Abstract
Computational protein design (CPD) algorithms that compute binding affinity,
Ka
, search for sequences with an energetically favorable free energy of binding. Recent work shows that three principles improve the biological accuracy of CPD: ensemble-based design, continuous flexibility of backbone and side-chain conformations, and provable guarantees of accuracy with respect to the input. However, previous methods that use all three design principles are single-sequence (SS) algorithms, which are very costly: linear in the number of sequences and thus exponential in the number of simultaneously mutable residues. To address this computational challenge, we introduce BBK*, a new CPD algorithm whose key innovation is the multisequence (MS) bound: BBK* efficiently computes a single provable upper bound to approximate
Ka
for a combinatorial number of sequences, and avoids SS computation for all provably suboptimal sequences. Thus, to our knowledge, BBK* is the first provable, ensemble-based CPD algorithm to run in time sublinear in the number of sequences. Computational experiments on 204 protein design problems show that BBK* finds the tightest binding sequences while approximating
Ka
for up to 105-fold fewer sequences than the previous state-of-the-art algorithms, which require exhaustive enumeration of sequences. Furthermore, for 51 protein–ligand design problems, BBK* provably approximates
Ka
up to 1982-fold faster than the previous state-of-the-art iMinDEE/
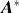 /
/
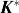 algorithm. Therefore, BBK* not only accelerates protein designs that are possible with previous provable algorithms, but also efficiently performs designs that are too large for previous methods.
algorithm. Therefore, BBK* not only accelerates protein designs that are possible with previous provable algorithms, but also efficiently performs designs that are too large for previous methods.
