
Abstract
Abstract
The regulatory genome controls genome activity throughout the life of an organism. This requires that complex information processing functions are encoded in, and operated by, the regulatory genome. Although much remains to be learned about how the regulatory genome works, we here discuss two cases where regulatory functions have been experimentally dissected in great detail and at the systems level, and formalized by computational logic models. Both examples derive from the sea urchin embryo, but assess two distinct organizational levels of genomic information processing. The first example shows how the regulatory system of a single gene, endo16, executes logic operations through individual transcription factor binding sites and cis-regulatory modules that control the expression of this gene. The second example shows information processing at the gene regulatory network (GRN) level. The GRN controlling development of the sea urchin endomesoderm has been experimentally explored at an almost complete level. A Boolean logic model of this GRN suggests that the modular logic functions encoded at the single-gene level show compositionality and suffice to account for integrated function at the network level. We discuss these examples both from a biological-experimental point of view and from a computer science-informational point of view, as both illuminate principles of how the regulatory genome works.
1. Introduction
“There exists today a very elaborate system of formal logic, and specifically, of logic as applied to mathematics. This is a discipline with many good sides, but also with certain serious weaknesses. …Everybody who has worked in formal logic will confirm that it is one of the technically most refractory parts of mathematics. The reason for this is that it deals with rigid, all-or-none concepts, and has very little contact with the continuous concept of the real or of complex number, that is, with mathematical analysis. Yet analysis is the technically most successful and best-elaborated part of mathematics. Thus formal logic is, by the nature of its approach, cut off from the best cultivated portions of mathematics, and forced onto the most difficult part of mathematical terrain, into combinatorics.”—John von Neumann
Mechanisms to annotate genomic sequences encoding RNAs and proteins are well established, but the term “regulatory genome” refers to parts of the genome that provide information not for the structure of molecules but for when and where molecules are produced within an organism (Davidson, 2006; Peter and Davidson, 2015). What will it take to annotate the regulatory genome? Which structural and functional definitions will be adequate to describe the regulatory genome? Typically, regulatory DNA encodes binding sites for transcription factors that in turn control gene expression. At the sequence level, however, it is so far not possible to distinguish regulatory from nonregulatory sequences, since transcription factor binding sites are small and occur throughout the genome in regulatory as well as nonregulatory DNA. Thus at the structural level, the rules by which regulatory DNA encodes gene expression patterns are not clearly understood, and the current definition of regulatory sequences relies on the observed function in gene regulation.
From a computational point of view, the function of the regulatory genome is to execute highly complex information processing functions at several levels of organization. At a basic conceptual level, the function of regulatory DNA is to control the expression of individual genes. Even at this level, regulatory systems associated with individual genes display a complex modular form, with clusters of transcription factor binding sites encoded in multiple cis-regulatory modules that all contribute to the correct gene expression output. With the discovery of gene regulatory networks (GRNs), it became clear, however, that the function of the regulatory genome goes beyond the control of individual genes. Thus, from a systems-level perspective, the regulatory genome also provides the information system for the development of the animal body plan. GRNs are networks of regulatory genes encoding transcription factors and signaling molecules, and of regulatory sequences encoding their interactions. Cis-regulatory sequences controlling the expression of transcription factors and signaling molecules affect not just the activity of single genes, but they also affect all other genes expressed downstream of these regulators. Regulatory sequences that control the expression of transcription factors contribute directly to the interpretation of the regulatory genome, since they determine the combination of expressed transcription factors, the regulatory state (Peter, 2017). At this level, the regulatory genome encodes information for genome activity in all different developmental and physiological contexts, throughout the life of an organism.
How are the different levels of informational organization encoded in the regulatory genome? Important insights into how the regulatory genome works have been generated in the sea urchin embryo by detailed system-level dissection of regulatory systems at both the single-gene and GRN levels. One of the first and best understood cis-regulatory control systems for an individual gene controls the expression of endo16, a gene expressed in the midgut of sea urchin embryos (Soltysik-Espanola et al., 1994; Yuh and Davidson, 1996). The expression of endo16 is controlled by several cis-regulatory modules, as is typical for any gene, and each module includes binding sites for several transcription factors. The impact of the individual modules, and even individual transcription factor binding sites within these modules, on the gene expression output of endo16 has been analyzed experimentally and shows a complex code for information processing even at the single-gene level. At the GRN level, one of the most complete experimental analyses of a network has also been conducted in the context of endomesoderm development in the sea urchin embryo (Davidson et al., 2002a; Oliveri et al., 2008; Croce and McClay, 2010; Peter and Davidson, 2010, 2011; Sethi et al., 2012; Materna et al., 2013; Cui et al., 2014). This GRN consists of ∼50 regulatory factors that control gene expression and the specification of several distinct endomesodermal cell fates during 30 hours of development.
Curiously, the computational function of the regulatory genome, at both the single-gene and GRN levels, has been made accessible through computational logic models. The observation that the function of the regulatory genome can be approximated by computational logic formulas, not unlike the logic gates used in computer science, indicates that formal logic approaches successfully capture the information processing functions of the regulatory genome at different scales. We discuss the insights that have been generated by experiments and computational models, and that illuminate the functional properties of the regulatory genome.
2. Control of a Single Gene: the endo16 Regulatory System in Experiment and Model
The cis-regulatory system controlling the expression of the sea urchin endo16 gene was one of the first to be experimentally dissected in great detail, and as of today is probably still one of the best understood regulatory control systems (Soltysik-Espanola et al., 1994; Yuh et al., 1994, 1996; Kirchhamer et al., 1996; Yuh and Davidson, 1996). The regulatory function of this sequence was first experimentally discovered, and then captured in an elegant computational model (Yuh et al., 1998). But to be able to appreciate this model, we will first revisit some of the experimental work that served as its foundation.
The sequence fragment that encodes sufficient information to recapitulate the expression pattern of endo16 during sea urchin endoderm development was identified by reporter assays (Yuh et al., 1994, 1996; Yuh and Davidson, 1996). A DNA fragment of 2300 bp just upstream of the transcription start site of endo16, when placed upstream of a reporter gene and injected into sea urchin embryos, drives expression at first in endodermal progenitors and in the midgut during later development. Similar experiments have been performed with many cis-regulatory sequences, demonstrating that regulatory DNA encodes information for particular gene expression patterns. In the case of endo16, the individual regulatory functions encoded within the cis-regulatory system have been carefully dissected. First, the 2300-bp fragment was separated into seven modules (modules A–G; Fig. 1A) by restriction enzymes. Each module contains clusters of binding sites that are recognized and bound by transcription factors, which regulate gene expression (Yuh et al., 1994). All modules together encode a total of >30 binding sites for 13 transcription factors. The seven modules were then tested for transcriptional activity, either alone or in various combinations (Yuh and Davidson, 1996). These experiments revealed that of the seven modules, three contributed to the activation of gene expression in the endoderm (modules A, B, and G) and four contributed to the repression of endo16 in the ectoderm (modules E, F) and skeletogenic mesoderm (modules C, D).
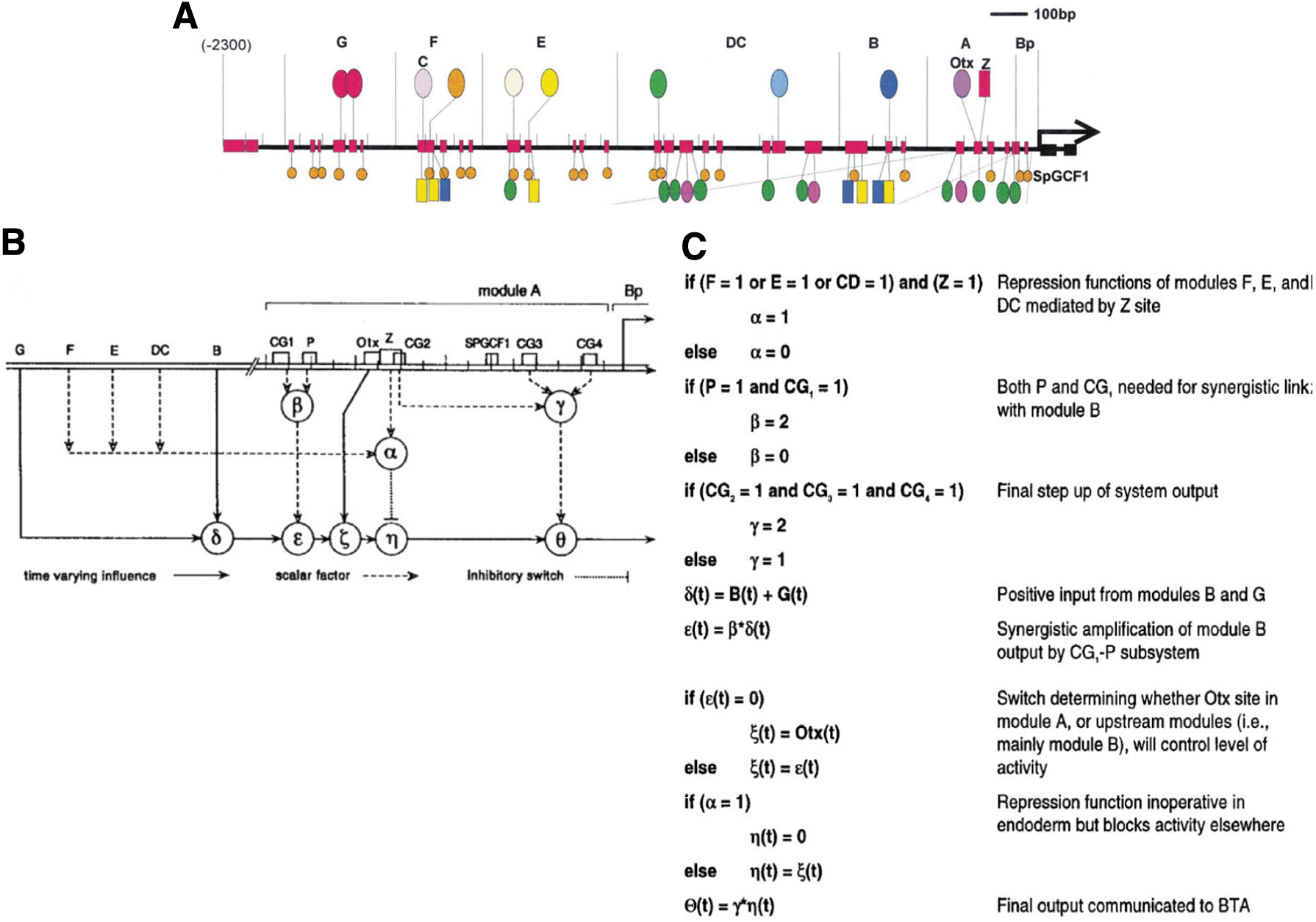
The endo16 cis-regulatory control system.
The key to understanding the regulatory logic of the endo16 cis-regulatory system is that neither the modules nor the transcription factor binding sites operate by simple linear addition or subtraction of individual regulatory functions. Instead, individual transcription factor inputs and individual modules operate by nonlinear combinatorial synergism (Yuh et al., 1998). Thus, the complete construct with all seven modules produces specific expression in the endoderm during development of the sea urchin embryo (Yuh and Davidson, 1996). Furthermore, modules A, B, and G are each capable of driving endoderm expression on their own, although they also show some expression in the ectoderm and skeletogenic mesoderm that is not observed with the full construct (Yuh and Davidson, 1996). Similarly, a construct with all three modules A, B, and G produces correct endodermal expression in addition to ectopic expression in ectoderm and skeletogenic mesoderm. Of the three activating modules, module G shows only weak activity, while module A drives endodermal expression predominantly during early development and module B drives expression predominantly during later development in the midgut. Adding either module E or module F to the construct with all three activating modules suppresses the ectopic expression in the ectoderm, meaning that both modules encode binding sites that are sufficient to repress expression of endo16 in the ectoderm. Modules C and D on the contrary are both required simultaneously to repress ectopic expression of the ABG construct in the skeletogenic mesoderm (Yuh and Davidson, 1996).
The interesting feature of this regulatory system that is perhaps common to many transcriptional control systems is that the modules, when present together, do not operate independently despite the fact that each module also shows activity when tested individually. Thus, a construct containing the two activating modules A and B shows more transcriptional activity, then the sum of the activity of the two constructs carrying modules A and B alone. The data indicate that the contribution of module A can be described as a linear amplification function by a factor of 4 of the output of module B (Yuh et al., 1996). Similarly, adding module A to a construct including both modules B and G will lead to an amplification of the output of construct BG by a factor of 3. Thus, module A, although active on its own, functions as a modulator of activity when placed in combination with other modules. Even more remarkable, module A is also required to mediate the repressive activity of modules CD, E, and F. Thus, when the repressive modules are combined with the GBA activator modules or with module A alone, they turn off gene expression in nonendodermal cell fates. However, if modules CD, E, and F are placed in combination with modules G and B, without module A, the repressive modules have no effect on gene expression. This means that module A contributes to both activation and repression of endo16 in response to alternative regulatory modules, in a Janus-like function, while activating gene expression by itself. So how does an activating module mediate the function of repressive modules?
The model for endo16 regulation shown in Figure 1B summarizes the logic operations that are executed by module A (Yuh et al., 1998). This model captures the function of individual transcription factor binding sites within module A that perform the computation of gene activity. Thus, binding sites CG1 and P are both required for the interaction of module A with module B. The mutation of either CG1 or P will lead to gene expression comparable with module A alone, even when module B is present, and to a reduction of gene activity by a factor of 2. When by itself, module A drives gene expression in the endoderm of early sea urchin embryos. This expression pattern is mediated by the binding site for Otx, and Otx is absolutely required for endodermal expression. Without a functional Otx binding site, the endodermal activity of module A is abolished (Yuh et al., 1998). However, even though mutation of the Otx binding site abolishes module A function in the early endoderm, it does not interfere with the ability of module A to interact with module B through binding sites CG1 and P. Furthermore, module A interacts with modules F, E, and DC through binding site Z. The interaction of module A with the basal transcription apparatus (BTA) is mediated by binding sites CG2, CG3, and CG4, and mutation of these sites leads to a reduction in gene expression by a factor of 2. Thus, interactions between modules A and B and between module A and the BTA are equally contributing to the fourfold increase of module B activity in the presence of module A. If we keep in mind that each site is just a few nucleotides long, then this entire complex operation is encoded in just a few short sequence elements with no particular apparent organization other than being part of module A.
The computation performed by module A in the control of endo16 expression can be approximated by the logic model shown in Figure 1C (Yuh et al., 1998). Interestingly, this computational model is a hybrid model including both discrete logic functions and the response to continuous inputs. The overall gene output function in this model is shown as a continuous function in time that is modulated by discrete repression or amplification functions. Each term represented by Greek letters in Figure 1B and C captures the regulatory impact of either transcription factor binding sites or cis-regulatory modules based on experimental observations. For some inputs, this impact is modeled as strictly Boolean, such as for the repressor modules F, E, and DC, where presence of any one of the repressing transcription factors dominantly turns off gene expression (e.g., if the integrated repressor function α = 1, then η(t) = 0, and thus the final output Θ(t) = γ * η(t) = 0). In a Boolean logic statement, this would correspond to a dominant NOT logic function. On the contrary, for other inputs, the impact is represented by an amplifier function, where presence of an activating transcription factor amplifies the gene output by a factor of 2 (e.g., if P = 1 AND CG = 1, b = 2, else b = 0). A few inputs are represented in this model as time-dependent continuous regulators determining the kinetics of the gene output.
There are many lessons to be learned from the work on the endo16 regulatory system. One is that even a relatively simple expression pattern requires complex computation of regulatory inputs. These regulatory inputs occupy their respective binding sites within regulatory DNA wherever they are expressed in the embryo, but whether this interaction leads to a gene expression output depends on the computation of the overall output based on the information encoded in the proximal cis-regulatory module A. Module A integrates the response to all other modules and the function of regulatory inputs binding to these modules. The developmental functions of this regulatory system, that is, activation in endoderm or repression in ectoderm, are mediated by separate DNA sequence modules, which makes it necessary to determine the final output through a proximal element responsive to all other modules. In addition, the endo16 model suggests a more profound truth, which is that the function of cis-regulatory sequences can be thought of in the context of a logic framework, each module contributing a unique function.
This function is mediated by transcription factors that bind to regulatory modules, and can be described by a combination of discrete or continuous functions. In the endo16 case, a few binding sites were identified, which determined the dynamic change in expression levels while the repressive modules were better approximated as Boolean ON/OFF switches. But regardless of the qualitative contribution of each input, the overall gene expression output is computed by the integration of individual regulatory functions according to strict logic rules. This idea was further explored by Istrail and Davidson (2005). This work found that by comparing the function of many different cis-regulatory systems, several logic operations could be defined, which were commonly executed by regulatory sequences. Thus, the regulatory systems controlling expression of single genes can be described as a repertoire of logic gates that is valid and applied across organisms, and a fundamental feature of the regulatory genome that is also used in the following to describe higher level GRN functions.
3. Linking Regulatory Systems: the Operation of Regulatory Circuits
The endo16 example shows a view of the regulatory genome that we are perhaps most familiar with, which is the function of cis-regulatory systems to control gene expression in response to transcription factor inputs. But the information contributed by the regulatory genome goes beyond the regulation of single genes in response to a regulatory state. The regulatory genome is also responsible for generating the regulatory states, thereby controlling the activity of the genome. The information for the control of genome activity is stored in the genome in the form of GRNs (Peter and Davidson, 2015). GRNs consist of genes encoding regulatory factors and of cis-regulatory sequences controlling gene expression. GRNs control the expression of transcription factors and signaling molecules that in turn regulate the expression of all other genes.
Interestingly, the regulatory systems controlling expression of transcriptional regulators are in principle no different from the regulatory systems controlling expression of any other gene, although perhaps slightly more complicated in design. However, what is substantially different is that cis-regulatory systems controlling expression of transcriptional regulators are connected with one another through regulatory interactions, whereby the transcription factor expressed as the output of a regulatory gene will serve as an input into other cis-regulatory systems. As a result, the regulatory circuits that are formed by multiple regulatory genes and their regulatory interactions have properties that go beyond regulating the expression of individual genes in response to transcription factor inputs. By connecting multiple regulatory systems, these circuits are able to execute more complex multigene logic functions.
An example of a small regulatory circuit is shown in Figure 2A. Here, the three genes gcm, gatae, and six1/2 are connected by a positive feedback circuit that is active in the sea urchin mesoderm (Ransick and Davidson, 2006, 2012; Peter and Davidson, 2017). In this circuit, Gcm activates the expression of gatae, and Gatae activates the expression of six1/2. The two regulatory feedbacks are provided by Gcm, activating its own expression, and by Six1/2 activating gcm expression. The cis-regulatory system of gcm encodes the response to both Gcm and Six1/2, and both transcription factors are required to ensure late expression of gcm (Ransick and Davidson, 2012). The initial activation of this positive feedback circuit comes from Delta/Notch signaling, which activates the expression of gcm (Ransick and Davidson, 2006; Croce and McClay, 2010). In the presence of Delta/Notch signaling, gcm expression is activated despite the absence of Gcm and Six1/2. And vice versa, Delta/Notch signaling is only present during early development and then turns off. At this point, Gcm and Six1/2 regulate gcm expression even in the absence of Delta/Notch signaling (Ransick and Davidson, 2012). Thus, Delta/Notch operates in OR logic to the other two inputs, while Gcm and Six1/2 regulate gcm expression in AND logic (Fig. 2A). This regulatory logic is reflected in the cis-regulatory system of gcm. The initial input, Delta/Notch signaling, activates a cis-regulatory module (CRM) that is independent of the module responding to Gcm and Six1/2. The two CRMs function independently, and therefore constitute an OR logic gate, while the two inputs regulating the second CRM have to be both present to activate the AND logic gate.
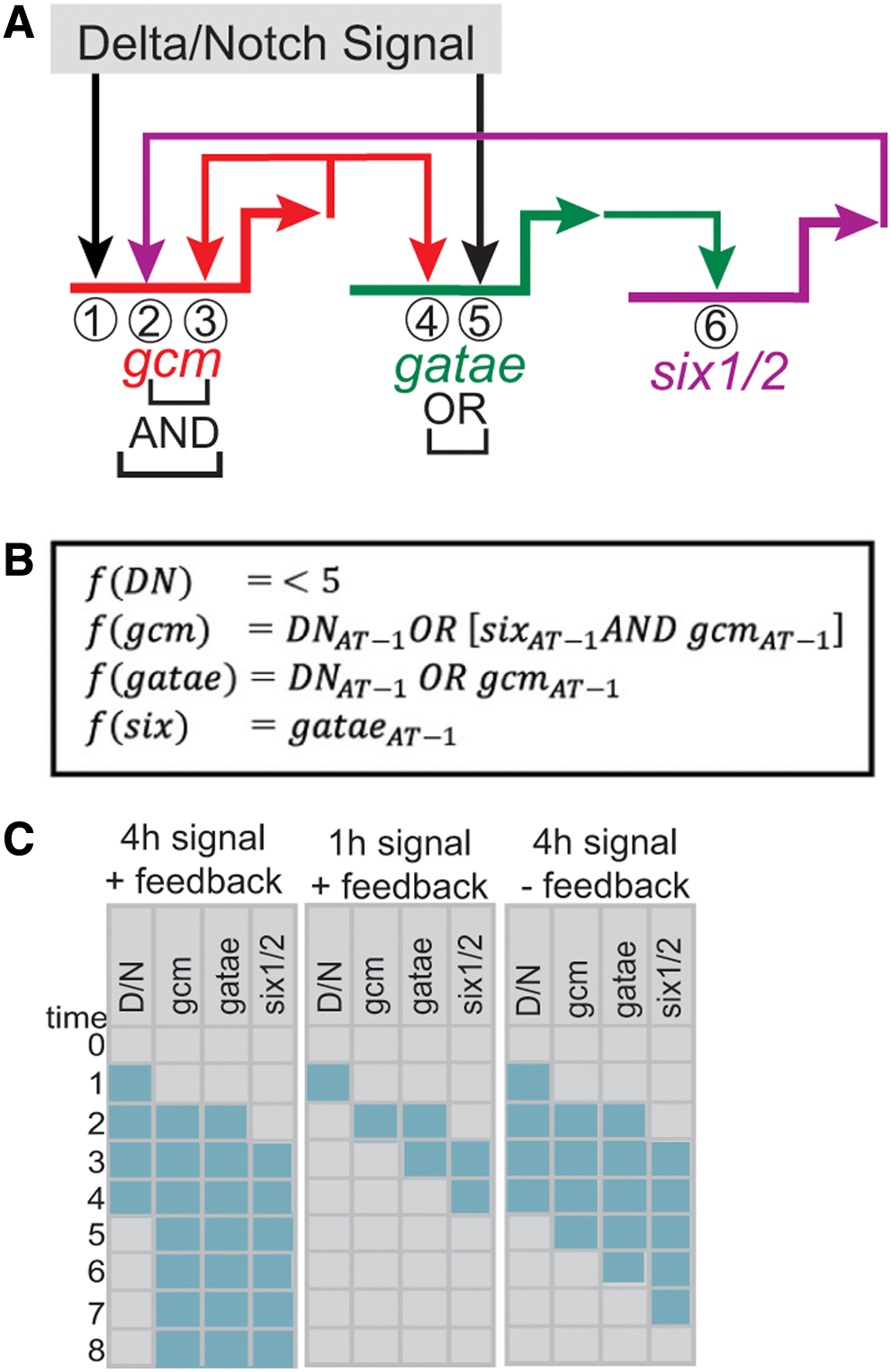
Structure and function of a positive feedback circuit.
The function of this regulatory circuit can be captured in a Boolean logic model (Fig. 2). In this model, we assume that a gene is either expressed (1) or not expressed (0), and that expression of the gene will lead to the production of functional levels of the corresponding transcription factor. Furthermore, we take into account that there is a time delay between expression of a regulatory gene and activation of its target gene, which is defined by the time it takes to produce sufficient amounts of transcription factor product to activate target gene expression (Bolouri and Davidson, 2003). If we assume that the delay time in this example is 1 hour, and we assume the regulatory logic for the three genes as given in Figure 2B, then all three regulatory genes are expressed within 2 hours after turning on the initial input Delta/Notch signaling (Fig. 2C). Moreover, the positive feedback into gcm is active after 3 hours (three regulatory steps at 1 hour each). If Delta/Notch signaling is turned off after 4 hours, all three genes remain being expressed because of the operation of the positive feedback circuit. However, if the Delta/Notch input lasts only for 1 hour, this does not provide enough time to activate the positive feedback circuit, and the three regulatory genes are only expressed transiently for as long as their inputs are present (Fig. 2C). Similarly, if the positive feedback circuit in this model is removed, expression of the three regulatory genes depends exclusively on the Delta/Notch signaling input, and gene expression turns off once the input is no longer available (Fig. 2C).
This example shows how the regulatory inputs controlling expression of gcm do not just operate in isolation but are part of a regulatory circuit with a function beyond the control of gcm expression. Activating gene expression is a function of Delta/Notch signaling, but this signal lasts only for a few hours in the sea urchin embryo, and the positive feedback circuit is required for continued gene expression. The function of this positive feedback circuit is not to turn on expression of the three regulatory genes, but to maintain their expression once the initial input is no longer available. Similar positive feedback circuit configurations have been discovered in many GRNs that control very different developmental processes (Narula et al., 2010; Peter and Davidson, 2015). Very often, positive feedback circuits occur downstream of transient developmental signaling inputs, implying that they function in a way similar to the example discussed here. Since in each developmental context these positive feedback circuits are composed of cell-fate-specific sets of transcription factors, the similarity in circuit function must be caused by the similarity in regulatory circuit architecture and not because of the specific molecular properties of the transcription factors involved. Since the architecture of regulatory circuits and GRNs is encoded in the regulatory genome, this means that important developmental functions are encoded in the regulatory genome in addition to protein coding sequences. We will now turn to the function of the regulatory genome at the GRN level, which is responsible for the control of entire developmental processes.
4. Regulatory Logic at the Level of Gene Regulatory Networks: the Endomesoderm Gene Regulatory Network
GRNs have been experimentally studied in many developmental contexts (Peter and Davidson, 2015). One of the most extensively characterized GRNs controls endomesoderm development in pregastrular sea urchin embryos (Davidson et al., 2002a, 2002b; Oliveri et al., 2008; Peter and Davidson, 2010, 2011; Materna et al., 2013). About 50 transcription factors and signaling ligands/receptors are involved in the specification of endoderm and mesoderm during the first 30 hours of sea urchin development. These regulatory factors have been identified based on a systematic analysis to be expressed in either endodermal or mesodermal cell fates. The regulatory interactions between these transcription factors were analyzed by systematically perturbing the expression of each transcriptional regulator and by monitoring the effect on the expression of all other regulatory genes in the system. The results of gene expression analyses and perturbation experiments were used to reconstruct the GRN that connects these regulators into a functional program for endomesoderm development.
The function of the endomesoderm GRN is to determine that skeletogenic cells will form at the vegetal pole in every embryo of this species, and that these cells will be surrounded by other mesodermal cell fates and the endoderm that gives rise to the gut. The developmental organization of these cell fates within the embryo is an important function of the GRN. Thus, the GRN ensures that the set of transcription factors associated with each endodermal and mesodermal cell fate are expressed in the correct position within the embryo. The GRN also controls which downstream differentiation genes are expressed in each cell fate. For example, proteins involved in synthesizing a skeleton are expressed in skeletogenic cells (Rafiq et al., 2014), whereas proteins with digestive enzymatic functions are expressed in the gut. The purpose of experimentally dissecting GRNs is therefore to obtain a causal understanding on how the genome controls the developmental organization of an embryo (Peter and Davidson, 2015).
The endomesoderm GRN model shown in Figure 3A shows how the expression of transcription factors is regulated in each endomesodermal cell fate (Davidson et al., 2002a; Longabaugh et al., 2005; Oliveri et al., 2008; Peter and Davidson, 2011; Longabaugh, 2012; Materna et al., 2013). Each of the colored boxes represents a distinct cell fate, and the genes shown in each box together compose the regulatory state of the corresponding fate. For each gene, the regulatory inputs that regulate its expression are shown as linkages into the associated cis-regulatory system, while the regulatory functions of the transcription factor outputs are shown as linkages into the regulatory systems of target genes. In a GRN, individual cis-regulatory systems are therefore connected through the transcription factors with which they are associated. We have seen in the example of endo16 how individual cis-regulatory binding sites and modules are computed to control expression of a single gene. But if we extrapolate this to the level of a GRN, how does the logic that is encoded in the regulatory systems of different genes operate when connected within a network? Do these systems operate independently, or is there an intrinsic logic to combining several genes into a network circuit? Is there a degree of freedom to connect cis-regulatory systems, or are there specific rules for the compositionality when combining the logic of cis-regulatory sequences?
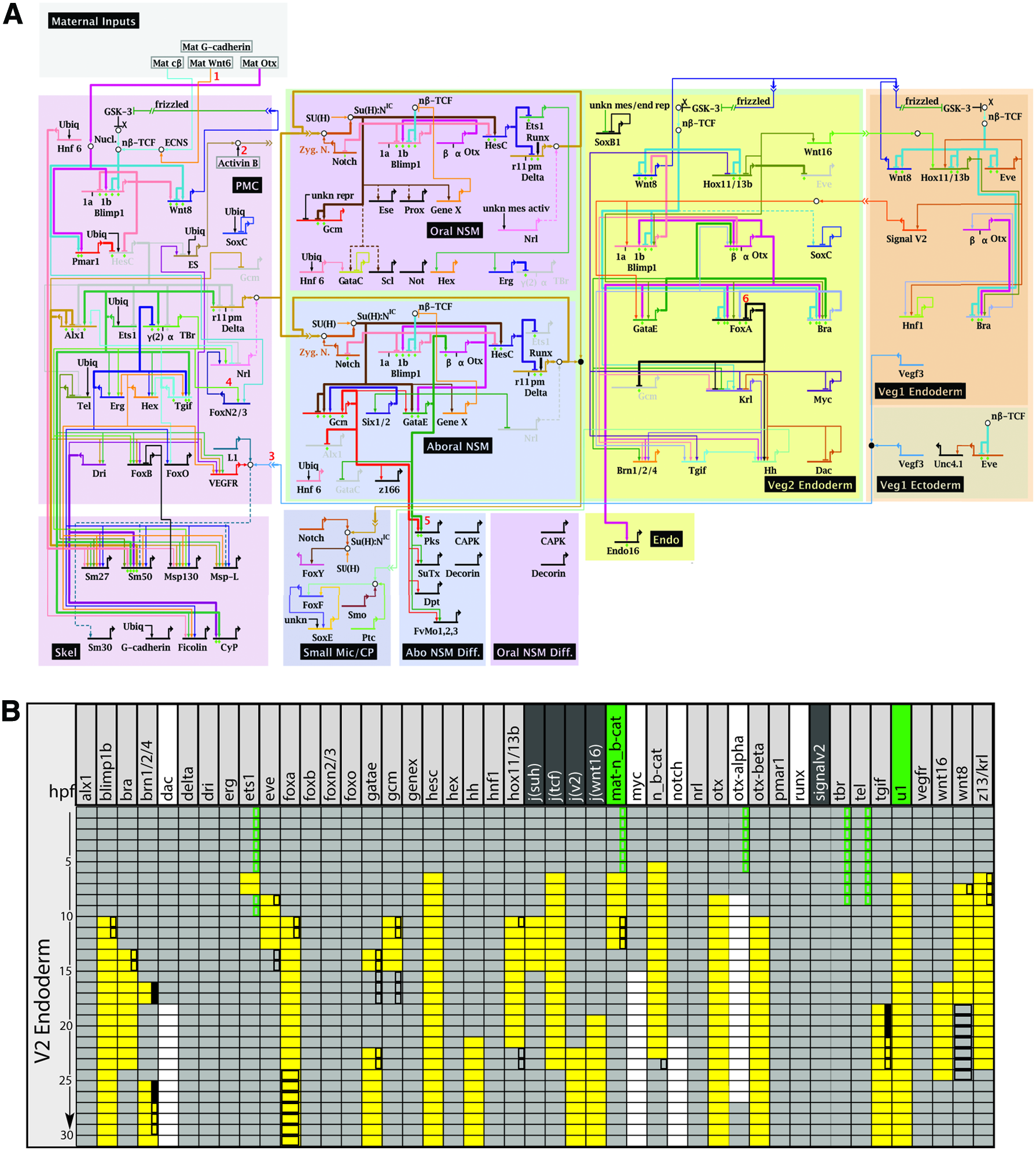
Regulatory logic of the endomesoderm GRN.
A computational model of the endomesoderm GRN provides perhaps some answers to these questions. Thus, in an attempt to capture the dynamic behavior of a system of interconnecting regulatory genes, a Boolean logic model was mathematically defined based on the experimental observation of the endomesoderm GRN (Peter et al., 2012). The purpose of this computational model was to test whether the GRN that was reconstructed as shown in the topological model in Figure 3A would suffice to explain the developmental control of gene expression and the specification of different endomesodermal cell fates in the sea urchin embryo. The basic components of this model are (1) the regulatory logic controlling each gene in the GRN, identified based on the effect of experimental perturbation of its transcription factor inputs; (2) a temporal delay function associated with each regulatory interaction; and (3) maternal inputs that initiate the activation of the zygotic developmental program.
The regulatory logic of each gene in this model was captured by Boolean logic statements that were formulated based on the regulatory inputs controlling expression of each gene and based on the logic operation computed by the cis-regulatory system (Peter et al., 2012). For instance, if perturbation experiments indicate that two transcription factors A and B regulate the expression of gene C, and that perturbation of either A or B leads to a strong reduction of expression of gene C, this would indicate that the presence of both A and B is required for activation of gene expression. The Boolean logic statement that captures the regulatory logic for gene C would therefore be C = A AND B. The time function in this model derives from the time it takes from starting transcription of an upstream regulatory gene to producing levels of transcription factor sufficient to regulate target gene expression. This time was calculated based on RNA and protein synthesis rates to be ∼3 hours in sea urchins developing at 15C (Bolouri and Davidson, 2003). Surprisingly, using a temporal step function of 3 hours for almost all regulatory interactions was a correct assumption to reproduce the temporal and spatial expression of almost all genes in the system. And finally, the maternal inputs are transcription factors that are present in the egg and in the model these factors are turned ON by default for the first few hours of development.
This Boolean computational model was used to compute expression or absence of expression for each gene in the endomesoderm GRN based on a Boolean logic statement and based on the presence or absence of its inputs. Except for the maternal factors, these inputs are present only if the corresponding regulatory genes are computed as expressed based on their own associated logic statements. The computed expression for all regulatory genes in the GRN model is shown in Figure 3B for the endoderm domain during 30 hours of development. A comparison of the gene expression patterns computed by the Boolean logic GRN and the gene expression experimentally observed in the sea urchin embryo demonstrates that the information captured in the GRN is sufficiently complete to recapitulate the embryonic gene expression patterns. Thus, this system behaves as an automaton, where early maternal inputs initialize a program that is self-sufficient to operate without any further inputs from outside the system. This analysis shows that it is possible to obtain a complete explanation for developmental gene activity based on the experimental analysis of a GRN. In addition, these results suggest that the cis-regulatory systems at each network node can be combined without further instructions to reproduce the correct system-level output of an entire GRN both in terms of gene expression and in terms of differential cell fate specification.
The observation that a system of interconnected cis-regulatory modules is sufficient to capture an entire developmental program provides a powerful confirmation of the information processing function of the regulatory genome. Of course, although the examples here derive from the sea urchin embryo, the computation of logic functions by regulatory sequences must represent a general property of the regulatory genome that applies to sea urchins as well as to any other organism, in development and beyond. It demonstrates that the regulatory logic controlling individual genes can be viewed as a system of logic gates that compute developmental gene expression. Returning to von Neumann's quote, although mathematical analysis is applicable to many areas of biology, the system-level information processing functions of the regulatory genome might be better approximated by formal logic. Thus, the logic encoded in regulatory DNA provides a unifying concept that defines the function of the regulatory genome, from the modular regulatory systems controlling individual genes to the networks controlling genome activity throughout biological processes.
Footnotes
ACKNOWLEDGMENTS
In memory of Eric Davidson, who 50 years ago envisioned the importance of gene regulation and who together with Roy Britten built a theoretical foundation (“Gene regulation for higher cells: a theory” Science, 1969), many years before the regulatory genome became accessible to experimental exploration in animals. We are grateful to Deanna Thomas for her help in generating the figures. This work was supported by National Institutes of Health Grant HD 037105 (to I.S.P.).
Author Disclosure Statement
The authors declare there are no competing financial interests.