
Abstract
The Robinson-Foulds (RF) distance, one of the most widely used metrics for comparing phylogenetic trees, has the advantage of being intuitive, with a natural interpretation in terms of common splits, and it can be computed in linear time, but it has a very low resolution, and it may become trivial for phylogenetic trees with overlapping taxa, that is, phylogenetic trees that share some but not all of their leaf labels. In this article, we study the properties of the Generalized Robinson-Foulds (GRF) distance, a recently proposed metric for comparing any structures that can be described by multisets of multisets of labels, when applied to rooted phylogenetic trees with overlapping taxa, which are described by sets of clusters, that is, by sets of sets of labels. We show that the GRF distance has a very high resolution, it can also be computed in linear time, and it is not (uniformly) equivalent to the RF distance.
1. Introduction
Trees are simple mathematical structures that are used to represent or model a relation on individuals. When those individuals are species and the relation is the sequence similarity of their genomes, the tree-like representation becomes a phylogenetic tree. Phylogenetic trees can also be inferred from metabolic pathways (Forst and Schulten, 2001; Chor and Tuller, 2007), protein–protein interaction networks (Erten et al., 2009), tumor clones of cancer (Miura et al., 2020), languages (Pompei et al., 2011), music (Bomin et al., 2016), etc., with appropriate distance or similarity relations. At the beginning, phylogenetic trees were designed to infer evolutionary relationships based on some species appearance (mainly morphological and physiological traits). However, the explosion of sequencing technologies yielding a vast amount of DNA and RNA sequence data has generated different methodologies to obtain such a phylogenetic tree, such as maximum parsimony and maximum likelihood approaches; see Bruyn et al. (2014) and Kapli et al. (2020) for an overview on these phylogenetic reconstruction methods), Bayesian inference (Rannala and Yang, 1996, 1997; Mau et al., 1999; Li et al., 2000), and the neighbor-joining method (Saitou and Nei, 1987; Studier and Keppler, 1988). Each of them reconstructs a phylogenetic tree from a set of sequences; however, those phylogenetic trees may differ from one methodology to another, providing different trees for the same input data.
As a first solution to the disagreement on the different methodologies, consensus trees have been also defined and consensus tree methods have been implemented as well (Jansson et al., 2014, 2016). Nevertheless, one step further is when different experiments are performed, yielding different sequence data. In this case, and to analyze the evolutionary relationship of such sequences, the reconstructed phylogenetic trees must be compared. Reconstructed phylogenetic trees can be of any type: binary or multifurcating, labeled only on the leaves (taxa) or on all the nodes, with no repeated labels (injectively labelled) or with repeated labels, etc. Hence, different experiments may yield different phylogenetic trees of different types, such as phylogenetic trees with overlapping taxa as in the case of the experiment described later, where trees share some of the leaves labels but not all of them. Regarding a comparison of phylogenetic trees, several metrics have been defined as dealing with different types of trees. For phylogenetic trees injectively labeled on a set of taxa, that is, with no repeated labels, the most widely used distance is the Robinson-Foulds (RF) distance, which compares the clades (or clusters) of every node in the trees and counts how many of them are not shared by both trees. Many other metrics on phylogenetic trees have been proposed in the past few years; see Kuhner and Yamato (2015); Wang et al. (2020) for recent reviews.
Unlike phylogenetic trees reconstructed by different experiments on the same input data, phylogenetic trees reconstructed from different input data usually have overlapping taxa and thus, the comparison of phylogenetic trees with overlapping taxa is also of utmost importance. Figure 1 shows two alternative phylogenetic trees with overlapping taxa for several species of tomato (genus Solanum): a chloroplast DNA phylogeny, adapted from (Palmer and Zamir, 1982), and a mitochondrial DNA phylogeny, adapted from (McClean and Hanson, 1986); see also Baum and Ragan (2004). They overlap in all taxa but S. juglandifolium and S. rickii, shown to be highlighted. Both the chloroplast DNA phylogeny and the mitochondrial DNA phylogeny have 19 clusters each. They differ in all clusters but those of their nine common taxa,
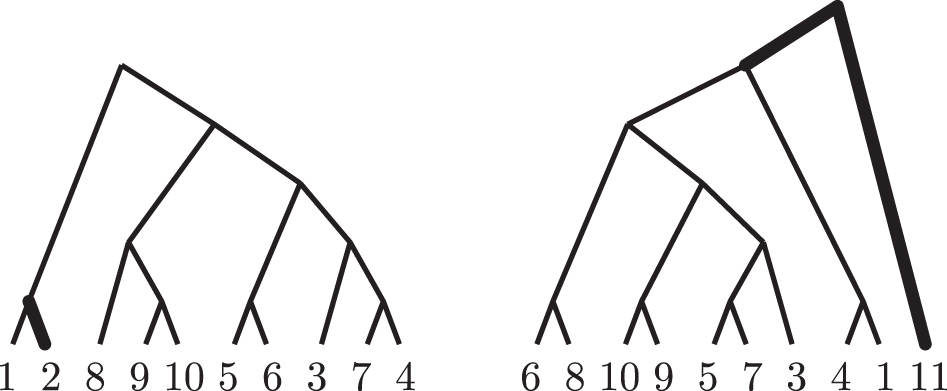
Chloroplast DNA phylogeny (left) and mitochondrial DNA phylogeny (right) of several species of the genus Solanum. 1: S. lycopersicoides; 2: S. juglandifolium; 3: S. peruvianum; 4: S. chilense; 5: S. pennellii; 6: S. hirsutum; 7: S. chmielewskii; 8: S. esculentum; 9: S. pimpinellifolium; 10: S. cheesmaniae; 11: S. rickii.
2. Background
2.1. The RF distance
Recall that the cluster (also, the clade or the monophyletic group) associated with a node in a phylogenetic tree is the set of descendant leaf labels of the node in the tree, and the cluster representation of a phylogenetic tree (Steel, 2016, §2.2) is the set of clusters for the nodes in the tree.
The RF distance (Robinson and Foulds, 1981), a widely used metric for comparing phylogenetic trees, can be computed in linear time in the size of the trees (Day, 1985; Pattengale et al., 2007). It was originally defined as the cardinality of the symmetric difference between the sets of clusters of the phylogenetic trees. When normalized to the unit interval, it becomes the Jaccard distance on these sets (Levandowsky and Winter, 1971): the one-complement of their Jaccard index (Jaccard, 1912).
Despite the wide acceptance over several decades now, the RF distance has some shortcomings. On the one hand, it has a very low resolution, because it can only take a small number of different values—the total number of leaves in the pair of compared trees—and it only takes into account whether two clusters are equal or not. Hence, when two clusters differ in only one label, this is equally counted as when they differ in all their labels (Section 3). On the other hand, and as a consequence of the latter, it may become trivial for phylogenetic trees with overlapping taxa, which differ in all their clusters except at most those consisting only of common labels.
2.2. Previous generalizations of the RF distance
Several generalizations of the RF distance have been proposed over the past few years, in an attempt to address the shortcomings discussed earlier. One approach consists of considering the distance between two phylogenetic trees as an optimal matching problem on a weighted complete bipartite graph, where the vertices correspond to the clusters of descendant node labels of the two phylogenetic trees. In this setting, in the RF distance (Robinson and Foulds, 1981), the edges are weighted by 1 (for different clusters) or 0 (for identical clusters). In the
A related approach consists of matching each cluster in one tree to the most similar cluster in the other tree. In the cluster dissimilarity (CD) (Shuguang and Zhihui, 2015), the edges are also weighted by the size of the symmetric difference of the two clusters, and the distance between two phylogenetic trees is the sum of the minimum edge weights for the clusters of the first tree and the non-trivial clusters of the second tree, averaged with the sum of the minimum edge weights for the clusters of the second tree and the non-trivial clusters of the first tree.
Similar generalizations of the RF distance based on matching have also been proposed for unrooted phylogenetic trees (Boc et al., 2010; Bogdanowicz and Giaro, 2012a; Lin et al., 2012; Shuguang et al., 2014; Smith, 2020), and for trees with labeled internal nodes (Briand et al., 2020; Jahn et al., 2020). Further generalizations based on matching that take into account not only the clusters but also the structure of the phylogenetic trees have been proposed (Böcker et al., 2013; Borozan et al., 2019).
We have presented (Llabrés et al., 2020) a different generalization of the RF distance, based on the distances between sets of sets defined in Fujita (2013) and generalized to distances between multisets of multisets, which is a metric for the clonal trees (Govek et al., 2018; Karpov et al., 2019; DiNardo et al., 2020; Jahn et al., 2020) and the mutation trees (Kim and Simon, 2014; Aguse et al., 2019) that model tumor evolution under perfect phylogeny, phylogenetic trees, and several classes of phylogenetic networks, such as binary galled trees (Cardona et al., 2011), tree-child time-consistent phylogenetic networks (Cardona et al., 2008c, 2009a,b), and semi-binary tree-sibling time-consistent phylogenetic networks (Cardona et al., 2008a).
In this article, we further study the generalization of the RF distance when applied to phylogenetic trees, including theoretical properties, further empirical results, and more efficient algorithms. In comparison to previous generalizations, the latter has the advantage that it can be applied to phylogenetic trees over the same taxa and with overlapping taxa as well, that it has a much higher resolution, and that it, similar to the RF distance, can be computed in linear time.
2.3. Notation and basic results
Except when otherwise explicitly stated, all phylogenetic trees considered in this article are rooted phylogenetic trees. For every phylogenetic tree T, let:
For every pair of sets
For every pair of phylogenetic trees
3. The GRF Distance
In this section, we introduce the GRF distance, and we establish some properties of this distance comparing it with the RF distance.
3.1. Definition
In the original formulation, the GRF distance allowed for comparing any structures that can be described by sets or multisets of sets of multisets of labels (Llabrés et al., 2020). When restricted to phylogenetic trees (with possibly different sets of taxa), which are described by sets of sets of taxa, the GRF distance is defined as follows.
This GRF distance is, indeed, a metric, in the sense, that, for any phylogenetic trees
The proof of this fact is a simple application of Fujita, 2013, Theorem 1, taking into account that the cardinality of symmetric difference is a metric on sets and that every phylogenetic tree is characterized by its set of clusters.
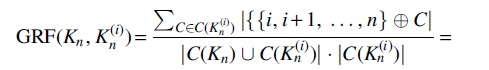
where
So, returning to Eq. (1), the numerator of its right-hand side is
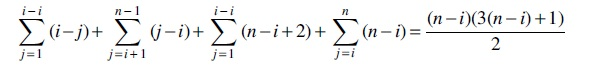
and hence, finally,
Therefore, contrary to what happens with the RF distance, all phylogenetic trees
The contraction of edge
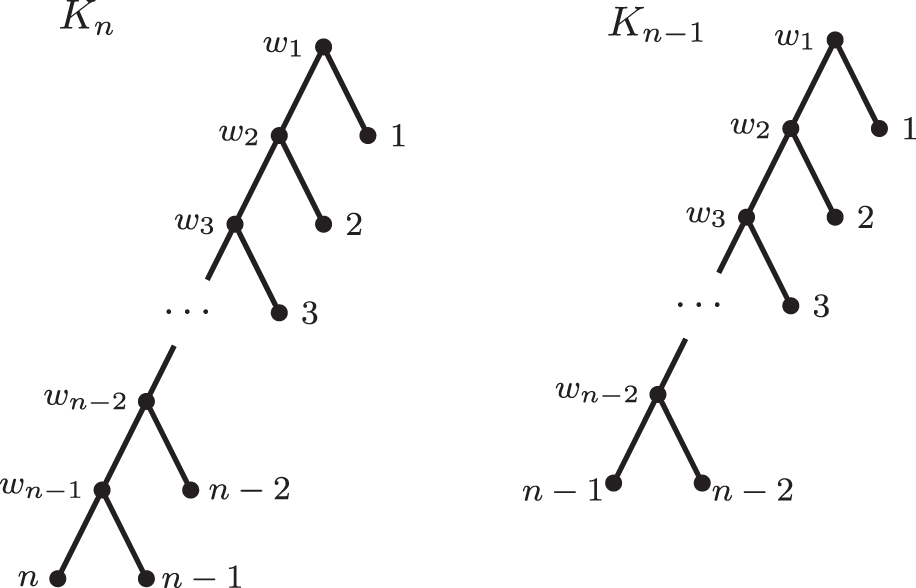
The caterpillars with n and
and hence,
Now,
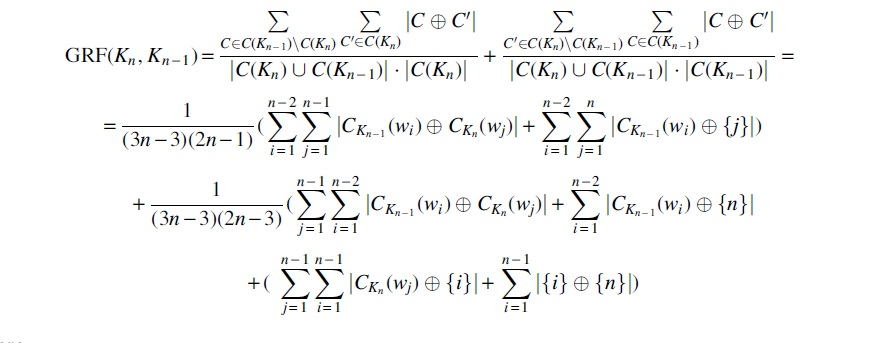
where
Therefore,
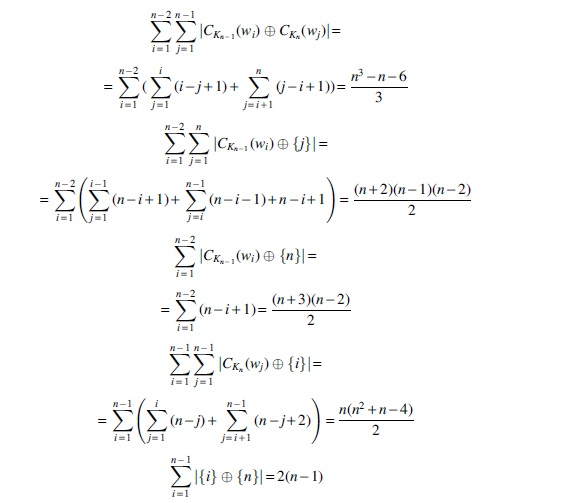
and, finally,
Then,
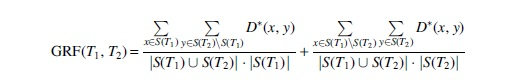
defines a GRF distance for unrooted phylogenetic trees.
The proof that this is a metric is again an application of (Fujita, 2013, Thm. 1), stating that every unrooted phylogenetic tree is characterized by its set of splits and that
is a metric on ordered pairs of sets. Now, each split
3.2. Theoretical properties
Our first lemma deals with the metric equivalence between GRF and RF. Recall that two metrics d1 and d2 defined on a space X are equivalent when there exist a pair of non-negative real numbers
This definition captures the intuitive idea that both metrics define the same notion of “closeness” on X.
(b) There is no constant
Proof. (a) Notice that
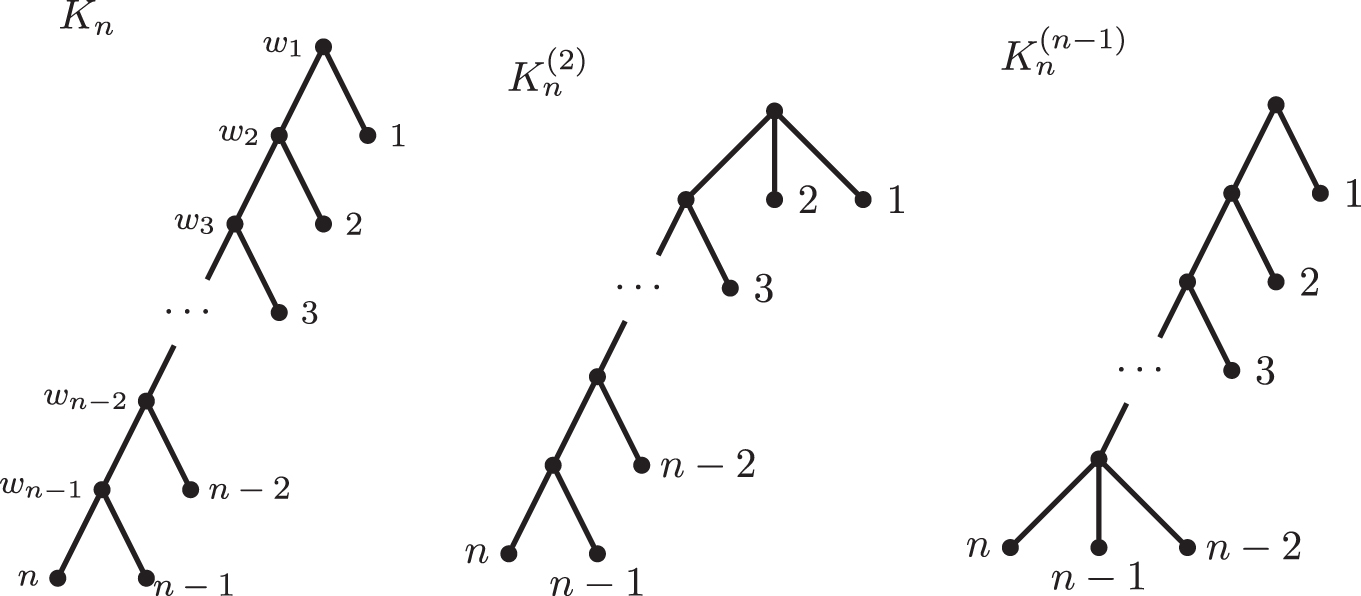
(b) Let Kn be the caterpillar with n leaves and
whereas
and there is no
The previous lemma entails that the GRF distance is not equivalent to the RF distance on the whole space of phylogenetic trees with any number of leaves. That is, although for every set of taxa, GRF and RF are equivalent metrics on the space of phylogenetic trees on this set of taxa, because all metrics on a given finite set are equivalent, any factor that transforms RF into a metric greater than GRF must depend on the number of taxa, being impossible to find a real number that works for every number of taxa. This is usually phrased by saying that RF and GRF are not uniformly equivalent (with respect to the number of leaves).
The following results will be used to show that, much like the RF distance, the GRF distance can also be computed in linear time in the size of the phylogenetic trees. Recall that the Sackin index of balance, introduced in Shao and Sokal (1990), for a phylogenetic tree T is
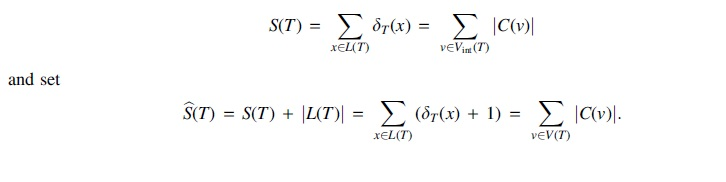
and set
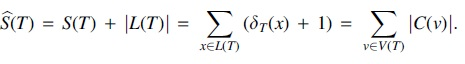
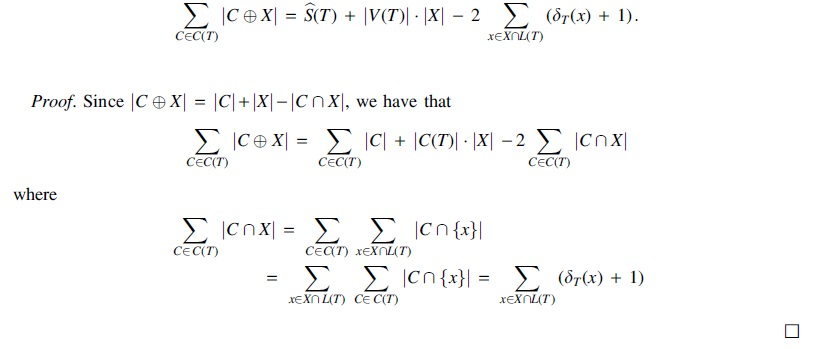
Proof. Since
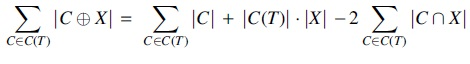
where
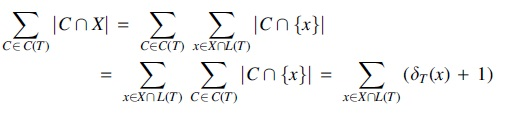
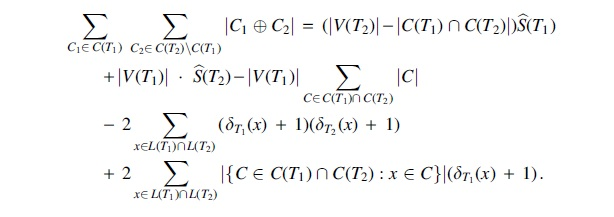
Proof. By the previous lemma,
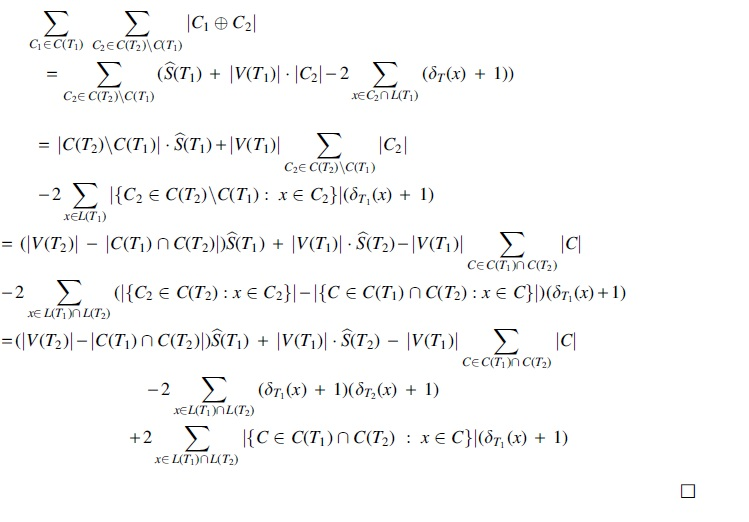
3.3. Computation in linear time
Let T1 and T2 be phylogenetic trees, let
With a sorted list representation of the clusters, which uses
With a bit-vector representation of the clusters, which uses
However, both the cluster representation of the trees and the intersection of the sets of clusters of the trees can be computed in
Proof. Let T1 and T2 be phylogenetic trees, let
4. Experimental Results
To study the resolution of the GRF distance and to compare it with the RF distance, we have performed a series of experiments on phylogenetic trees.
First of all, we have generated the 3 binary phylogenetic trees with 3 labeled leaves, the 15 binary phylogenetic trees with 4 labeled leaves, the 105 binary phylogenetic trees with 5 labeled leaves, and the 945 binary phylogenetic trees with 6 labeled leaves, using the algorithm described in Valiente (2009, §5.3.3), as implemented in Bio::Phylo (Cardona et al., 2008b; Vos et al., 2011).
Further, we have also generated 10,000 pairs of random binary phylogenetic trees with n labeled leaves, for
Then, we computed both the RF distance and the GRF distance for every generated pair of phylogenetic trees.
4.1. Resolution of the metric
The resolution of a distance between phylogenetic trees on a set of labels is the number of different values taken by the distance upon all pairs of phylogenetic trees on that set of labels. When divided by the number of phylogenetic trees on that set of labels, it is the recognition ratio defined by Shao and Sokal (1986) for consensus indices between phylogenetic tree shapes.
We have computed the RF distance and the GRF distance between each pair of binary phylogenetic trees with the same number
The resolution of the RF distance on binary phylogenetic trees with
Number of Different Values Taken by the
CD, cluster dissimilarity; GRF, generalized Robinson-Foulds.
Table 1 also shows the number of different values for two previous generalizations of the RF distance based on matching, the
4.2. Refinement of the RF distance
In this second test, we wanted to determine whether the GRF distance is a refinement of the RF distance. Hence, we first checked whether any triplet of phylogenetic trees, such that two of them are at the same GRF distance of the third one, is also at the same RF distance of the third one. We shall call GRF-equidistant triplets and RF-equidistant triplets those triplets of phylogenetic trees such that two of them are at the same GRF distance and RF distance of the third one, respectively. Thus, for all triplets of binary phylogenetic trees with
As shown in Table 2, the first ratio (i.e., the number of GRF-equidistant triplets that are not RF-equidistant over the number of GRF-equidistant triplets) quickly converges to one as the number of labeled leaves increases, meaning that almost none of the GRF-equidistant triplets are RF-equidistant. This result reinforces the statement in the previous section that the GRF distance has a much higher resolution than the RF distance. On the other hand, the second ratio (i.e., the number of RF-equidistant triplets that are not GRF-equidistant over the number of RF-equidistant triplets) turned out to be zero except for phylogenetic trees with 6 leaves. Indeed, in the example given next, we show a triplet of phylogenetic trees with 6 leaves that is a GRF-equidistant triplet but it is not RF-equidistant. This means that the GRF distance is not a refinement of the RF distance. However, we can also observe in Table 2 that almost all the RF-equidistant triplets are GRF-equidistant as well.
Ratio of Generalized Robinson-Foulds-Equidistant Triplets to Robinson-Foulds-Equidistant Triplets and Vice Versa, for All the Triplets of Binary Phylogenetic Trees with
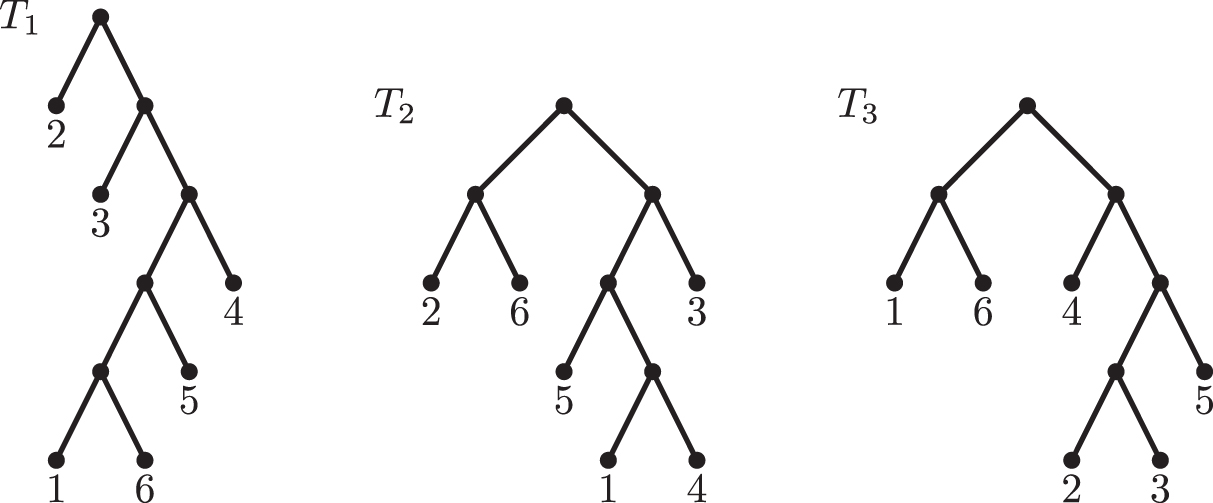
Triplet of phylogenetic trees
5. Discussion
Even though the RF distance is the most widely used distance for phylogenetic trees with no repeated labels, it has some drawbacks. First, it is only defined for pairs of trees on the same set of taxa. Second, it counts how many clades are shared by a pair of trees but, for the non-shared clades, it does not take into account how similar they are and, consequently, it has a very low resolution. In contrast to the RF distance, the GRF distance studied in this article happens to solve these shortcomings while keeping the advantages of the former one. First of all, the GRF distance allows for the comparison of any structures that can be described by multisets of multisets of labels. Thus, in the phylogenetic trees setting, phylogenetic trees are not restricted to be defined on the same set of taxa. In addition, for every pair of phylogenetic trees, it considers their shared clades but also, for the non-shared ones, it considers their dissimilarity, thus producing a distance with a high resolution. When restricted to phylogenetic trees on the same set of taxa, the tests presented in this study to compare both distances show that the GRF distance is nearly a refinement of the RF distance and it has a much higher resolution. As it is the case of the RF distance, the GRF distance can be computed in linear time and keeps the advantage of being intuitive, with a natural interpretation in terms of common splits.
Our current agenda involves the analysis of the topological behavior of the GRF distance, such as the metric diameter, that is, those phylogenetic trees at the maximum distance, the phylogenetic trees at minimum distance as well as the effect of elementary edit operations such as contracting an edge or removing a leaf, and rearrangement operations such as nearest-neighbor interchange, subtree pruning and regrafting, and tree bisection and reconnection (Allen and Steel, 2001) on the distance.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This research was partially supported by the Spanish Ministry of Science, Innovation and Universities and the European Regional Development Fund through project PGC2018-096956-B-C43 (FEDER/MICINN/AEI), and by the Agency for Management of University and Research Grants (AGAUR) through grant 2017-SGR-786 (ALBCOM).