimizers. Bioinformatics, 2020b;36(Suppl_1):i119–i127; doi: 10.1093/bioinformatics/btaa472