
Abstract
Background:
Expanded analysis of tumor genomics data enables current and future patients to gain more benefits, such as improving diagnosis, prognosis, and therapeutics.
Methods:
Here, we report tumor genomic data from 1146 cases accompanied by simultaneous expert analysis from patients visiting our oncological clinic. We developed an analytical approach that leverages combined germline and cancer genetics knowledge to evaluate opportunities, challenges, and yield of potentially medically relevant data.
Results:
We identified 499 cases (44%) with variants of interest, defined as either potentially actionable or pathogenic in a germline setting, and that were reported in the original analysis as variants of uncertain significance (VUS). Of the 7405 total unique tumor variants reported, 462 (6.2%) were reported as VUS at the time of diagnosis, yet information from germline analyses identified them as (likely) pathogenic. Notably, we find that a sizable number of these variants (36%–79%) had been reported in heritable disorders and deposited in public databases before the year of tumor testing.
Conclusions:
This finding indicates the need to develop data systems to bridge current gaps in variant annotation and interpretation and to develop more complete digital representations of actionable pathways. We outline our process for achieving such methodologic integration. Sharing genomics data across medical specialties can enable more robust, equitable, and thorough use of patient’s genomics data. This comprehensive analytical approach and the new knowledge derived from its results highlight its multi-specialty value in precision oncology settings.
INTRODUCTION
The field of precision oncology has been significantly transformed by advances in high-throughput sequencing and the identification of targetable tumor-specific molecular changes. As a result, tumor genetic testing has become an integral component of diagnosis, prognosis, and therapeutic decision-making (Malone et al., 2020). However, health care professionals face the complex task of translating and interpreting genetic reports obtained from in-house or commercial testing laboratories.
To aid in the interpretation of variants of uncertain significance (VUS), genomic tumor boards play a crucial role. The conclusions drawn by these boards naturally depend on the data aggregated for review. For instance, one tumor board may classify a variant as Tier II, utilizing established guidelines (Li et al., 2017), if they consider a recently published study surveying a rare cancer type, while another tumor board that does not consider the same study may classify it as a VUS. Thus, the determinations of tumor boards depend on the breadth of aggregated data and how it is presented, posing a practical and common data science requirement in genomics labs. Unfortunately, genetic VUS is often ignored and not iteratively reassessed as new knowledge emerges (Deignan et al., 2019). This process leads to a significant potential for overlooking essential genomic findings, resulting in the underutilization of precision medicine (Esteva et al., 2019; Norgeot et al., 2019). Furthermore, while tumor genomics predominantly focuses on identifying indications for therapies and clinical trials, many genetic variants relevant to patients and the precision oncology field fall outside these considerations. Hence, this study aims to evaluate the potential for gaining additional knowledge by reanalyzing tumor variants reported as VUS while incorporating less commonly used genomics data in oncology settings. We assembled a substantial cohort of 1146 tumors from patients evaluated by a multi-disciplinary Precision Oncology clinic. The reanalysis involved developing an enhanced variant interpretation approach, utilizing semantic-based algorithms that link knowledge of pathogenicity derived from heritable cancer syndromes, inherited non-cancer disorders, and mutations recurrent in other tumor types (i.e., “off-tumor” mutations). Our findings reveal that 44% of the cohort had tumor-reported VUS that align with potentially actionable pathways or are considered pathogenic in a germline setting. These results support the notion that integrating information on genetic variants across rare diseases and cancer enhances the classification of variants and leads to new insights relevant to precision oncology.
METHODS
Genomics data annotation
Reanalysis of tumor profiling data obtained from Foundation Medicine for the first 1146 patients seen at Froedtert Hospital (2007 and 2018) was performed. Extensible markup language files, a human-readable and computable format, for each case were obtained with all clinically reported elements identified with specific tags. The data received were reviewed in multiple ways including mapping of patient identifiers to ensure homogeneity and ability to link back to our electronic medical record (EMR)-based data warehouse. Our analytic pipeline leveraged the BioR annotation engine (Kocher et al., 2014) to annotate variants with population allele frequencies from gnomAD, histology and pathology from COSMIC (Catalog of Somatic Mutations in Cancer; Forbes et al., 2011), germline phenotype records from ClinVar (Landrum et al., 2014) and HGMD (Human Gene Mutation Database; Stenson et al., 2012), and disease inheritance patterns from OMIM (Online Mendielian Inheritance in Man; McKusick-Nathans Institute of Genetic Medicine, June 2019). Unless otherwise stated, gene and protein symbols are used according to HGNC (Human Genome Organization, Gene Nomenclature Committee) annotations. The annotated and deidentified dataset of 12,830 sample-variant pairs, from which the analyses of the current work are derived, is included (Supplementary Table S3).
Ethics statement
EMR data were deidentified for research using our in-house honest broker system that is sanctioned and monitored by the Medical College of Wisconsin IRB protocol PRO00013874. The Institutional Review Board (IRB) has waived individual consent for research studies leveraging this institutional deidentified research resource. This study was conducted with approval of IRB PRO00037572.
Defining cancer pathways
DNA damage repair (DDR) pathways were used as defined by The Cancer Genome Atlas Analysis Working Group (Knijnenburg et al., 2018). Cell cycle and checkpoint pathways were defined by cell cycle and checkpoint pathways from Reactome (Croft et al., 2011), BioCarta (Nishimura, 2001), and KEGG (Kyoto Encyclopedia of Genes and Genomes; Kanehisa et al., 2012), plus the genesets derived by Fischer (Fischer et al., 2016) and Whitefield (Whitfield et al., 2002), and indexed by MSigDB (Subramanian et al., 2005). We defined Growth Factor Receptor (GFR) genesets as Reactome, BioCarta, and KEGG pathways for EGFR (epidermal growth factor receptor), VEGFR (vascular endothelial), FGFR (fibroblast), PDGFR (platelet-derived), ERBB2 (erythroblastic oncogene B; also known as human epidermal growth factor receptor 2, HER2), and TGFBR (transforming growth factor-β receptor) signaling, indexed by MSigDB.
RESULTS
Tumor-reported VUS are common across the cohort, and a subset exhibits potential of germline origin
We initiated the current study due to the need to understand how to better analyze genomic data from tumor testing. The cohort of 1146 individuals averages 61 ± 13 years of age and evenly split between males and females (Table 1). The most common cancer types were colon (399 patients), lung (304 patients), and pancreas (191 patients), reflective of our catchment and practice (Supplementary Table S4). Significant clinical variables such as stage, grade, and cancer type were often unknown (not provided on the clinical report), emphasizing the need for patient data to be integrated within institutional information management systems. Importantly, we quantify the potential impact of re-annotation in three ways—the numbers of affected genetic variants, patients, and variant reports, which we defined as variant–patient pairs (Table 2). Across our cohort, 8818 distinct tumor genomic variants were clinically reported, with 1413 reported as actionable and 7405 as VUS. Some tumor variants were reported from multiple samples, leading to 2442 total actionable report instances and 8884 VUS report instances. Then, 548 (48%) patients received these actionable report instances and 595 (52%) patients received VUS report instances. Tumor mutation burden was assessed and moderately associated with the number of reported VUS (Spearman’s rho = 0.33 with p < 4.5 × 10−10; Supplementary Fig. S1 and Supplementary Data S1). Thus, this data formed the baseline for benchmarking the current and future methods for genomic interpretation in precision oncology.
Cohort Demographics Among 1146 Cases
Cohort Demographics Among 1146 Cases
Patient numbers are calculated from distinct patient/diagnosis combinations. That is, a patient may be counted for more than one diagnosis if they have multiple samples with different diagnoses.
Unique patients may have multiple tumors. The eight most frequent tumor types are shown; tumor types with fewer samples are grouped into Other.
COAD, colon adenocarcinoma; EAD, esophagus adenocarcinoma; LCA, Liver cholangiocarcinoma; LNSCC, lung non-small cell carcinoma; LUAD, lung adenocarcinoma; PAAD, prostate acinar adenocarcinoma; PDAC, pancreas ductal adenocarcinoma; RAD, rectum adenocarcinoma.
Number of Tumor Variants, Variant Reports, and Patients Affected by Reported Evidence and After Re-Annotation
Variants are reported as actionable if they have potential to change therapies (Rx) or clinical trial (CT) recruitment. Otherwise, they are of uncertain significance (VUS).
Patients were counted in the most extreme table cell for which they had variants, with preference top right < bottom left < top left < bottom right.
Because germline disease data are less often used references for oncology laboratories, compared with other somatic resources, we first address the likelihood that a VUS observed in our tumor cohort could be of germline origin. Our purpose is to understand the nature of tumor-reported data and motivate inclusion of germline annotations. We analyzed the distribution of variant allele fraction (VAF) and found 53.6% of variants at >0.3 VAF. Further, previous large-scale studies (Mandelker et al., 2019) demonstrated that some genes have a high rate of germline validation compared with others. Using the subset of genes with high germline validation rates, we identified 538 variants (4.2%) from 426 patients (37%). Additionally, 54% of reported variants have 0.45 < VAF < 0.55. Therefore, we consider these variants as having a high likelihood of being from germline origins. To further test this likelihood, we assumed that germline variants reported in population genetics studies are more likely to be germline when reported in a tumor. Thus, we correlated tumor VAF with the variant minor allele frequency (MAF) in the general population. Pearson’s correlation between VAF and MAF was non-significant (p value = 0.61) while Spearman’s was significant(p = 4.5 × 10−3). Interestingly, we observed a strong trend toward a 50% VAF across the range of MAF (Fig. 1A). The ratio was most for variants also previously observed in germline diseases (Fig. 1B). All Mann–Whitney tests were statistically significant, indicating that novel VUS identified during tumor testing are more often of low VAF, supporting somatic origins, while VUS that are previously reported from germline genetic conditions have VAFs compatible with being heterozygous from population genetics databases. Specifically, previously reported variants from publicly available sources were closer to MAF = 0.5 compared with the entire distribution of tumor-reported variants(p < 1 × 10−16), previously reported variants with a disease association from public sources were closer to MAF = 0.5 compared with those with any previous report including those lacking a phenotype (p < 2 × 10−10), and previously reported variants in association with a germline condition were closer to MAF = 0.5 compared with those with any previous disease association(p = 9 × 10−3). Thus, the possibility of a subset of these tumor variants being within the germline of patients cannot be ignored, opening a door for future investigations.
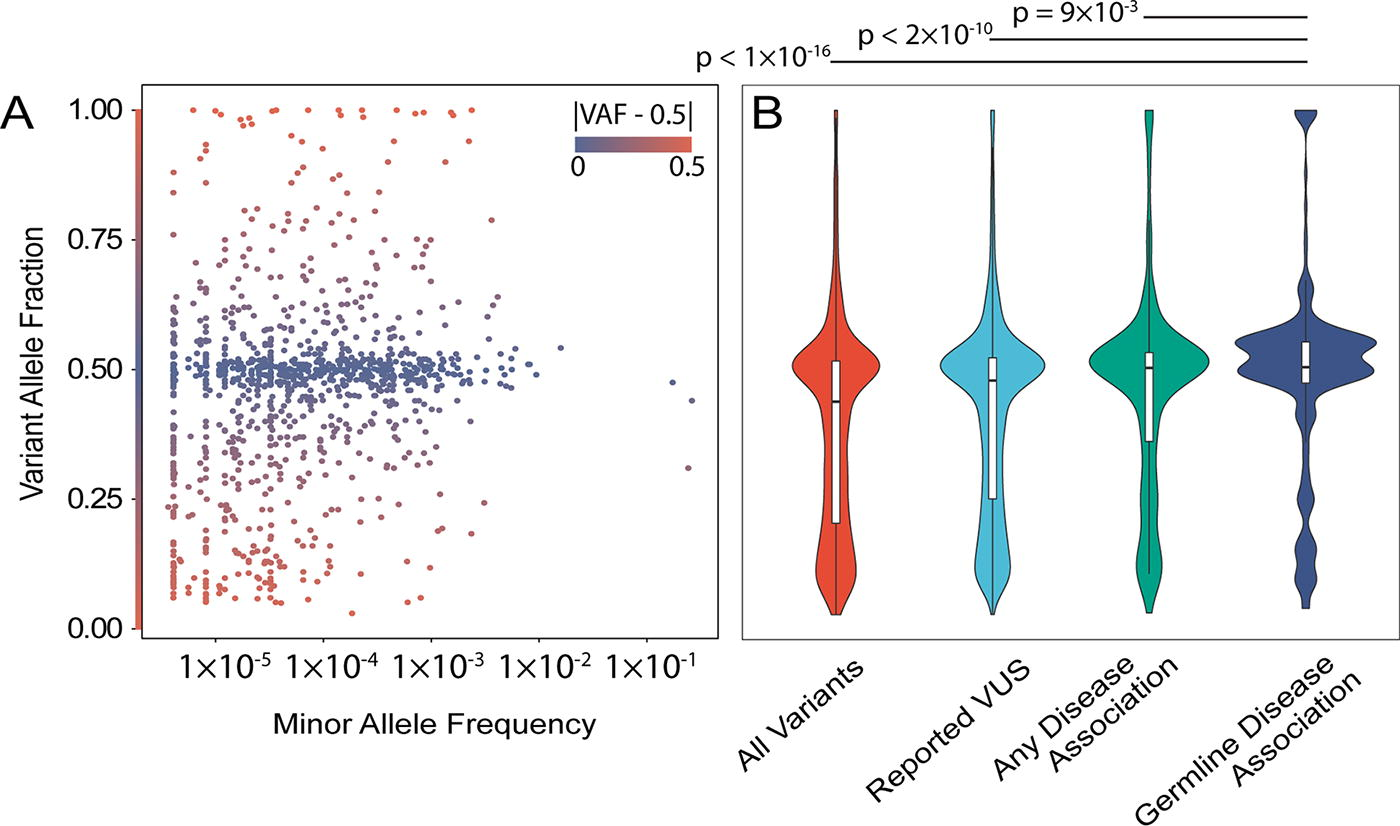
Most genomic variants identified in tumor genomics testing are suggestive of germline origin.
Subsequently, we evaluated which tumor-reported VUS were observed recurrently in cancer or previously classified as (likely)pathogenic germline disease alleles. This analysis highlighted 462 unique variants (6.2%; Table 2) across 499 patients (44% of the cohort; 710 report instances). Their presence in a sample was independent of the year the test was ordered, with a median of 42% of cases per year (Supplementary Fig. S2). We calculated the fraction of these variants that were reported in public databases at the testing time. We found 36%–79% across testing years were reported in the previous calendar year’s database. The 462 variants occur in specific genes more often than others, and the pattern is different when variants were previously observed in cancer versus heritable conditions (Fig. 2). We identified NTRK1, MAP3K1, ARID1A, AR, and MSH6 as the five most frequently altered genes (Fig. 2A). In addition, variants previously reported for germline cancer predisposition most often occurred in MAPK1, MSH6, and APC (Fig. 2B), which is essential information for families with unknown congenital risk. Variants previously reported for non-cancer germline conditions most often occurred in AR, ARID1B, and ARID1A (Fig. 2C), while NTRK1, SPEN, and EPHB1 were the most frequently detected recurrent somatic variants (Fig. 2D). Importantly, variants observed in some genes, such as MAP3K1, ARID1A, and MSH6, were previously observed as altered in both germline and somatic contexts.
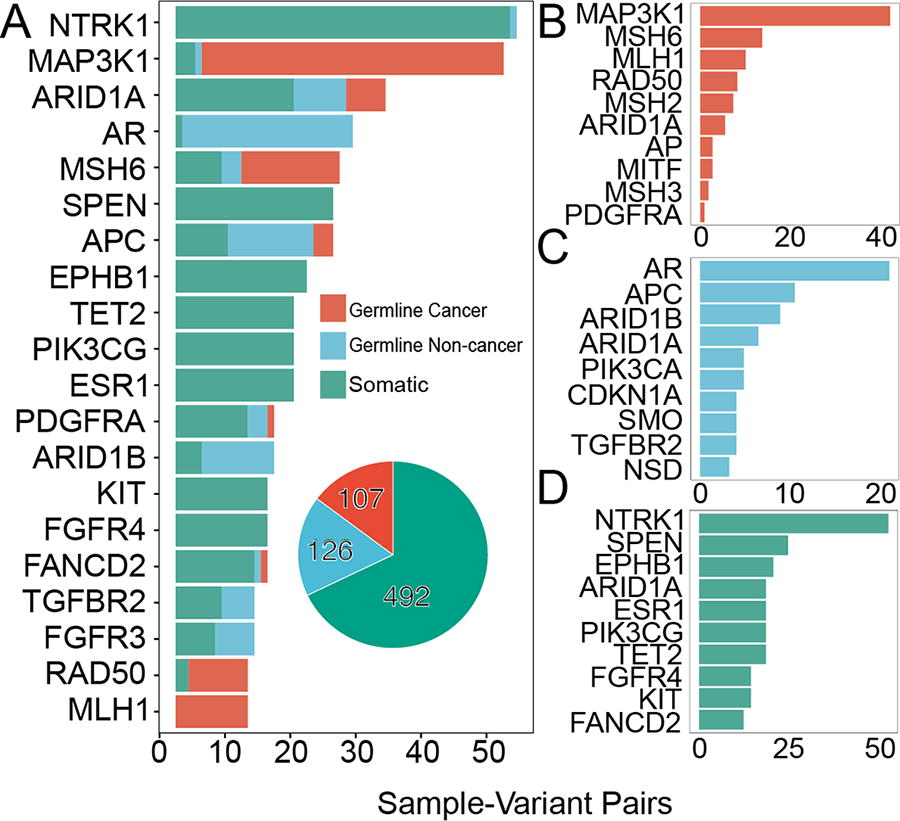
Variants of uncertain significance (VUS) reported in tumor sequencing frequently alter critical genes. Different genetic resources contribute distinct information about variants, together building a more complete context for describing the tumor genome. We plot the most frequently occurring genes containing variants that were reported to be VUS, but upon reanalysis have known disease associations or have been reported in other cancers. Variants are colored by the class of disease associated with them. The affected genes are displayed as those most common
Among the 462 variants found in our cohort, 387 (83.7%) had been previously reported in cancer (Fig. 3A). These variants are observed across 362 samples (32% of the cohort). Notably, the organ of origin where the variant was previously observed was frequently not the same as our patient’s diagnosis (i.e., “off-tumor”; Fig. 3B). For instance, we find many variants in pancreas, colon, breast, or lung tumors that were previously reported in blood cancers. We find colon adenocarcinoma tumors harbored variants previously reported in renal, lymphoid, glial, and other cancers. We mapped specific diagnoses to a simplified histological ontology and found many such off-tumor scenarios (Fig. 3C). Thus, a conceptual framework combining germline and somatic data can significantly enhance information, which otherwise would remain obscured.
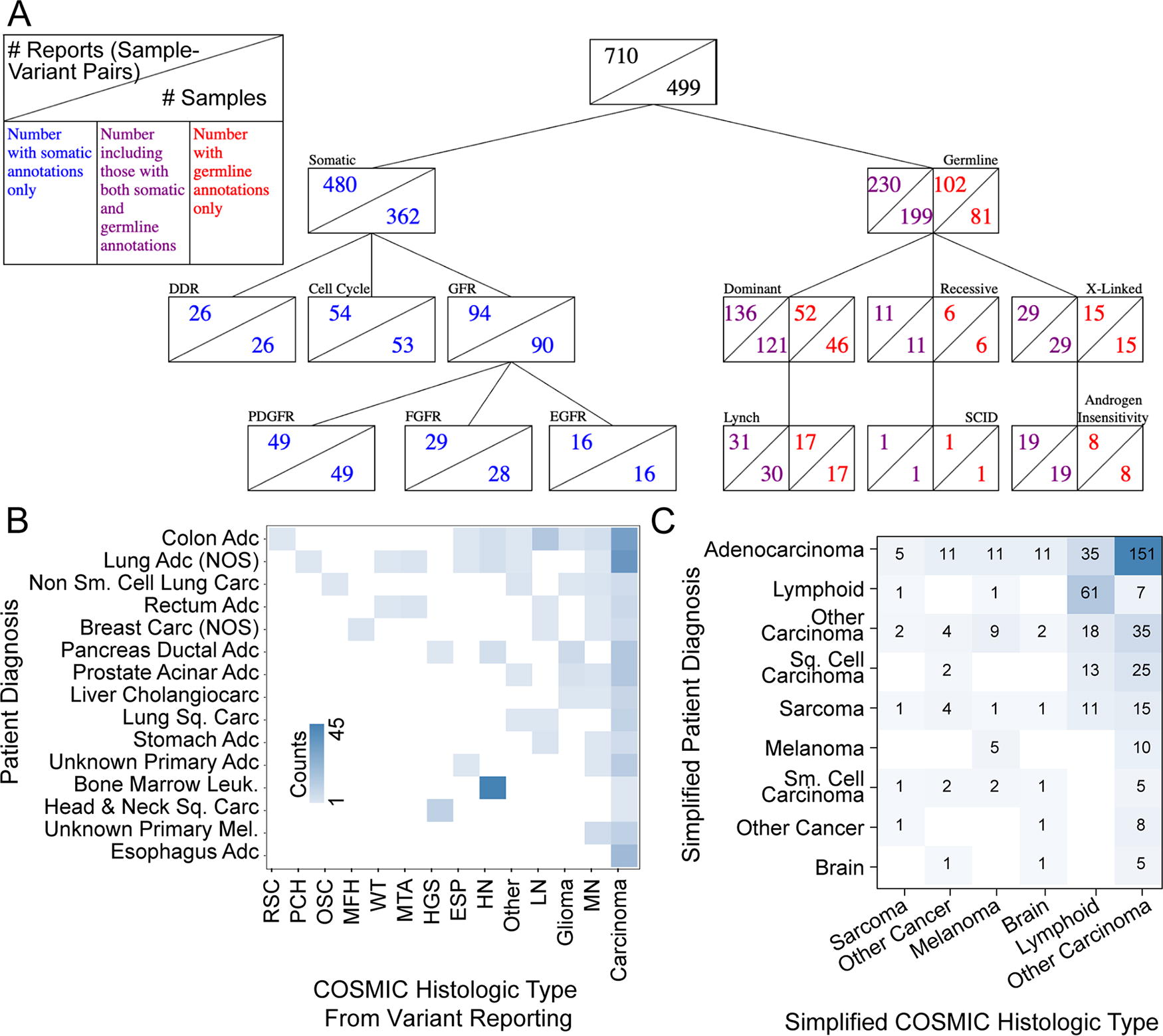
Diversity among patient diagnoses and the previously observed disease context of tumor VUS.
Our cohort obtained highly heterogeneous germline testing, with many patients receiving none, a small subset receiving germline exome sequencing, and a larger subset receiving gene panel testing (Supplementary Fig. S3). Many of these tests either do not overlap or share only a subset of genes in common with each other or with the tumor test (Supplementary Fig. S4), leaving origins ambiguous. In total, we had germline results for 115 patients, 84 of which were negative reports (no variants reported), 35 revealed (likely) pathogenic variants, and 48 returned VUS. In this sub-cohort, 29 patients (25.2%) had tumor variants confirmed as germline. Each of these results was assisted by genetic counseling specialists. From this work, we describe three scenarios that epitomize possibilities for how these data were followed: (1) no germline follow-up left family members at potentially unknown risk, (2) the germline allele was confirmed but no verifiable follow-up with family members, or (3) the germline allele was communicated to and tested for across the family resulting in changes to family member’s clinical risk management. Thus, a higher-than-recognized proportion of tumor-reported variants may originate from the germline, underscoring the value of comparing tumor genomics with germline reference data.
Multi-disciplinary impact of clinical genomics results
We next considered selected case examples to further investigate the potential for added value of more broadly understanding tumor-reported genomic variants. These cases are additionally summarized in Table 3.
Three Selected Clinical Cases with Germline and Somatic Genomic Testing
Three Selected Clinical Cases with Germline and Somatic Genomic Testing
BRCA, breast carcinoma; COAD, colorectal adenocarcinoma; EAC, esophageal adenocarcinoma; IHC, immunohistochemistry; PDAC, pancreatic ductal adenocarcinoma; STAD, stomach/gastric adenocarcinoma; VUS, variants of uncertain significance.
A male in his 70s was diagnosed with advanced esophageal adenocarcinoma. Reoccurrence occurred after 4 years and pembrolizumab was started. Positron emission tomography (PET) scan after eight cycles demonstrated mild radiographical response and improvement in clinical symptoms. Tumor gene panel profiling was then ordered and identified two actionable variants in the MSH6 gene, p.E807* and splice site c.3439-2A>G. This tumor test also reported microsatellite instability (MSI)-intermediate and high tumor mutational burden (TMB). Germline pathogenic variants in MSH6 are associated with Lynch syndrome; a condition that significantly increases the risk for colorectal, stomach, endometrial, ovarian, and other cancers (Kohlmann and Gruber, 1993). Esophageal cancer is not commonly associated with Lynch syndrome. The patient’s family history was significant for tw0 first degree relatives with gynecological cancers at unknown ages. Without more information on his family history of cancer, this patient did not meet clinical criteria for germline genetic testing.
Our reanalysis revealed that the splice site c.3439-2A>G variant in MSH6 was seen at an allele fraction of 0.48, absent from the gnomAD database, and listed as pathogenic in ClinVar by multiple submitters. Further, the p.E807* variant is absent from the gnomAD database and listed as pathogenic in ClinVar by two submitters. In addition, recent studies found that MSH6 pathogenic variants are more likely to be of germline origin versus somatic (∼60% vs 40%; Meric-Bernstam et al., 2016). It is highly suspicious that this variant is of germline origin and would be recommended for germline genetic testing. Unfortunately, the patient was never referred to genetic counseling for confirmatory germline genetic testing and hence passed away. This case highlights the missed opportunity to identify a hereditary cancer syndrome that would significantly impact medical management recommendations for at-risk family members.
Case example 2
A male in his 60s was diagnosed with advanced colorectal adenocarcinoma and referred to genetic counseling due to his MSI-high tumor with immunohistochemistry staining showing loss of MLH1 and PMS2. Family history was significant for an unknown primary cancer in a parent and lung cancer in family members who were smokers. BRAF V600E and MLH1 promotor hypermethylation were ordered and neither marker was detected. Subsequently, a germline multi-gene hereditary cancer panel was ordered and revealed a pathogenic variant in the MLH1 gene, c.1667G>C (p.S556T), which is consistent with a diagnosis of Lynch syndrome. Following these results, pembrolizumab was initiated, but unfortunately there was disease progression. Subsequently, tumor gene panel profiling was ordered which also reported the S556T variant in MLH1 but as VUS. This tumor test also reported MSI-intermediate and high TMB. This case highlights the potential to miss clinically relevant variants due to reporting practices (no mention of potential germline significance) and use of tumor-only testing, rather than paired samples for somatic testing.
Case example 3
A male in his 70s, diagnosed with metastatic gastric invasive adenocarcinoma, poorly differentiated, with signet ring cell features, had tumor gene panel profiling ordered soon after diagnosis. Most notably, tumor profiling identified a variant in the CDH1 gene at p.Arg63* and reported the sample MSI-stable with low TMB. Germline pathogenic variants in CDH1 are associated with hereditary diffuse gastric cancer (HDGC)—a condition that is associated with an increased risk of diffuse gastric cancer and lobular breast cancer. The average age of HDGC onset is 38 years, with a range of 14–69 years (Kaurah and Huntsman, 1993). Although the age of onset in this patient was older than expected for HDGC, a referral was placed to genetic counseling for evaluation of this CDH1 variant. The patient reported two relatives diagnosed with breast cancer in their 50s, another diagnosed with pancreatic cancer in their 70s, and further three diagnosed with breast cancer at unknown ages. The patient did not know the type of breast cancer (lobular vs ductal) in these family members. A germline multi-gene hereditary cancer panel was ordered and revealed a pathogenic variant in CDH1, c.187C>T (p.Arg63*). Germline genetic testing also found a possibly mosaic, pathogenic variant in the TP53 gene at c.734G>A (p.Gly245Asp) and VUS in APC at c.2307A>T (p.Leu769Phe); in BLM at c.968A>G (p.Lys323Arg); in POLE at c.2773T>C (p.Ser925Pro); and in STK11 at c.464 + 5G>A (Intronic). Each of these variants were previously reported on the tumor profiling report except for the STK11 c.464 + 5G>A (Intronic) variant.
Subsequently, germline genetic testing was recommended for family members. One child tested positive for the familial pathogenic variant in CDH1 and a prophylactic gastrectomy was recommended. This case highlights the importance of recognizing potential germline variants on tumor profiling and the significant clinical impact on families when a hereditary cancer syndrome is identified.
Pathway mapping bears potential implications for therapeutics and genetic counseling
Because genomic information is used to decide cancer therapy, we investigated how many VUS from the original reports fell within actionable pathways (Fig. 3A). We considered DDR, cell cycle, and key growth factor receptors (GFRs). We found 26 samples affected by DDR pathway alterations. Three variants were specifically within the homology-directed repair, which may suggest therapies that inhibit single-strand repair. Additional variants within this group affected MSH6, MSH2, MSH3, and MLH1, which participate in microsatellite instability. Cell cycle variants were observed across 53 samples. Tyrosine kinase receptor pathways were more commonly altered, represented by 73 variants across 90 samples. They were most frequent in PDGFR (49 samples), FGFR (29), EGFR (16), ERBB2 (13), TGFBR (9), and VEGFR (8) pathways. Genes in these pathways have been the most represented in tissue-agnostic basket trials, making their further assessment highly relevant to research (Offin et al., 2018; Park et al., 2020). Therefore, VUS identified in tumors warrants further consideration for their potential to affect actionable pathways.
Finding associations with other genetic diseases for VUS reported in tumors
Additionally, among the 462 variants, 122 (26%) had been previously reported, at the time of cancer diagnoses, as pathogenic in a germline setting. These 122 variants are observed in 17% of our cohort. Interestingly, 47 (39%) of these variants were also reported in other cancers (Fig. 3A). The most frequent germline phenotypes associated with these 122 variants (Supplementary Fig. S5A) are risk for gynecological malignancies, colorectal and breast cancers, and Lynch syndrome (Supplementary Fig. S5B). Most (68 variants) follow an autosomal dominant inheritance pattern, a moderate number (15) are X-linked, and few (10) are recessive (Fig. 3A). The most common disease with a dominant inheritance pattern was Lynch syndrome (26). We also observed three Fanconi anemia variants. Additional variants were previously reported for non-cancer diseases, including autism, Coffin-Siris, infertility idiopathic, and aortic aneurism syndrome (Supplementary Fig. S5C). X-linked diseases include androgen insensitivity syndrome, idiopathic infertility, and Reifenstein syndrome (another androgen insensitivity). Recessive conditions included severe combined immunodeficiency disease. Thus, many variants reported as VUS in tumor sequencing are described as germline pathogenic variants conferring risk for heritable cancers and warranting their further consideration for both patients and family members.
Comparative performance of the current approach with state-of-the-art computational tool for variants annotation
We sought to compare our reanalysis with other state-of-the-art computational tools for assessing genomic variants. We used VIC (He et al., 2019) and CHASMplus (Tokheim and Karchin, 2019) to annotate our cohort’s genomics results. We again split the variant reports into those that were reported as actionable or VUS, and then by our re-annotation results, and finally by each tool’s classification. VIC classifications were partly concordant with re-annotation, yet more conservative (Supplementary Table S2A). Specifically, of the 426 actionable variant reports with re-annotation results, VIC identified 20 as having “strong evidence of clinical significance” and 235 with “potential” evidence. Then, among the 710 VUS reports with re-annotation results, VIC identified none as strong and 73 as potential evidence. All the variants with strong evidence by VIC had a prior germline pathogenic or cancer-associated annotation in our reanalysis. CHASMplus classifications similarly had overlap with re-annotation and were also more conservative (Supplementary Table S2B). Specifically, of the 426 actionable variant reports with re-annotation results, CHASMplus identified 145 as more likely to be driver mutations. Then, among the 710 VUS reports with re-annotation results, CHASMplus identified 12 as more likely to be driver mutations. Finally, we compared the numbers of unique protein coding variants prioritized by all three methods. We found that 196 (22%) are shared by at least two approaches, while 909 are prioritized by only one (Supplementary Fig. S6). We interpret this finding as the two types of analysis, somatic prioritization algorithms and enhanced annotation, complementing and enhancing each other. Therefore, current state-of-the-art tools leave most variants as VUS, and different approaches are not highly concordant.
DISCUSSION
The current article contributes to the field of genomics by proposing a broader bioinformatic analytical approach to reanalysis, which maximizes the yield from tumor genetic testing results in precision oncology. This multi-tier approach involves several key components: (1) the reclassification and annotation of tumor-derived variants for their potential role in cancer, (2) reclassification of VUS in tumors using germline information, (3) exploration of pathway associations with implications for therapeutics and genetic counseling, and (4) investigation of the association of other genetic diseases with tumor VUS. The data obtained from this approach emphasize the importance of untapped information in tumor genomics testing results, which can be effectively extracted through an enhanced annotation approach. Genomic data can provide valuable insights for patient care and family planning beyond the initial purpose of the test. Therefore, we argue that an enhanced annotation approach is necessary to maximize the yield from genetic testing.
Based on this knowledge, we propose three activities to enhance standard practices in genomics data interpretation and better support precision oncology: (1) critical evaluation of practice emphasis, (2) assessment of assays, and (3) integration across practices. The first recommended activity involves carefully evaluating the emphasis placed on different aspects of practice. In the context of congenital diseases, the primary concern lies in understanding etiology and making a diagnosis (Richards et al., 2015). In contrast, in oncology, the primary focus is on therapeutics and prognosis (Li et al., 2017). Consequently, the current reporting conventions, resources leveraged, and prioritization algorithms employed in oncology differ from those in inherited disorders (Fig. 4A). We demonstrate that a broader approach to variant annotation could enhance patient care by capturing information about alleles that are relevant to clinical disciplines beyond oncology, in line with recent guidelines (Li et al., 2017; Mandelker et al., 2019; Richards et al., 2015). While the primary goal of tumor profiling is to guide treatment decisions, it is important to note that up to 17% of patients may harbor an inherited (likely) pathogenic variant in a cancer susceptibility gene (DeLeonardis et al., 2019; Mandelker et al., 2017; Meric-Bernstam et al., 2016; Neben et al., 2019; Schrader et al., 2016). Moreover, data indicate that patients undergoing tumor profiling often desire to be informed of incidental findings (Meric-Bernstam et al., 2016). Therefore, we believe that these two paradigms must merge (Fig. 4B). In our view, all patients can benefit from the generation of systems that will uniformly and comprehensively evaluate their data because the goal of medicine has always been about personalized approaches. In the era of systems biology, it is increasingly appreciated that a genetic diagnosis is critical to cancer treatment (beyond the organ of origin), that treatments can be developed for congenital genetic diseases, and more. To achieve this convergence and practical implementation, institutions must establish processes that enable dynamic sharing and annotation of data. This can be accomplished through retrospective research (as demonstrated in this study), real-time discussions at tumor boards (with adequate data science support), or institution-wide implementation of robust and well-vetted systems. Thus, two approaches to software architecture can be used—to build either a centralized system that can ensure compliance and uniformity or a distributed network of vetted software applications that can be accessed on demand. In either case, computational tools and systems must be established to account for diverse types of genomic annotations and bring them together to support decision-making. By diverse types of annotations, we refer to oncology and heritable conditions as in the current work and information beyond DNA annotations, such as from functional genomics, biochemistry, biophysics, and advanced computational modeling of gene products (Chi et al., 2023; Haque et al., 2023; Jorge et al., 2023; Ratnasinghe et al., 2023), for example. Properly implemented with clear guidelines paired with narrow-AI-based automations, adopting a more robust and integrated approach will undoubtedly add value to patient care.
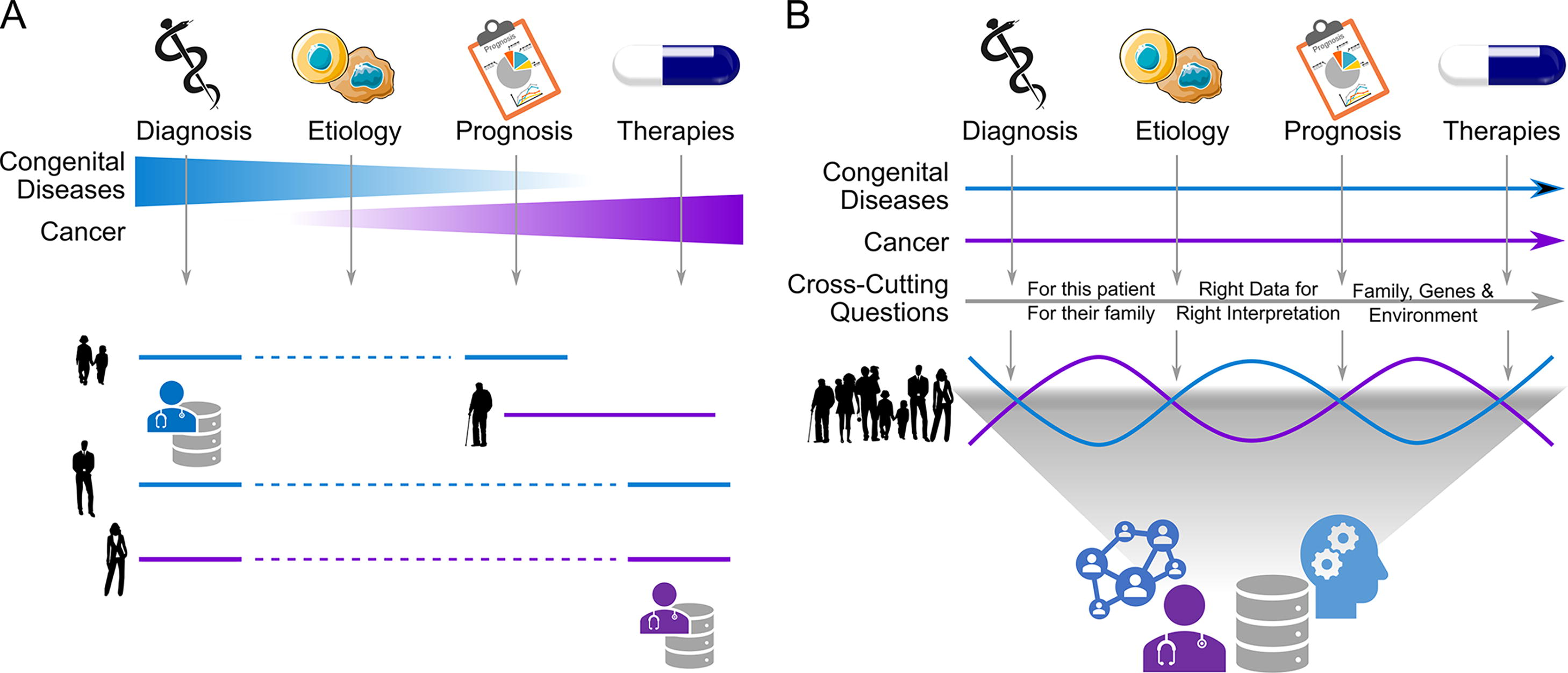
Intersection and integration between practice emphases are necessary for comprehensive patient care. This figure diagrams what we view as key challenges in the field that, with the help of additional data systems including enhanced annotation and more data science to support oncology clinics, can be addressed to improve care.
Clinical genomics testing exhibits variability in reporting, even when the same sample is sent to different laboratories (Harrison et al., 2017; Madhavan et al., 2018). Tumors often display heterogeneity, resulting in the presence of different variants in different regions of the tumor tissue. Additionally, bioinformatics analysis pipelines may be optimized to identify certain types of mutations more effectively than others. The technical details of genomic assay design, which vary significantly among different vendors (Supplementary Fig. S4), further contribute to the complexity. These challenges are independent of the testing laboratory or the specific approach employed to enhance tumor variant interpretation.
The field continues to recognize the value that approaches such as ours can provide to patients. During this study, other research groups have proposed guidelines for interpreting tumor-derived genomics data (Mandelker et al., 2019). We have taken additional steps to identify phenotypic trends within the growth factor pathway, off-tumor associations, and non-cancer associations of tumor VUS observed in our patient cohort. Through a series of clinical vignettes, we further demonstrate how enhanced information could have benefited patients or their families. However, the increased integration of genomics data raises important considerations related to informed consent and the scope of data processing and linking that should be routinely addressed in clinical care (Fisher and Layman, 2018) and in support of guidelines for secondary findings (Miller et al., 2021). Above all, we strongly believe that an integrated approach will provide better support to medical practice, resulting in cost savings through a reduction in manual curation time and improved patient care by enabling increased access to and utilization of accurate data for addressing the relevant clinical questions.
Footnotes
ACKNOWLEDGMENTS
The authors thank the CTSI grant National Institutes of Health CTSA award, UL1TR001436, for resources, services, and facilities. This research was completed in part with computational resources and technical support provided by the Research Computing Center at the Medical College of Wisconsin.
AUTHORS’ CONTRIBUTIONS
E.D. performed formal analysis, data curation, and wrote the article. H.V.R. reviewed the article. B.W.T. supervised staff, facilitated resource access, contributed to data curation, and reviewed the article. S.S. performed formal analyses and investigation. J.L.G. supervised staff and reviewed the article. B.G. facilitated resource access and contributed to data curation. R.S. facilitated resource access. R.U. facilitated resource access, funding acquisition, study conceptualization, and wrote the article. M.T.Z. conceived the study, contributed to formal analyses and data curation, supervised staff, and wrote the article.
DATA AVAILABILITY STATEMENT
This work uses data derived for clinical care and anonymized for research by our IRB-approved investigative team. The authors have made selected data available through the current publication with broader access to the data possible on individual bases, with requisite IRB approvals. The deidentified primary dataset used in this study is available in ![]() .
.
AUTHOR DISCLOSURE STATEMENT
The authors report no conflict of interest.
FUNDING INFORMATION
This publication was supported in part by
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.