
Abstract
Current work for multivariate analysis of phenotypes in genome-wide association studies often requires that genetic similarity matrices be inverted or decomposed. This can be a computational bottleneck when many phenotypes are presented, each with a different missingness pattern. A usual method in this case is to perform decompositions on subsets of the kinship matrix for each phenotype, with each subset corresponding to the set of observed samples for that phenotype. We provide a new method for decomposing these kinship matrices that can reduce the computational complexity by an order of magnitude by propagating low-rank modifications along a tree spanning the phenotypes. We demonstrate that our method provides speed improvements of around 40% under reasonable conditions.
1. Introduction
Understanding the etiology and biological pathways involved in health and disease requires a multivariate analysis of phenotypic data in genome-wide association studies (GWAS). This sort of analysis is becoming more common due to increased compute power (Cox et al., 2018). Only recently have such analyses become tractable through improvements to the efficiency of linear mixed models and related models (Lippert et al., 2011; Listgarten et al., 2012, 2013; Dahl et al., 2016). Further multivariate analysis is required for the study of complex diseases such as cancer (Knox, 2010) and groups of complex phenotypes such as brain imaging phenotypes (Elliott et al., 2018), and the analysis of large consortia involving multimodal data such as the U.K. Biobank (Bycroft et al., 2018).
The use of multiphenotype (or multivariate) data often requires that a scaled version of the kinship matrix (or genetic similarity matrix, or genetic relationship matrix: Patterson et al., 2006; Balding et al., 2007) K be used as a covariance matrix of a multivariate distribution or matrix variate normal distribution, defined on the phenotype vector in a GWAS (Dutilleul, 1999). And so, solutions to
Phenotype matrices may be partially observed (e.g., outliers are removed leading to column-specific missingness, or experimental paradigm varies among subjects leading to blockwise missingness in phenotype categories). To compute 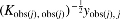 for each j. This is equivalent to marginalizing out the missing samples under the assumption of Gaussianity (i.e., it does not add additional assumptions beyond those already assumed by mixed models). Here
for each j. This is equivalent to marginalizing out the missing samples under the assumption of Gaussianity (i.e., it does not add additional assumptions beyond those already assumed by mixed models). Here
In this article, we present an efficient algorithm for computing the Cholesky decomposition (Benoıt, 1924) of Kj, for use in calculation of
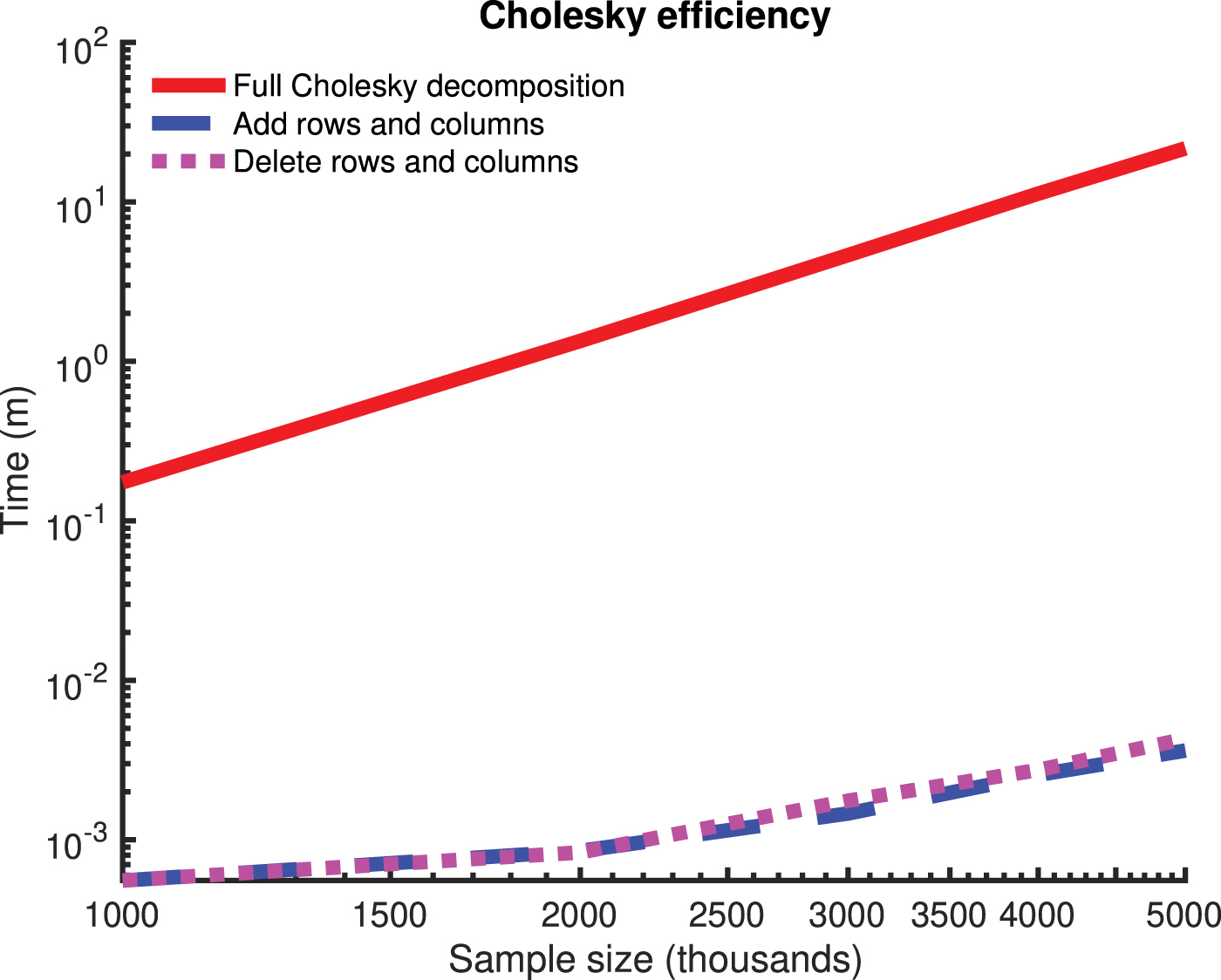
Runtimes for Cholesky decomposition and modifications for 10,000 to 50,000 samples. The log/log scale shows the asymptote and the order of magnitude relative speed of the modifications over the full Cholesky decompositions. For each condition, 15 random positive-semidefinite matrices are considered. The Cholesky decomposition is performed (mean given by red line) and then modifications are performed on a random row and column. Computations are performed using the Intel Math Kernel Library (MKL) implementation of the Netlib library.
The asymptotic complexity of performing Cholesky decompositions in a naive way for all Kj is
Here r is defined as
1.1. Related work
To reduce the computational complexity of genetic similarity matrix operations, several research programs have been conducted to store and manipulate sparse representations of the genetic similarity matrix (Shor et al., 2019). In these representations, researchers set a threshold and then zero out elements of the genetic similarity matrix with absolute value less than this threshold. The computational gains of such an approach may be large, but in theory, such an approach could lead to loss of power. Furthermore, this approach would be less suitable to data originating from pedigrees or small isolated populations. To our knowledge, our work is the first to leverage Cholesky low-rank modifications for improving efficiency of genetic similarity matrix-based inference.
2. Methods
In this section, we describe the genetic similarity matrices for which the kgen algorithm is appropriate and then provide the details of the kgen algorithm.
2.1. Genetic similarity matrices
The definition of the kinship matrix we use is that of a genetic similarity matrix centered at population-level minor allele frequencies. This definition is based on Patterson et al. (2006) but note that it involves population-level normalization instead of sample-level normalization. Let 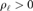 ). Then, the genetic similarity matrix is an
). Then, the genetic similarity matrix is an
The Cholesky decomposition of an
2.2. The kgen algorithm
A meditation on these asymptotes suggests the kgen algorithm. The Cholesky modifications can be designed to add and remove rows and columns of K (Osborne et al., 2010), and so, the computation of
Procedures to arrange Cholesky modifications in a way that adds and removes rows and columns of K are provided in Osborne et al. (2010). We refer to these operations as delete and insert. These operations are described formally as follows. Let v be an
Then,
Conversely, let L be the Cholesky decomposition of the matrix
These insert and delete operations can be performed in
We now describe the kgen algorithm in detail. The kgen algorithm is listed in Algorithm 1 in this article. This algorithm assumes that an
The kgen algorithm works by first finding a phenotype j0 such that the number of missing entries for the j0-th column of Y is less than or equal to the number of missing phenotypes in any other column. And then, we find a tree spanning all phenotypes. The tree is chosen such that the sum of the number of samples that must be added or removed for each edge of the tree (the sum of the weights of the edges) is minimized. For an edge from phenotypes j1 to j2, a sample must be added if it is in
After this minimum spanning tree is created, a breadth-first enumeration of the edges of this tree is constructed, such that the first edge includes the root of the tree. This enumeration must be breadth-first, because propagation of Cholesky decompositions along an edge may involve hard-drive reads and writes. The finished decompositions may have to be written and read to disk, as RAM (random access memory) provisions on supercomputers often cannot store the Cholesky decompositions of >1,000 phenotypes with 20,000 samples. So, software implementing the kgen algorithm will read the decomposition from the “source” vertex unless that decomposition has been recently read. Ensuring that the decomposition for the “source” vertex has most often been recently read is equivalent to providing the path through the spanning tree in a breadth-first way.
After the enumeration is created, the kgen algorithm computes the Cholesky decomposition of
2.3. A worked example of the kgen algorithm
In Figure 2, we provide a worked example of the kgen algorithm involving 10 samples and 8 phenotypes. In this example, the root of the minimum spanning tree is phenotype two and a breadth-first enumeration of the edges of the minimum spanning tree (such that the root is given by the first edge) is
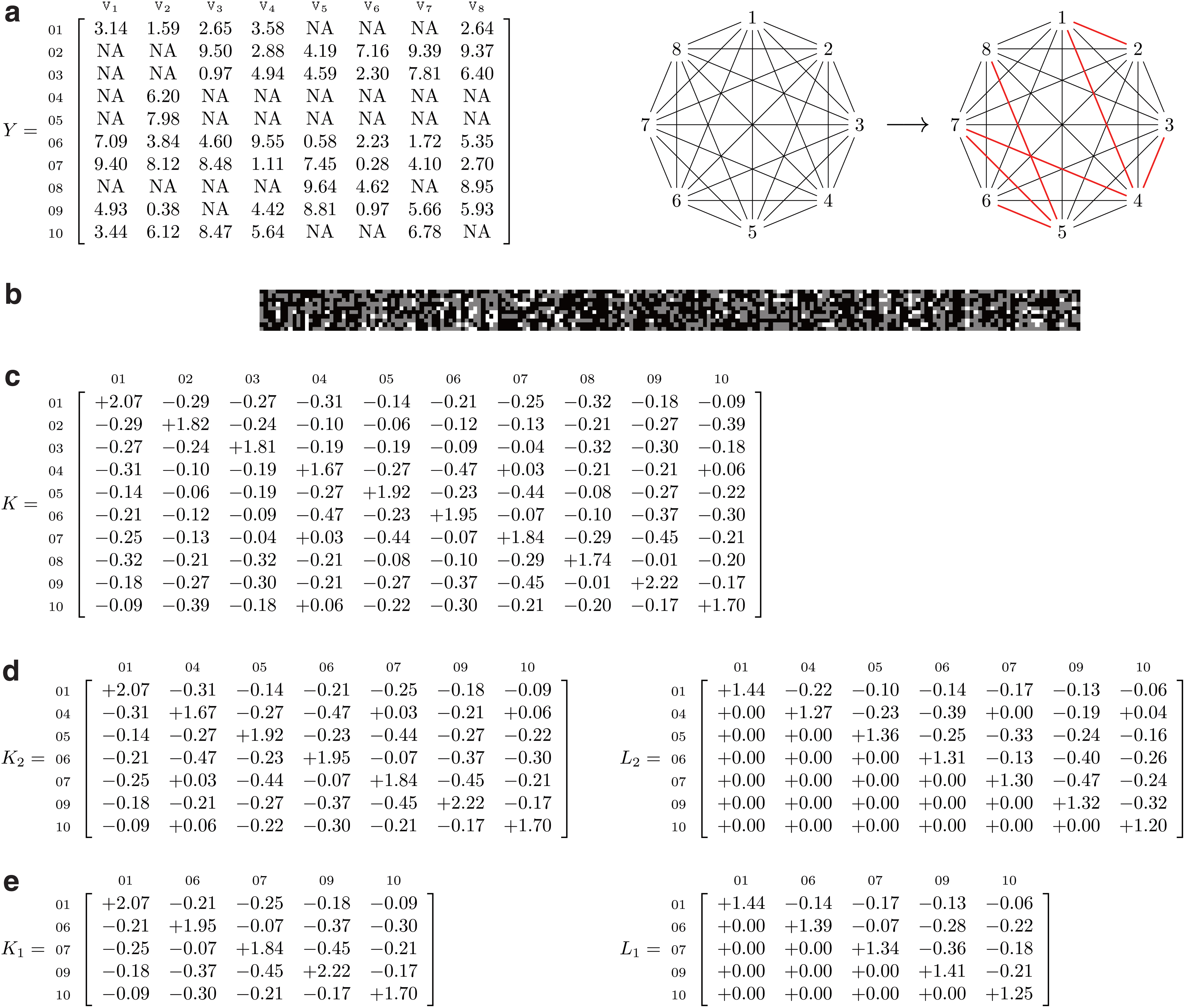
A worked example for the kgen algorithm.
3. Experiments
We now consider two experiments on simulated data and compare the speed and accuracy of the kgen algorithm to that of a naive algorithm in which the full Cholesky decomposition is computed for each phenotype. In the first experiment, we vary the number of samples and the missingness rate of the phenotype measurements, and assume that the data are missing-at-random, and assume a fixed number of 100 phenotypes per condition. In the second experiment, we examine a blockwise missingness pattern.
These experiments (and the results are displayed in Fig. 1) were conducted on Intel Xeon E5-2683 CPUs, and the numerical matrix operations were performed using Intel's MKL (math kernel library) implementation of the Netlib library (Anderson et al., 1999). The machine epsilon on this CPU was
3.1. Experiment 1
We consider missingness rates of 0.01%, 0.05%, 0.1%, 0.15%, and 0.2%, and a missing-at-random missingness pattern over 100 phenotypes. We consider n = 10,000, 15,000, 20,000, 25,000, and 30,000 samples. For each condition, we consider five independent replicates, and we sample the kinship matrix from the same Wishart distribution that was used in Figure 1 (i.e., with a mean given by the identity matrix and with 10,000 degrees of freedom). The difference in the runtime between kgen and the naive method (in which a full Cholesky decomposition is done for each phenotype), averaged over the five independent replicates for each condition, is displayed in Figure 3. The maximum entrywise absolute difference between the two methods over all phenotypes and conditions was 1.1768e − 14 (in the units of the Cholesky decomposition space), indicating close alignment and low numerical imprecision.
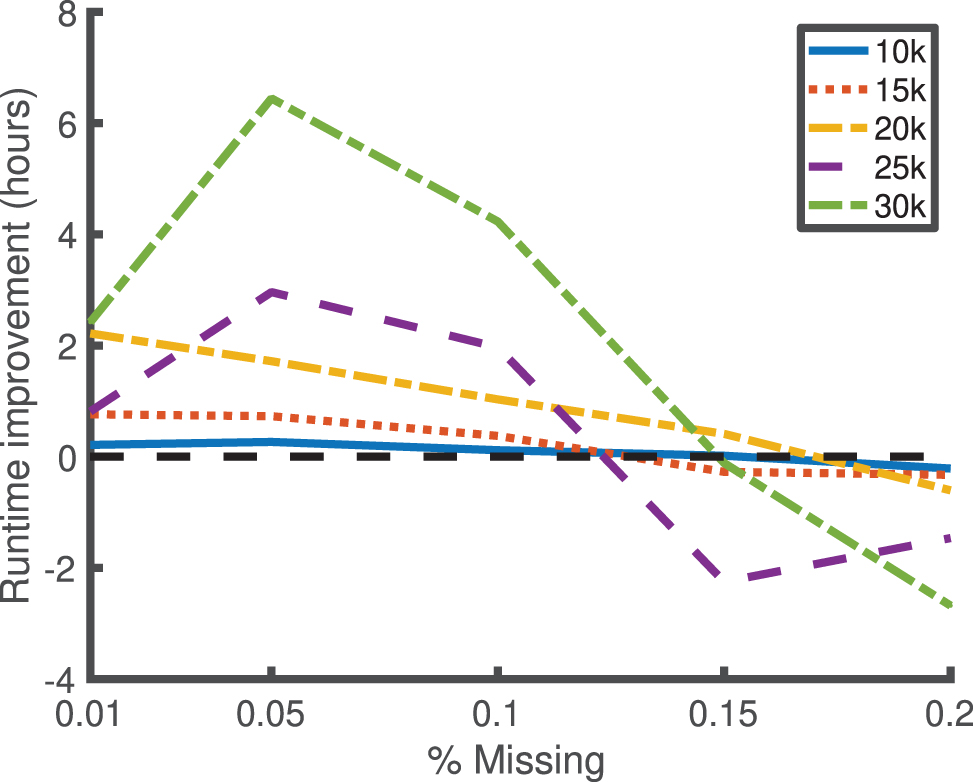
Improvement in runtime for the kgen software over naive Cholesky decompositions for low values of missingness (x-scale) for 100 phenotypes, and varying numbers of samples (indicated by legend). The y-scale indicates the median runtime for the kgen software minus the runtime of naive Cholesky decompositions, over five replicates per condition. The kgen software is better for all values of missingness
A histogram of all of the elementwise absolute differences is displayed in Supplementary Figure S1 in the Supplementary Material. The runtime of each replicate (for both the kgen algorithm and the naive method) and the maximum entrywise absolute difference between the Cholesky decompositions of each method are shown in Supplementary Table S1 of Appendix B in the Supplementary Material.
3.2. Experiment 2
In our second experiment, we consider a blockwise missingness pattern and vary the number of phenotypes. We fix the number of samples at
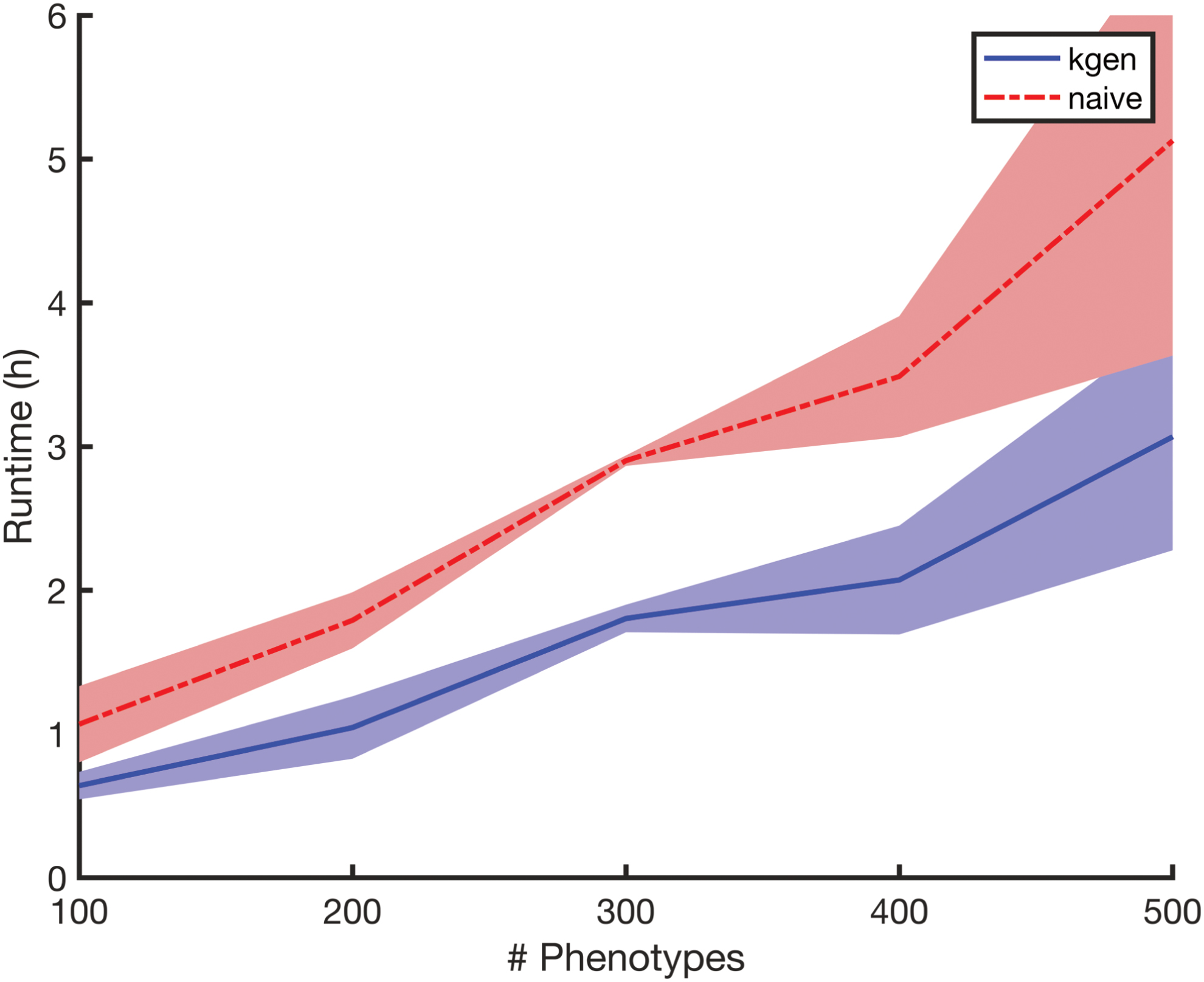
Runtimes for Experiment 2 versus number of phenotypes, displaying some linearity in d (varied in the set
Improvements shown in Figure 3 are only generally realized for
4. Results
In Figure 3, we see a between 1- and 6-hour improvement for sample sizes greater than 15,000 and missingness rates <0.1%, and for 100 phenotypes. In theory, and in Figure 4, we see an indication that these improvements are linear in the number of phenotypes. And so a study with 10,000 phenotypes under the right conditions may benefit from a 25-day reduction in compute on our hardware through the kgen algorithm.
Since Figure 3 indicates the difference in runtimes between kgen and the naive method, we also provide means and standard deviations for the runtime for both methods in Table 1. This table indicates that the kgen algorithm provides between 0% and 40% improvement in runtime for missingness at random rates of less than 0.1%. Figure 4 indicates around a 40% improvement for situations similar to The Wellcome Trust Case/Control Consortium (2007).
Detailed Results for Experiment 1
The “n” column indicates number of samples, “rate” column indicates the missingness rate (in basis points), “mean (kgen)” and “std (kgen),” “mean (naive)” and “std (naive)” column indicates means and standard deviations over five independent replicates for kgen and the naive method (in seconds) resp.
5. Conclusion
Multivariate GWAS are limited by computational resources. We have provided a new method to create and manipulate the genetic similarity matrices required for linear mixed models for multivariate GWAS. On our hardware, our method provides an improvement of around 40% under reasonable simulation settings. Software implementing our methods are released under an open source license.
Footnotes
Acknowledgments
We thank Kevin Sharp, Shufei Ge, Shijia Wang, and Winfield Chen for helpful discussion. We also thank Fred Popowich and the Big Data Hub at Simon Fraser University for help with computational resources.
Author Disclosure Statement
The author declares there are no conflicting financial interests.
Funding Information
This research was supported by NSERC grant numbers RGPIN/05484-2019 and DGECR/00118-2019.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.