
Abstract
Current expression quantification methods suffer from a fundamental but undercharacterized type of error: the most likely estimates for transcript abundances are not unique. This means multiple estimates of transcript abundances generate the observed RNA-seq reads with equal likelihood, and the underlying true expression cannot be determined. This is called nonidentifiability in probabilistic modeling. It is further exacerbated by incomplete reference transcriptomes where reads may be sequenced from unannotated transcripts. Graph quantification is a generalization to transcript quantification, accounting for the reference incompleteness by allowing exponentially many unannotated transcripts to express reads. We propose methods to calculate a “confidence range of expression” for each transcript, representing its possible abundance across equally optimal estimates for both quantification models. This range informs both whether a transcript has potential estimation error due to nonidentifiability and the extent of the error. Applying our methods to the Human Body Map data, we observe that 35%–50% of transcripts potentially suffer from inaccurate quantification caused by nonidentifiability. When comparing the expression between isoforms in one sample, we find that the degree of inaccuracy of 20%–47% transcripts can be so large that the ranking of expression between the transcript and other isoforms from the same gene cannot be determined. When comparing the expression of a transcript between two groups of RNA-seq samples in differential expression analysis, we observe that the majority of detected differentially expressed transcripts are reliable with a few exceptions after considering the ranges of the optimal expression estimates.
1. INTRODUCTION
Despite the improvements of transcript expression estimation methods based on RNA-seq data (Li and Dewey, 2011; Hensman et al., 2015; Bray et al., 2016; Patro et al., 2017), the estimated transcript expression can still be inaccurate and uncertain. One source of uncertainty in expression estimation is that multiple sets of expression estimates can optimally explain the observed RNA-seq reads. Therefore, the “best” estimation cannot be uniquely identified. The state-of-the-art methods to quantify transcripts' expression are based on probabilistic models, and, in probabilistic model inference terminology, the phenomenon of nonuniqueness in optimal parameters under infinite data is called model “nonidentifiability.”
In this work, we relax the concept and use this term to refer to the nonuniqueness of optimal parameters under a given finite data set. See Figure 1 for a toy example of model nonidentifiability in expression quantification. The two main problems for evaluating the accuracy of transcript expression estimates under nonidentifiability are (1) detecting the transcripts whose expression estimates are nonidentifiable and (2) bounding the range of the uncertain expression of the transcripts.
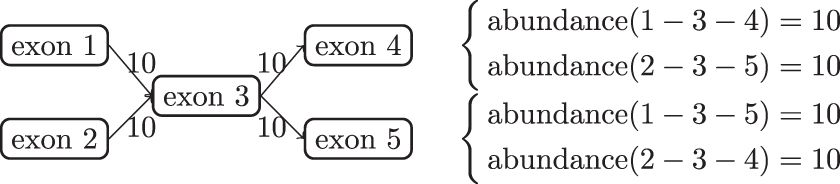
Example of a nonidentifiable quantification model. Transcripts are the paths in the splice graph, denoted by the concatenation of exon indices. The number on each edge indicates the number of observed reads mapped to this splice junction. The set of transcript abundances is optimal if it perfectly explains the observed reads. That is, for each junction, the total abundances of transcripts containing that junction sum up to the number displayed on the edge. The right side of the figure shows two co-optimal expression abundances. It can be verified that both solutions explain the observed reads perfectly as they both predict 10 reads on each junction.
Expression of transcripts is used in many analyses, and understanding the accuracy and uncertainty of the estimated expression helps us evaluate the confidence of the conclusions of such analyses. Transcript expression estimates are used for detecting splicing isoform switching (Nowicka and Robinson, 2016; Guo et al., 2017; Vitting-Seerup and Sandelin, 2017), for identifying differential expression (McCarthy et al., 2012; Love et al., 2014; Ritchie et al., 2015; Soneson et al., 2015; Pimentel et al., 2017), and for predicting disease status and treatment outcome (Morán et al., 2012; Hoadley et al., 2014). Ranges of uncertain expression estimates provide useful insights into the reliability of these studies.
We focus on the uncertainty of expression estimates due to model nonidentifiability, but there are other causes of estimation uncertainty or inaccuracy. The small sample sizes (low sequencing depth of RNA-seq) also lead to estimation errors in transcripts' expression. Statistical methods such as bootstrapping and Gibbs sampling (Turro et al., 2011; Glaus et al., 2012; Al Seesi et al., 2014) provide an estimate on the error in expression levels due to the sample size. This estimation error can be reduced by increasing the sequencing depth. In contrast, the estimation uncertainty due to model nonidentifiability is more fundamental because it cannot be addressed even under infinite sample size.
In RNA-seq data, exon sequences are usually shared among multiple transcripts, and RNA-seq reads usually cannot be mapped to a unique transcript. Due to the high rate of multimapping events in RNA-seq data, the best set of transcripts' expression cannot be uniquely resolved, and the phenomenon of nonidentifiability occurs. Multimapped reads are prevalent in RNA-seq data regardless of the sequencing depth. Thus, the uncertainty of expression due to model nonidentifiability cannot be easily addressed.
Previous works have analyzed the nonidentifiability phenomenon in transcript expression quantification. However, they mainly focused on the first problem, to identify the transcripts for which the expression estimates are nonidentifiable, but not the second, which is to bound the ranges of the true expressions for the transcripts with uncertain expression estimates. Lacroix et al. (2008) and Hiller et al. (2009) developed methods to list the transcripts that have nonunique expression estimates, but their methods do not provide information about the values of optimal abundances. Roberts and Pachter (2013) incorporated the detection of nonidentifiable abundances into their quantification method, and designed a tool to output an identifiability flag for each transcript along with a single-expression estimate.
In this work, we develop methods that address the range of optimal expression estimates for transcripts under model nonidentifiability. For each transcript, we calculate the minimum and maximum values across all optimal expression estimates. That is, for any expression value between the computed minimum and maximum, there exists a set of expression of the other transcripts such that the estimation of all transcripts' expression (combining both the expression of this transcript and the other transcripts) leads to the largest likelihood in the probabilistic model of expression quantification. Compared with a list of names of the transcripts for which the expressions are nonidentifiable, the range of optimal expression of a transcript provides more information on the accuracy (or inaccuracy) of the estimate.
Most widely used quantification software (Li and Dewey, 2011; Hensman et al., 2015; Bray et al., 2016; Patro et al., 2017) take a set of reference transcript sequences as input and assume that the reference transcripts are the complete set of sequences that can be expressed. This is called reference-transcript-based expression quantification. Another line of expression quantification models (LeGault and Dewey, 2013; Bernard et al., 2014; Ma et al., 2020) called “graph quantification” assumes that the current reference transcriptome database is incomplete. Instead, the splice graph that encodes the exon/exon connection relationships is assumed to be correct and complete, meaning every possibly expressed transcript corresponds to a path in the splice graph and vice versa. Those models infer the splice graph edge expression or edge selection propensity in the quantification probabilistic models.
We provide a more detailed overview in Section 2.3. Many transcript assembly methods also adopt a similar setup, assuming a mixture of reference transcripts and novel isoforms are expressed (Trapnell et al., 2010; Tomescu et al., 2013; Pertea et al., 2015; Liu and Dickerson, 2017; Gatter and Stadler, 2019).
We develop methods to bound the range of optimal expression estimates for both reference-transcript-based and graph quantification models. Our method for the reference-transcript-based quantification is based on linear programming over sufficient statistics (Section 2.2), and our method for graph quantification is based on max-flow formulations to “introspect” the graph quantification model (Section 2.4). Our introspection algorithm can not only bound the uncertain expression of full transcripts, but also extends to graph structures. For example, given a set of edges, our method computes the range of the optimal total expression of transcripts that cover any edge in the set.
Combining our methods for quantification models and interpolating between the complete and incomplete reference transcriptome assumption, we can additionally compute the range of optimal expression estimates under the assumption that a given percentage of the expression comes from the reference and the remaining expression comes from the full paths in splice graphs (Section 2.5).
Applying our method to 16 Human Body Map samples, we analyze to what degree the expressions of transcripts are estimated inaccurately due to nonidentifiability. We observe that around 35%–50% of transcripts potentially suffer from expression estimation error across the 16 samples. Most of these transcripts (or 20%–47% of total transcripts) have very uncertain expression estimates such that the ranking of expression between the transcript and its sibling isoforms from the same gene is inconclusive. Around half of the transcripts with uncertain expression estimates due to nonidentifiability are different from those due to finite sample size.
Applying our method on sequencing data sets of an MCF10 cell line and of CD8 T cells, we use the ranges of optima to evaluate the reliability of detected differentially expressed (DE) transcripts within each data set. A DE detection is unreliable if the ranges of optimal expression between the DE groups largely overlap. We observe that the majority of the DE calls are reliable and robust to the uncertain expression estimation due to nonidentifiability when the reference transcriptome contributes to >40% of expression. However, there are 5 unreliable DE calls (out of 257 detections) in the MCF10 data set, and 19 unreliable DE calls (out of 3152 detections) in the CD8 T cell data set. It requires further investigation to determine whether these transcripts are actually DE, and analyses based on the DE status of these transcripts require extra caution.
2. METHODS
This study does not collect new biological data, and thus IRB approval is not required.
We start with relevant definitions in Section 2.1. Section 2.2 provides a high-level overview of probabilistic modeling of transcript quantification, and the linear programming to derive a range of optimal abundance estimates for this setup. Section 2.3 provides an overview of graph quantification, and Section 2.4 describes our introspection algorithm to derive the ranges under this setup.
2.1. Definitions
A “splice graph” is a directed acyclic graph representing alternative splicing events in a gene. The graph has two special vertices: S represents start of transcripts and T represents termination of transcripts. Every other vertex represents a (partial) exon. Edges in the splice graph represent “splice junctions,” that is, potential adjacency between exons in transcripts. Each transcript corresponds to a unique
2.2. Ranges of optimal estimates for reference-transcript-based quantification
Recall the problem of reference-transcript-based expression quantification. We focus on paired-end reads for now, however, this formulation naturally extends to other types. We also focus on a particular probabilistic modeling, which lies at the heart of most modern transcript quantifiers (Li and Dewey, 2011; Hensman et al., 2015; Bray et al., 2016; Patro et al., 2017). Assume that the paired-end reads from an RNA-seq experiment are error-free and uniquely aligned to a reference genome as fragments. We denote the set of fragments as F, and the set of transcripts as
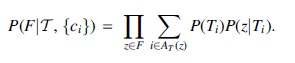
To generate a fragment, first a transcript is sampled, then the fragment is sampled from the selected transcript, and we only observe the resulting fragments.
We now introduce the idea of reparameterization. Assume, for now, each fragment spans exactly one junction. If two sets of quantified transcripts result in exactly the same total abundance on each junction, they yield the same generative model. In other words, these two sets will be indistinguishable from each other. Let
As this defines a linear system, we calculate the maximum and minimum possible value of
This argument no longer holds when fragments can span multiple junctions. However, as we have shown previously (Ma et al., 2020), to ensure that two sets of quantified transcripts yield the same fragment distribution, it suffices that for each phasing path (i.e., the set of junctions in a fragment, as a path on the splice graph), the total abundance of transcripts containing the phasing path in full is equal. In other words, the form of the linear program remains unchanged, with the only change being that Ji is now a phasing path instead of a single junction.
2.3. Bird's-eye view: graph quantification
We now provide a high-level overview of graph quantification, and draw similarities with its transcript-based counterpart. The problem of splice graph expression quantification is very similar to the transcript version of the problem, but with the variables associated with transcripts
To generate a fragment, first an edge in G is sampled, then the fragment is sampled from the selected edge, and we only observe the resulting fragments. Again,
After inferring the splice flow value from the set of observed fragments, we obtain the optimal splice graph flow that explains the observation. While the graph flow itself finds use in certain cases, for most analysis it is imperative that the flow will be decomposed into a weighted set of
In the next section, we describe methods that consider all possible flow decompositions of a given graph flow. As a particular use case, these methods are able to determine the minimal and maximal possible abundance for a given transcript, across all possible flow decompositions.
Similarly to the transcript-based quantification, the conclusion no longer holds when fragments span multiple junctions, and it is much harder to adjust the graph quantification model to properly take these into account. However, for graph quantification, G can be any supergraph of the splice graph, as long as each
2.4. Subgraph quantification
Nonidentifiability manifests itself in graph quantification as different flow decompositions from the inferred splice graph flow. Specifically for a transcript, the corresponding
In addition, we are also interested in the related problem of “local quantification,” that is, to estimate the abundance of a specific splicing pattern by aggregating expression of full-length transcripts containing the pattern. This is natural for analysis of complex gene loci, where certain splicing patterns within a region might be of larger interest than the patterns outside. Our reasoning for deriving a “confidence interval” can similarly apply for splicing patterns, that is, we are interested in the total weight of
For quantifying a full transcript, Ek consists of single-edge sets, each corresponding to an edge in the
To solve AND-
The OR-
If we know the minimum and maximum total “bad flow” (negative of good flow), we can obtain the answer to AND-
We now solve OR-
Constructing
See Figure 2 for an example of both problems. In the following three sections, we provide proofs to Theorems 1 and 2, as well as an algorithm to construct GB in linear time.
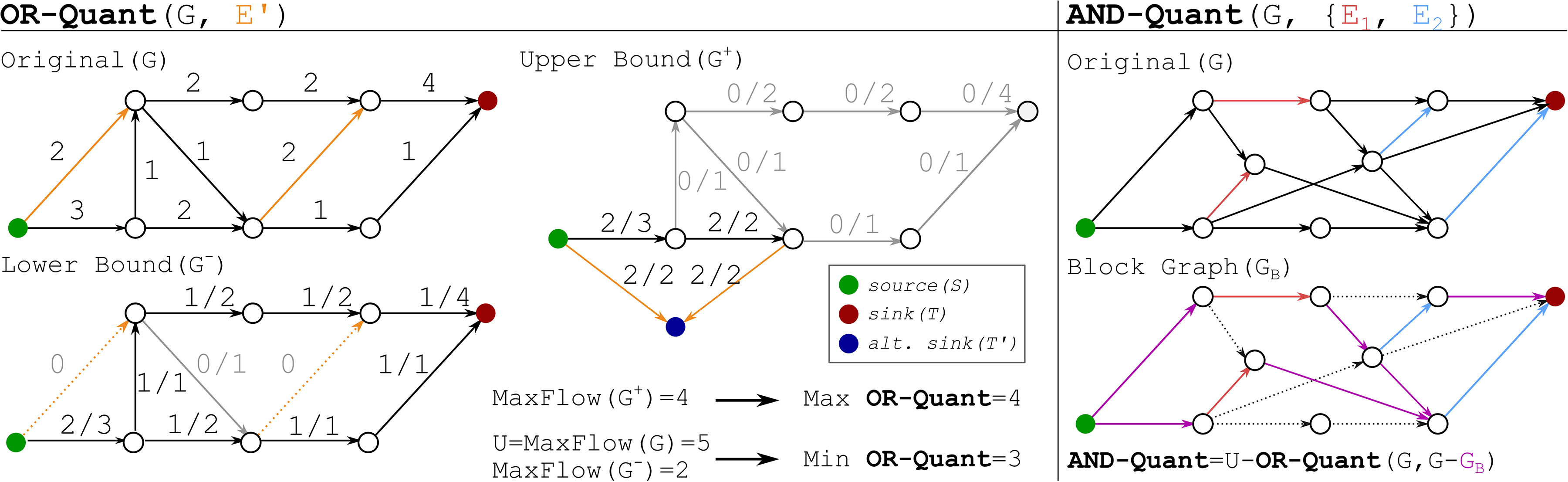
Examples for subgraph quantification. Left: Example for OR-
2.4.1. Algorithms for OR-Quant
In this section, we state the algorithms for OR-
We start with several basic definitions.
We define partial decompositions on arbitrary non-negative edge weights, as required for a later proof. We present the following lemma without proof.
We now restate the definition of OR-
We now state our algorithms and prove the correctness of the algorithms, starting from the lower bound.
Proof. We prove the theorem in two parts: First, we show there is a decomposition r such that
The key observation is that all
We have
Now for any decomposition R, we split it into two parts.
Proof. We prove the theorem in a similar style: First, we show any decomposition satisfies
For the first part of the theorem, for a decomposition R of F, we can write
The second part of the proof, that is, to show there is a decomposition R of F such that
2.4.1.1. Overview
This part of the proof involves actually constructing the “correct” decomposition of R such that
So, we may need to further split the weight of a path in the truncated decomposition when trying to complete it. Our constructive proof follows a strict protocol of doing these splits and completions, one path at a time, so flow capacity is respected in every step.
We start by building an arbitrary decomposition
2.4.1.2. Path mapping for
A good
Similarly, an
We apply an induction argument, formally defined as follows. Figure 3 provides a concrete example for the induction argument.
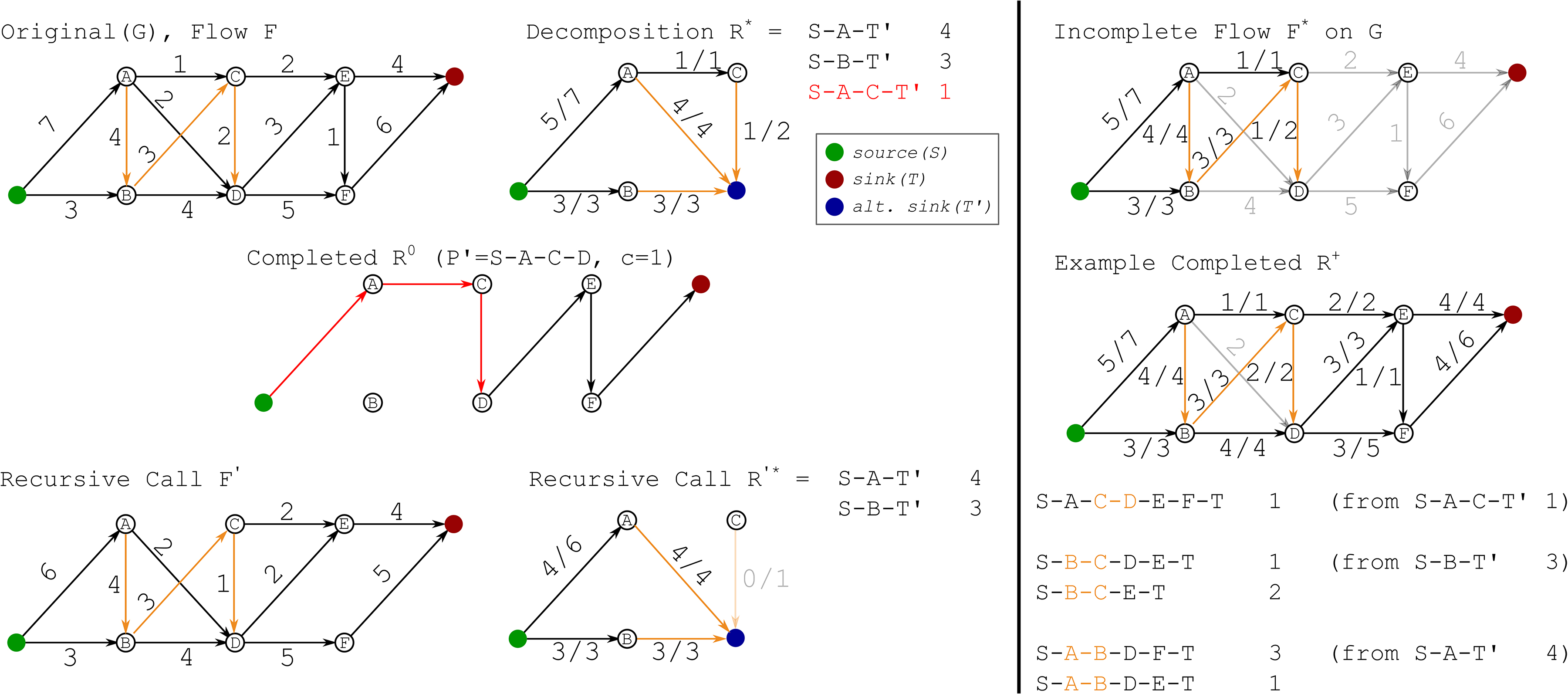
Example for flow reconstruction. Input graph G is shown on the top left, with edges in
2.4.1.3. Flow reconstruction instance
An instance for flow reconstruction has two inputs:
We will apply the induction on
2.4.1.4. Induction algorithm
We first pick the path
We next run a MaxFlow on G from v to T with total flow c. This call always returns a full flow (with total flow c), because there is at least c incoming flow for v from the existence of edge
We continue the process by recursively calling the procedure on
2.4.1.5. Proof of induction correctness
The new instance to be called is an instance with size
If the call is invalid, it means some edge in
If this is an edge not in P, we claim the edge has zero flow in
Given the recursive call is valid, we can prove the correctness of the algorithm by showing that
2.4.1.6. Completing the proof
With the induction algorithm, we can reconstruct a partial decomposition
With both bounds proved, we can formally finish the proof of the main theorem:
Proof. This is implied by Theorem 3 for the lower bound, and Theorem 4 for the upper bound.
2.4.2. Algorithms for AND-Quant
In this section, we prove that the complementarity argument mentioned above is correct. Our setup consists of a DAG G with predetermined source S and sink T, a flow
We start by discarding edges e in any of Ei such that no good
Now given G is a DAG and the edge sets satisfy the well-ordering property, we can define the following:
We define the natural order
The filtering steps and construction of the block graph can be done in linear time, as discussed in Section 2.4.3.
The rest of this section is dedicated to prove the following lemma, which directly implies the main theorem:
We now provide a quick overview before proceeding to the actual proof.
2.4.2.1. Overview
The proof of the above lemma is done in several parts. The if direction of the statement is straightforward, and the only-if direction is harder to prove. We start by deriving some helpful corollaries of the well-ordering property. Next, we show that any vertex x in the block graph and any edge set Ei in the query list, x is either strictly before Ei, meaning there is a path starting at x and ending at an edge in Ei (this includes the case where
Proof. We can derive this from the well-ordering property of
Proof. Recall that we removed all edges in Ei that do not belong to any good paths.
Proof.
We can prove the symmetric statements about Vi.
Proof. If x is in one of Uj,
We can similarly prove that the condition holds for all
If x is in none of Uj or Vj, it is in an edge in Bj, which by definition of Bj means both
We have proved for any vertex in x and any i, one of
Proof. We first prove that if a path is good, it is within GB. An edge
To prove the other direction, we show that removing any of Ei results in disconnection of S to T. By the previous lemma, the vertices of GB can be partitioned into two disjoint sets: If If
So we have
We can now finish the proof of the main theorem:
Proof. By Lemma 3, a path is a bad path if and only if the path intersects with
2.4.3. Constructing GB
In this section we discuss how to construct GB in linear time using notations from Section 2.4.2.
We first run two breadth-first searches from S and T to mark the set of vertices that are reachable from S and can reach T. All vertices that are unable to do both will never appear in an
We now show that the values of
By chaining
Similarly, we can obtain
The first part is possible if and only if
To construct GB, we need to construct the set of blocks Bi. This can be done using the values of
Combining the results, we have the following lemma for preprocessing (note that topological sorting is a linear time algorithm):
2.5. Structured analysis of differential expression
We have discussed nonidentifiability-aware transcript quantification under two assumptions. In this section, we model the quantification problem under a hybrid assumption. Some fragments are generated from the reference transcriptome, while others are generated by combining known junctions (valid under the setup of graph quantification). This model is more realistic than the model under the two extreme assumptions.
For each transcript Ti, let
In differential expression analysis, for each transcript we receive two sets of abundance estimates
When considering nonidentifiability, if the mean of
3. RESULTS
3.1. Implementation
In the following analyses, we use GENCODE annotation (version 26) (Frankish et al., 2018) as the reference transcriptome for constructing the splice graphs and for expression quantification. Salmon (Patro et al., 2017) and its corresponding graph quantification probabilistic model (Ma et al., 2020) are used to obtain the edge abundances of the splice graphs under complete and incomplete reference assumptions. We evaluate the expression estimation uncertainty due to nonidentifiability by determining whether the ranking of expression can be altered under different optimal abundances. Specifically, the ranking is computed within the same sample across isoforms in Section 3.2, and for the same isoform across samples in Section 3.3, which is statistically defined by DE analysis.
3.2. Expression of 20%–47% transcripts has inconclusive ranking compared with sibling isoforms
We applied our methods to the Human Body Map data set (The Illumina Body Map 2.0 data; 2011) (SRA accession ERX011205), which consists of 16 RNA-seq samples from 16 tissues. We are interested in evaluating the expression estimation uncertainty due to nonidentifiability, and focus on the transcripts for which the uncertainty of expression estimation is so large that the ranking of expression between the transcript and its sibling isoforms cannot be determined. We use the term “sibling isoforms” of a transcript to refer to the annotated alternative splicing isoforms that belong to the same gene. For each transcript, we enumerate its sibling isoforms, and compare the range of optimal expression estimates for the pair of transcripts to determine whether the two ranges overlap. An overlap between the two ranges indicates an indecisiveness in the ranking of expression between the two transcripts.
We observe that around 35%–50% of transcripts have uncertain expression estimates due to nonidentifiability. That is, the ranges of optimal abundances of these transcripts are not single points. The majority of them (around 20%–47% of total transcripts across all 16 samples) have a very uncertain expression estimate such that the ranking of expression between the transcript and at least one of its sibling isoform is inconclusive (Fig. 4A). The range is computed under various
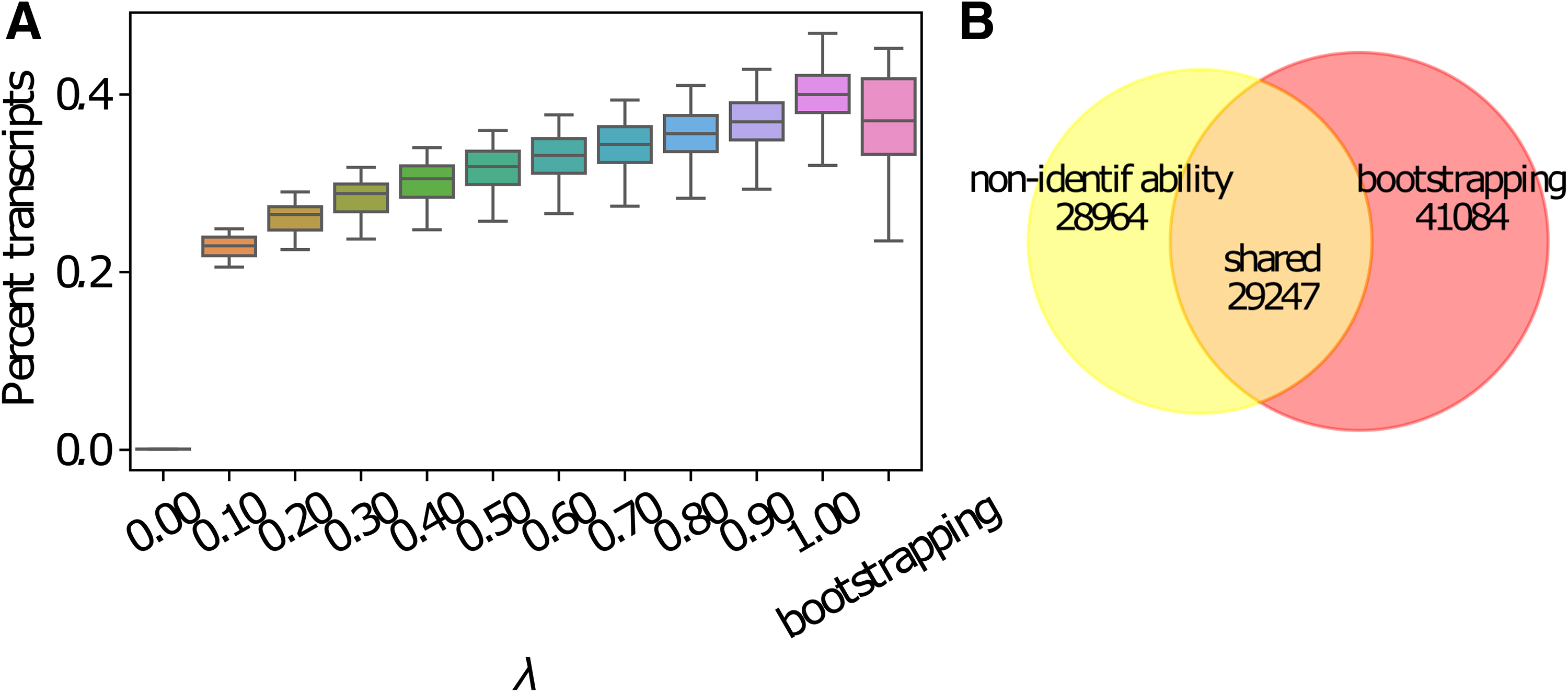
Transcripts with inconclusive ranking of expression among sibling isoforms due to nonidentifiability.
When compared with the expression estimation uncertainty caused by finite sample size (or finite sequencing depth), we observe that the sets of transcripts with inconclusive expression ranking due to the two sources of error do not have large overlap (Fig. 4B). In an arbitrarily chosen Human Body Map sample (a prostate sample, SRA accession ERR030877), we set the
The uncertainty caused by finite sample size is evaluated by bootstrapping in Salmon software (Patro et al., 2017). Terminus (Sarkar et al., 2020) identifies groups of transcripts that have smaller quantification variance when quantifying as a group compared with quantifying individually, and is based on bootstrapping or Gibbs sampling of expression estimation. Theoretically, the difference between Terminus and our approach for identifying uncertain expression estimates lies in the source of error and whether incomplete reference is considered. In Figure 5, we show the percentage of commonly identified transcripts with expression estimation uncertainty of the two methods in the Human Body Map data set.
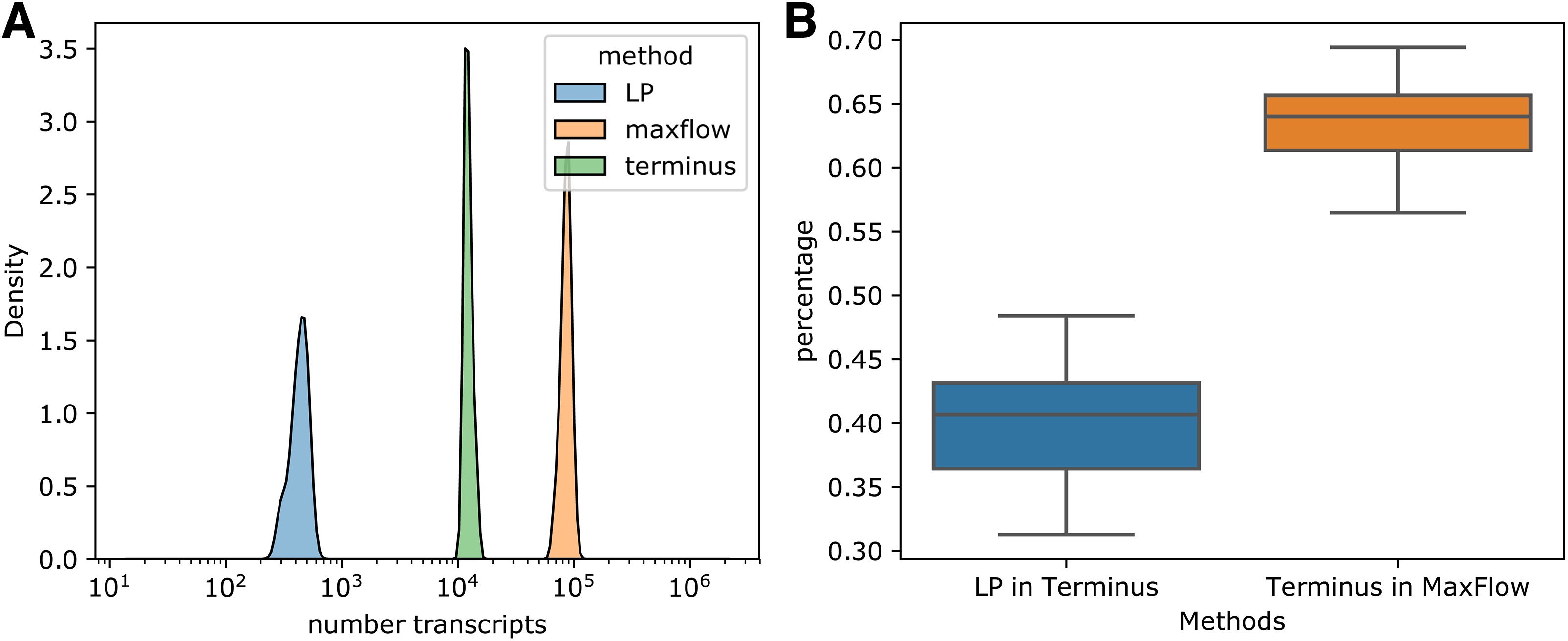
Counting the number of transcripts with uncertain expression estimates, and comparing with terminus.
The computed percentages of transcripts with inconclusive ranking of expression are upper bounds. Because the range of optimal expression estimate per transcript is calculated separately, arbitrarily selecting a pair of values from two ranges of optimal expression of two transcripts may not lead to an optimal pair of expression in the expression probabilistic model. For example, selecting the maximum values for both isoforms may lead to nonvalid estimates where the sum of estimated expression (before normalization) exceeds the number of observed RNA-seq reads.
However, we speculate that these upper bounds are close to the true percentages. Because reversing the ranking requires one to increase the expression of one transcript and decrease the expression of the other, and it is less likely to generate nonviable paired estimates under this operation.
3.3. DE transcripts are generally reliable when assuming the reference transcripts contribute >40% to the expression
Applying our method to the MCF10 cell line samples with and without epidermal growth factor (EGF) treatment (accession SRX1057418) (Kiselev et al., 2015), we analyze the reliability of the detected DE transcripts. We use Salmon (Patro et al., 2017), tximport (Soneson et al., 2015), and DESeq2 (Love et al., 2014) for the differential expression detection pipeline. This pipeline predicts 257 DE transcripts under a false discovery rate (FDR) threshold of 0.01. We use the overlap between the mean ranges of optimal expression estimated in the samples with and without EGF treatment as an indicator for unreliable DE detection (see Section 2.5 for details). The overlap is defined as the size of the intersection over the size of the smaller range of the two ranges of optima. We compute the overlap under various
We identify examples of reliable and unreliable DE predictions. There are five DE transcripts for which their differential expression statuses may change even when the reference expression proportion (
The five genes corresponding to the five transcripts are involved in the following cellular processes or pathways: mRNA degradation, cell apoptosis, glucose transportation, DNA repair, inhibition and transportation of certain kinetics (Pruitt et al., 2011). These genes contain between 6 and 22 isoforms. Further analyses based on the DE detection of these five transcripts require caution since they may be falsely detected to be DE due to nonidentifiability.
Other than these transcripts, the detected DE transcripts are generally reliable after considering the potential expression estimation uncertainty due to nonidentifiability. The ranges of optimal expression estimates between the two sample groups do not have large overlaps when the reference transcriptome is relatively complete and contribute >40% to the expression (Fig. 6E). This observation is supported by another data set, where replicates naive CD8 T cells, and four replicates of effector memory CD8 T cells are compared for differential expression detection, as detailed in Figure 7. There are 3152 DE transcripts under FDR threshold 0.01, 19 out of which are unreliable even when reference transcripts compose >90% expression. We observe the similar pattern that most DE predictions are reliable when the reference transcript expression is >40%.
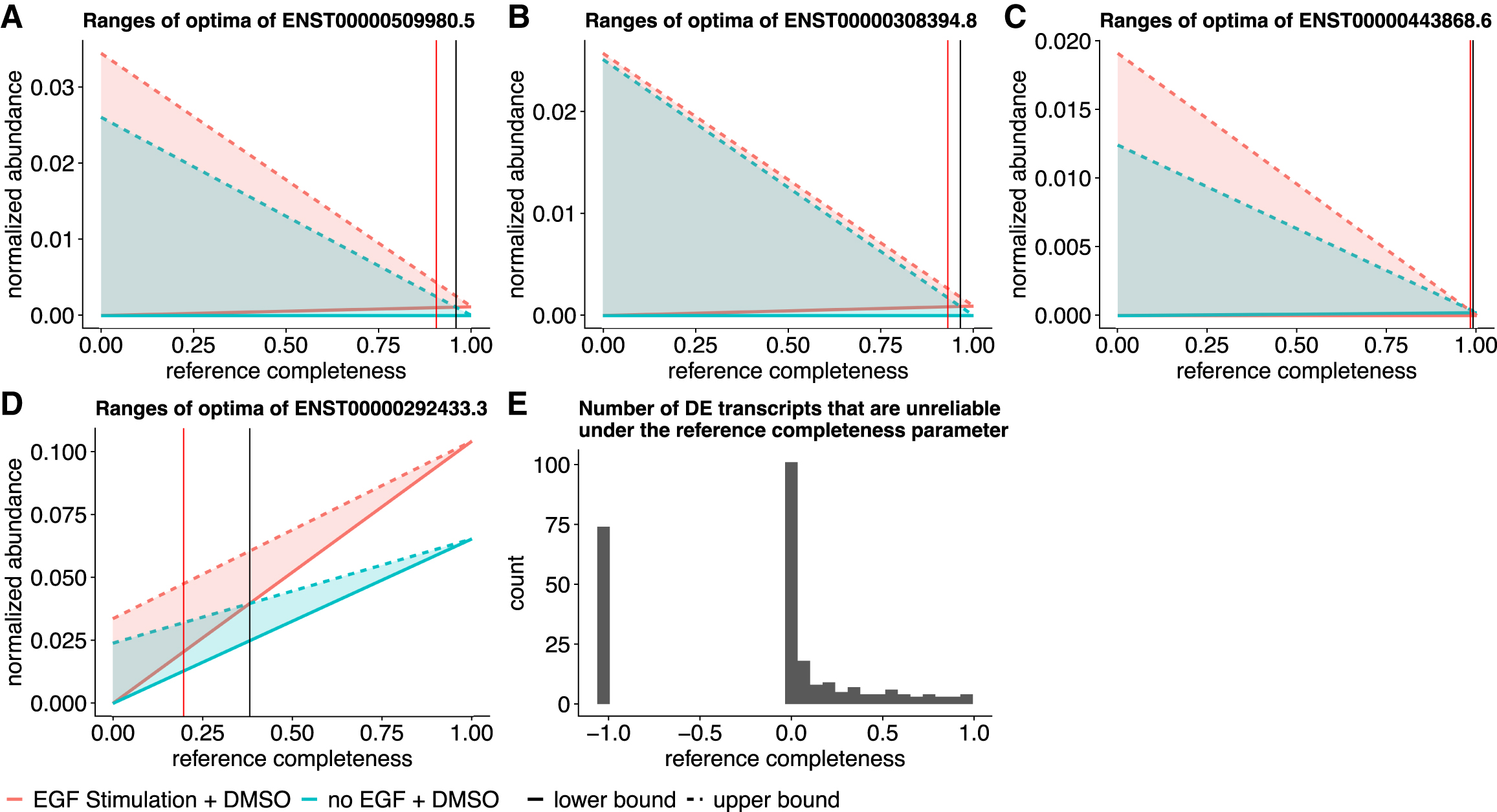
Overlap between ranges of expression indicates potentially unreliable DE detections in the MCF10 cell line data.
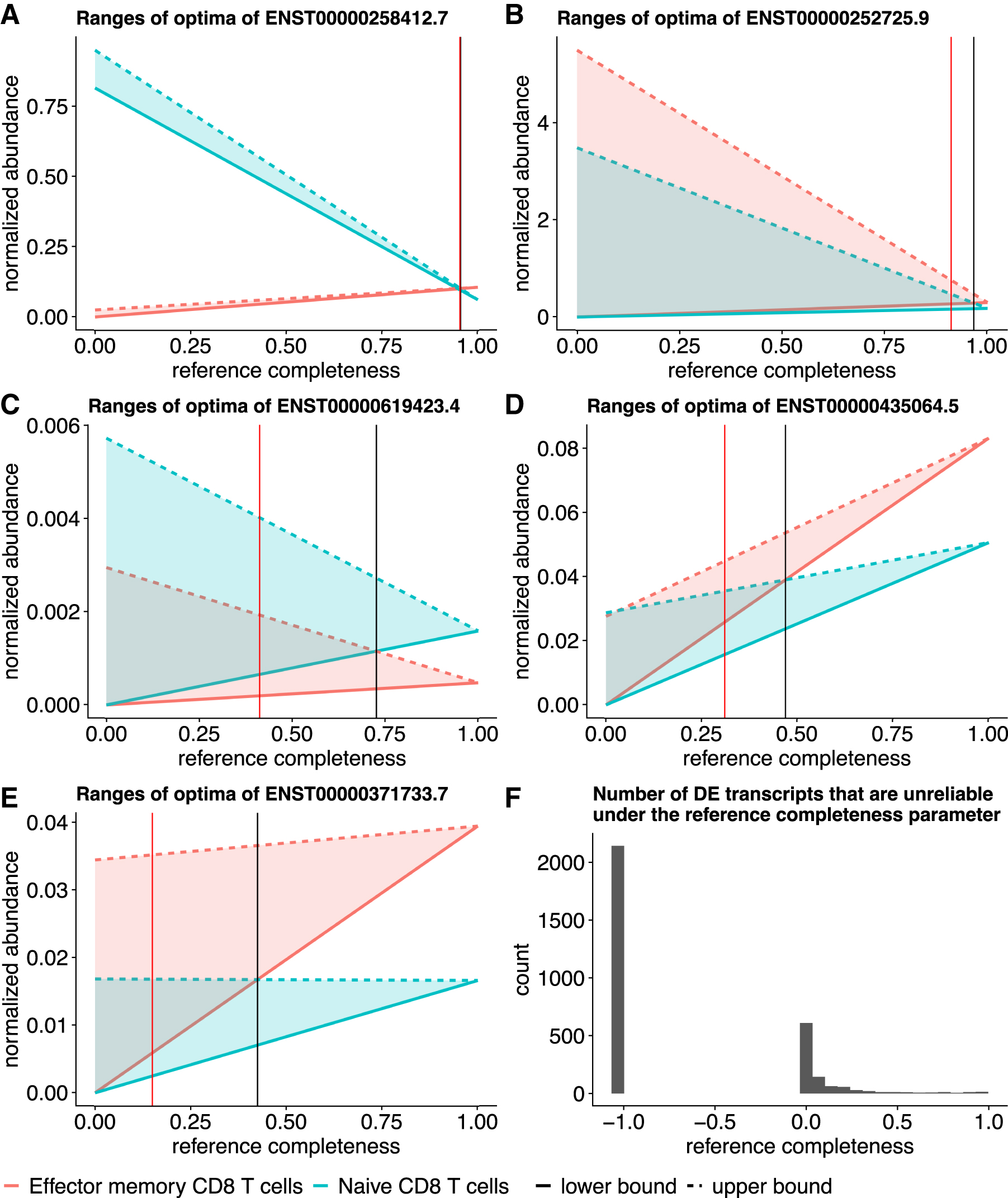
Overlap between ranges of expression indicates potentially unreliable DE detections in the CD8 T cell data.
A previous study (Everaert et al., 2017) showed that expression quantification software tends to make slightly more mistakes in deciding the relative expression of isoforms within one sample, compared with deciding the fold change of one isoform across multiple samples. Our results here and in the previous section show agreement with that observation, but with amplified errors in deciding relative expression of isoforms within one sample. Our model for bounding the range of uncertainty due to model nonidentifiability provides an explanation from the theoretical perspective: the short sequencing reads may not be sufficient to uniquely reveal the relative abundances of transcripts from complicated splice graphs.
4. CONCLUSION AND DISCUSSION
We develop algorithms to compute the range of optimal expression estimates due to nonidentifiability for each transcript. We consider both complete and incomplete reference transcript assumptions (quantified with reference-transcript-based quantification and graph quantification, respectively) and further provide the range of uncertain estimates under mixed assumptions: a certain proportion of expression is from reference transcripts and the rest (indicated by
Applying our methods on Human Body Map samples and two RNA-seq data sets for DE transcript detection, we observe the following expression uncertainty patterns: the ranking of expression between a transcript and its sibling isoforms in a given sample cannot be determined for many (20%–47%) transcripts if the expression estimation uncertainty is considered, but when comparing the expression estimates of a transcript across multiple RNA-seq samples, the detected DE transcripts are mostly reliable.
The
The nonidentifiability problem in expression quantification is partly due to the contrast between the complex splicing structure of the transcriptome and short length of observed fragments in RNA-seq. Recent developments of full-length transcript sequencing might be able to close this complexity gap by providing longer range phasing information. However, full-length transcript sequencing techniques suffer from problems such as low coverage and high error rate. It is still open whether full-length transcript sequencing is appropriate for quantification and how the current expression quantification methods, including this work, should be adapted to work with full-length transcript sequencing data.
Footnotes
ACKNOWLEDGMENTS
We thank Dr. Guillaume Marçais, Dr. Natalie Sauerwald, and Dr. Jose Lugo-Martinez for insightful comments on the article.
DISCLAIMER
The Department specifically disclaims responsibility for any analyses, interpretations, or conclusions.
AUTHOR DISCLOSURE STATEMENT
C.K. is the cofounder of Ocean Genomics, Inc.
FUNDING INFORMATION
This work has been supported, in part, by the Gordon and Betty Moore Foundations Data-Driven Discovery Initiative through Grant GBMF4554 to C.K., by the U.S. National Institutes of Health (R01GM122935), and the U.S. National Science Foundation (DBI-1937540). This work was partially funded by The Shurl and Kay Curci Foundation. This project is funded, in part, under a grant (No. 4100070287) with the Pennsylvania Department of Health.