
Abstract
Abstract
1. Introduction
Due to these complications, developing a probabilistic model for promoter sequences and describing the evolutionary dynamics of regulatory regions remains a challenge. However, there is growing body of experimental evidence that promoter regions are highly dynamic and that significant changes in gene regulation can occur on a microevolutionary time scale. For example, using ChIP-chip technology, Odom et al. (2007) inferred the binding sites of four tissue-specific TFs in human and mouse liver cells and found that despite the conserved function and motif of these TFs, 41–89% of the binding events are species specific. Taylor et al. (2006) investigated the evolutionary trends of mammalian promoters using large sets of experimentally supported transcription start sites and concluded that the evolution within mammalian promoters has been relatively rapid within approximately the last 25 Myrs.
In order to give a probabilistic explanation for the speed of cis-regulatory evolution, we address the following two questions: (1) How long does it take until a given TF binding site appears at random due to the evolutionary process of single nucleotide mutations? (2) How does the composition of a TF binding site affect this waiting time?
Stone and Wray (2001) tried to answer the first question by simulating the evolution of a promoter region of length 2,000 bp assuming a mutation rate of 10−9 per nucleotide per generation. After having estimated the waiting time for a 6-mer to emerge in a given promoter sequence, they divide this number by 106 · 2 (=effective population size times two DNA strands) yielding that on average it takes 6,000 years until a 6-mer appears in a promoter sequence of at least one individual in a human population. Their approach of simply dividing by the number of individuals has been critized by MacArthur and Brookfield (2004) and by Durrett and Schmidt (2007), especially because with this, Stone and Wray (2001) implicitly assume that the DNA sequences in a population evolve independently from each other. But indeed, two randomly chosen individuals differ only in 0.1% of their DNA as stated in Durrett and Schmidt (2007). Durrett and Schmidt (2007) tackeld the problem by using a proper population genetics model (the Moran model) and (Poisson) approximated the expected waiting time until a word of fixed length 6 or 8 appears in a promoter region of length 1000 bp in at least one individual in a population of effective size 104. Assuming a mutation rate of 10−8, Durrett and Schmidt (2007) computed that the expected waiting time for words of length 6 is 100,000 years and 375,000 years for words of length 8 given that there is a 7 out of 8 letter match in the population consensus sequence.
These results are helpful for getting a general idea of how fast TF binding sites can emerge - at least for binding sites of length 6 and 8. However, they rely on the assumption that once a TF binding site is created in one individual, it will confer a substantial benefit and hence, will spread rapidly through the population. But according to population genetics theory (Ewens, 2004) this event only occurs with a small probability: Let us assume for simplicity, that there are only two different individuals A and B in a population of size N where A symbolizes the individual with the new TF binding site appearing only once and B represents the remaining N − 1 individuals without the new TF binding. The fixation probability of A is then given by
where r is the relative fitness of individual A, i.e., the average number of surviving progeny of A compared to B after one generation; see e.g. section 6.3 in Nowak (2006). Setting N = 104 and assuming a relative fitness of 2 (100% selective advantage), the probability that the new TF binding site will get fixed in a population is only 0.5. Even when assuming a very high and unrealistic relative fitness of 10 the fixation probability is just 0.9. Thus, their assumption that a TF binding site once created will spread throughout the population is hard to justify.
In this work, we phrase the question differently. Instead of computing the waiting time until a given TF binding site emerges in a promoter sequence in at least one individual in a population, we are interested in determining the expected waiting time until a given TF binding site gets fixed in a species (assuming that fixation only occurs at the nucleotide/dinucleotide level) - either in one given promoter sequence or in at least one of several promoters, for example, in any or all of the human promoters. As mentioned above, two randomly chosen individuals differ only in 0.1% of their DNA. Therefore, it is reasonable to refer, for example, to the ”human genome.” With this in mind, in our model a DNA sequence should not be interpreted as a sequence of a single individual of a population but as a representative sequence of the considered species. Hence, waiting times relate to appearance in the species instead of appearance in one single individual. Starting with a muliple species alignment for the three species Homo sapiens, Pan troglodytes and Macaca mulatta, we can estimate the evolutionary substitution rates (=fixed mutation rates) for every nucleotide using the Maximum likelihood based tool developed by Arndt and Hwa (2005). As a consequence, for every k-mer, k ranging from 5 to 10, we can (almost) exactly compute its expected waiting time to appear in a species' promoter of a given length in dependence of its composition.
Since the CpG methylation-deamination process (CG → TG and CG → CA) is the predominant evolutionary substitution process, as a second step, we also incorporate neighbor dependent substitution rates into our model. For example, Wang et al. (1998) pointed out that single-nucleotide polymorphisms occur about 10 times more often at CpG dinucleotides than at other dinucleotides in the human DNA. Our approach of calculating waiting times in dependency of the promoter's and TF binding site's composition sheds new light on the process of TF binding site emergence and therefore, extends the previous knowledge about the dynamics of promoter sequence evolution.
As a last step, we compare k-mers which are predicted to appear rapidly according to our model with existing TF binding sites from the database JASPAR (Portales-Casamar et al., 2010). We show that k-mers with short waiting times are used more frequently as TF binding sites than those with long waiting times.
This article is organized as follows: in Section 2.1 we introduce two probabilistic models - an i.i.d. model (model M0) and a model taking the neighbor dependencies of nucleotides into account (model M1). Section 2.2 is devoted to the computation of the expected waiting time under our given models, and in Section 2.3, we explain how one can estimate the model parameters. Utilizing these parameter estimations, in Section 3 we provide the expected waiting times for k-mers, k ranging from 5 to 10, for model M0 and then present the more interesting results for model M1 in detail. Additionally, we relate these k-mers to existing TF binding sites from the database JASPAR (Portales-Casamar et al., 2010). Section 4 discusses the results and explains the impact of our findings.
2. Methods
2.1. The probabilistic model
In order to formalize the problem, one has to model two components: the initial promoter sequence
2.1.1. Modelling the initial promoter sequence
Let
In model M0, the probability of observing a sequence
where
In model M1, when
2.1.2. Modelling the time evolution of the promoter sequence
The promoter sequence evolution
In model M0, one only considers the independently evolving nucleotides (12 substitution rates). Since nucleotide substitutions on one strand of the DNA go along with nucleotide substitutions on the complementary strand in order to guarantee correct Watson – Crick base pairing, the number of free parameters is 6 (A → T/T → A, C → G/G → C, A → C/T → G, C → A/G → T, A → G/T → C, G → A/C → T). If Q denotes the 4 × 4 rate matrix, the matrix
In model M1, we incorporate one particular neighbor dependent substitution process: the CpG methylation-deamination process. That is, apart from the 12 substitution rates mentioned above (given by 6 parameters), one also has 2 other rates (CG → TG and CG → CA) which are assumed to be the same. This results into looking at trinucleotides (nucleotide plus left and right neighbor) whose dynamics are governed by a 64 × 64 rate matrix Q(3); for details see Duret and Arndt (2008). For t ≥ 0, the matrix
is given by
In model M1, referring to Arndt and Hwa (2005), Duret and Arndt (2008) the probability of sequence
2.2. The expected waiting time
Let
be a TF binding site of length k. We want to answer the question: provided that the binding site is not present in the initial sequence
given that b is not present in the initial sequence
Especially, one obtains
Let
In both models M0 and M1, the probability pl indeed only depends on ℓ since we have assumed stationarity. It is the probability of b appearing at ℓ given positions in generation 1 under the condition that it was not present in the generation before. Exact computation of pℓ becomes infeasible if the binding site can overlap with itself since computing the probability that b appears ℓ times in generation 1 given that it did not occur in generation 0 requires inspection of many possiblities: b can occur ℓ times with no overlap in generation 1, b can occur ℓ times in one big overlapping clump, b can occur m times in one clump and ℓ − m times in another clump etc. But, of course, for large ℓ the probability pℓ becomes very small, so q is dominated by the first summands. Since overlaps can be neglected for small ℓ, for ease of exposition, we neglect the possibility of b occuring self-overlapping. Thus, we use the following approximation
Due to the assumption that b cannot appear self-overlapping, b can occur at most
Now one can easily compute pl separately for the models M0 and M1 (notation: q0 and q1).
2.2.1. The expected waiting time in model M0
If |i − j| ≥ k for all
Hence, we can summarize the result:
Theorem 1
(Expected waiting time in model M0). Under the model M0 described in Section 2.1, the expected waiting time until a binding site b of length k occurs in a promoter sequence of length n is approximately given by
where
2.2.2. The expected waiting time in model M1
For model M1, we make the simplifying assumption that if b appears two or more times at once in generation 1, these occurrences of b are so far apart from each other that one can consider them as independent. Therefore, applying (3) and (7), one gets
Thus, one obtains:
Theorem 2
(Expected waiting time in model M1). Under the model M1 described in Section 2.1, the expected waiting time until a binding site b of length k occurs in a promoter sequence of length n is approximately given by
where
2.2.3. Waiting times for several promoters
In order to get an understanding of how fast regulatory regions can evolve, we do not only want to answer the question how long one has to wait until a new TF binding site appears in one particular promoter of a given size but until it emerges in at least one of several promoters, e.g. in one of all the human promoters. This might also induce a change in gene regulation which, in principle, could be crucial for the evolution of the whole species. Let
Analogously to Supplementary Material S1, one obtains that Tm has approximately a geometric distribution with parameter
Since the m promoters are independent and identically distributed, this yields
where q is given by q0 (see (16)) in model M0 and by q1 (see (19)) in model M1. Hence, for model M1,
2.3. Parameter estimation
For model M0, one has to estimate the parameters ν(a) and pa,b(1) (see (17)) and for model M1, one has to determine the parameters ν(a), πa,b and
The parameters ν(a),
where
To estimate the substitution probabilities pa,b(1) and
We downloaded multiple alignments to human DNA regions (hg18) of length 1 kb upstream of annotated transcription start sites for RefSeq genes with annotated 5′ UTRs from the USCS download server. For the estimation of ν(a) and πa,b,
Assuming a generation time of y = 20 and a speciation time between man and chimp of s = 4 Myrs (Hobolth et al. (2007)), one finally obtains estimations for pa,b(1) and
3. Results
Due to the fact that the CpG methylation-deamination process plays an important role which should not be neglected, model M1 is more general and realistic than model M0. Thus, we will concentrate on the expected waiting times in model M1. The waiting times in model M0 will be only stated briefly and will be used to pinpoint the characteristics of model M1 in comparison to the more simplistic model M0.
3.1. Results for model M0
Applying Theorem 1 and using the estimations for the parameters ν(a) and pa,b(1),
Expected waiting times (generations) for the ten fastest and the ten slowest emerging k-mers, 5 ≤ k ≤ 10, where the promoter length is n = 1000 bp.
In model M0, the expected emergence time of a k-mer only depends on the number of each nucleotide in the k-mer and not on the order of the nucleotides, e.g., the 5-mer CCCCG has the same waiting time as CCCGC, CCGCC and so on. In Table 1, it can be seen that for every k, 5 ≤ k ≤ 10, the k-mer only composed of Cs is the fastest emerging one (followed by CG-rich k-mers) while the k-mer only composed of As is the slowest one (preceded by AT-rich k-mers). For example, CCCCC is the fastest emerging 5-mer with an expected waiting time of 6,303,945 generations (=126 Myrs) to appear in a promoter of length 1 kb while AAAAA is the slowest emerging 5-mer with 7,653,814 generations (=153 Myrs). For 10-mers, the average expected waiting time is 72 billion years, the minimal and maximal waiting times are 51 billion (CCCCCCCCCC) and 99 billion years (AAAAAAAAAA).
3.2. Results for model M1
3.2.1. Waiting times for one promoter
Plugging in the estimators for the parameters ν(a), πa,b and
Expected waiting times (generations) for the ten fastest and the ten slowest emerging k-mers, 5 ≤ k ≤ 10, where the promoter length is n = 1000 bp.
First of all, when looking at the minimal, maximal and average waiting time for k-mers, one realizes that the expected waiting times increase exponentially with k, see Figure 1. Second, one observes the tendency that k-mers containing TG or CA, i.e. CpG methylation-deamination products (CG → TG and CG → CA), in combination with a high C-content are the fastest appearing TF binding sites. In contrast, k-mers containing the dinucleotides CG or TA are characterized by very long waiting times. Thus, taking this neighbor-dependent process into account changes the composition of the top ranking k-mers.
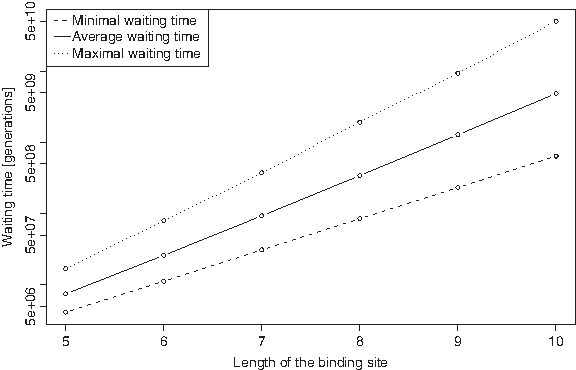
Minimal, maximal, and average waiting times in model M1 (log scale). These waiting times (generations) are computed based on the results in Table 2.
Let us focus on 5- and 10-mers. CCCTG is the fastest appearing 5-mer with an expected waiting time of 4,132,368 generations (≈83 Myrs) while ATATA is the slowest emerging 5-mer with a number of 16,906,864 generations (≈338 Myrs). This shows that incorporating the CpG methylation-deamination process into our model increases the variance in waiting times: on the one hand, the minimal waiting time for 5-mers in model M1 is much shorter than in model M0 (83 versus 126 Myrs) but, on the other hand, the maximal waiting time is much larger than in model M0 (338 versus 153 Myrs).
This variance is so high that for some (k + 1)-mers the waiting times are shorter than for some other k-mers. For example, the waiting time for the 5-mer ATATA is 338 Myrs and for the 6-mer CCCCTG it is only 224 Myrs. This confirms our approach of not just looking at waiting times in dependency of the length k of the TF binding site but also taking its composition into account. The effect gets even stronger for large k as can be seen in Figure 2: for small k, 5 ≤ k ≤ 7, the waiting times for k- and (k + 1)-mers are separated more clearly, for bigger k, 8 ≤ k ≤ 10, they overlap considerably.
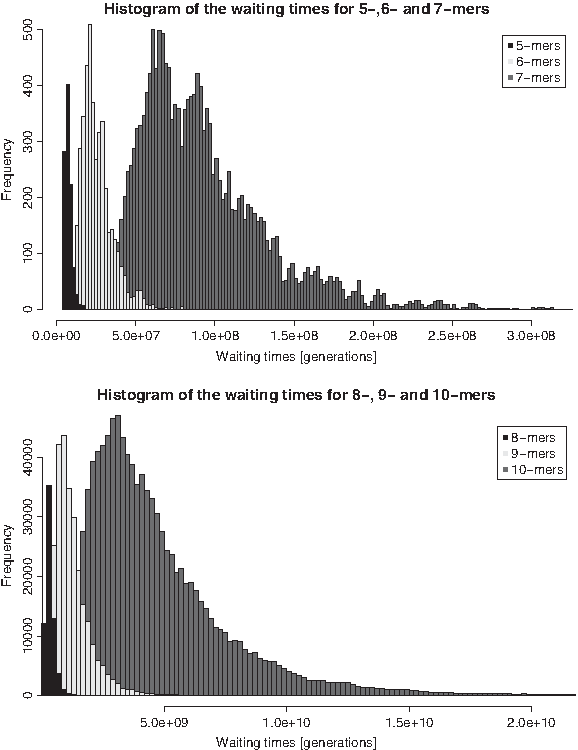
Histograms of the waiting times in model M1. The expected waiting times (generations) are taken from Table 2.
3.2.2. Waiting times for several promoters
The waiting times presented in the preceding section are quite long and by itself seem not to explain rapid evolutionary changes in regulatory regions. But answering the question how long one has to wait for one particular binding site to appear in one particular promoter may be too restrictive. Thus, we ask how long it takes until a new TF binding site emerges in at least one of all the human promoters. This could induce a change in gene regulation and hence, could be important for the evolution of the whole species (see Section 2.2.3). Assuming that there are around 20,000 human promoters, we computed the minimal, maximal and average waiting times
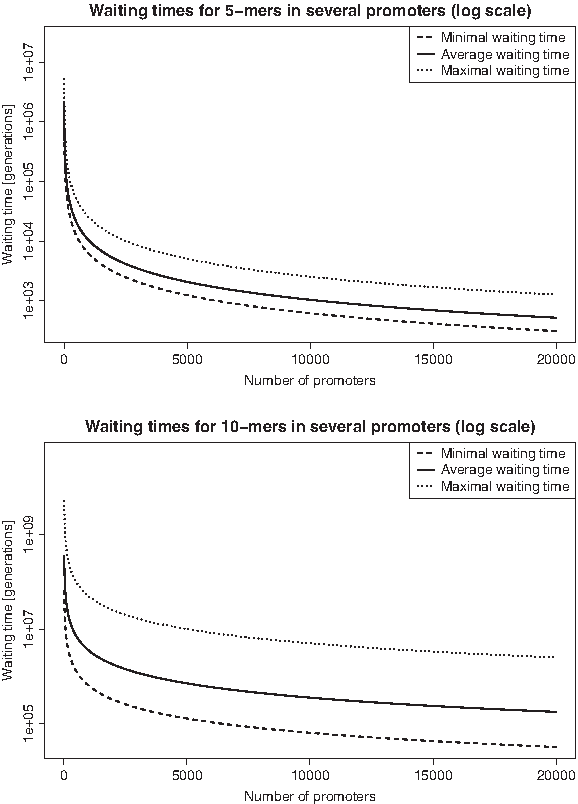
Waiting times in dependency of the number of promoters in model M1 (log scale). Minimal, maximal, and average waiting times (generations) for 5- and 10-mers to appear in at least one of several promoters.
The waiting times
The expected waiting times for k-mers to be created in at least one of all 20,000 human promoters are shown in Table 3. For example, on average, it only takes 7,467 years for a 5-mer to emerge (minimally 4,142 years and maximally 16,917 years). For 8-mers, on average one has to wait 341,104 years - a time span far below the speciation time of e.g. human and chimp (Hobolth et al. (2007)). And for 10-mers, the average waiting time is 4.8 Myrs implying that in a time comparable to the human-chimp split, on average one expects a given TF binding site of length 10 to be created at random in at least one of all the human promoters. But even after 700,000 years some particular new TF binding sites of length 10 are expected to be created (e.g. CCCCCCCCCC, CCCCCCCCCA, CCCCCCCCTG).
Minimal, maximal, and average waiting times (years) for k-mers, 5 ≤ k ≤ 10, to appear in at least one of all human promoters.
3.2.3. Comparison with existing TF binding sites
Given lists of k-mers ranked according to their waiting time till emergence, we are interested if one can observe top ranking k-mers in existing TF binding sites. We downloaded the non-redundant JASPAR CORE database for vertebrates, Version 4, (Portales-Casamar et al., 2010) and extracted all the human TF binding site position count matrices (PCMs) of length k, 5 ≤ k ≤ 10, i.e., 37 PCMs. In order to compare PCMs with k-mers, we converted a given PCM into a set of k-mers via the following procedure: after having computed a maximal score of a PCM by summing over the maximal column entries, we set a threshold of 0.95 of the maximal score and extracted all 10-mers with a score above this threshold.
For example, in case of the SP1 binding site (Fig. 4), 273 is the maximal score (CCCCGCCCC), the score threshold is 260 and the resulting 10-mer set of putative SP1 binding sites is given by {CCCCACCCCC, CCCCCCCCCC, CCCCGCCCCC, CCCCTCCCCC}. This set contains the top ranking 10-mers, e.g. CCCCCCCCCC is even the number 1 top ranking 10-mer, i.e. the fastest emerging 10-mer (see Figures 2 and 4). We repeated this procedure for all the PCMs extracted from the JASPAR database, also including the reverse complement for every k-mer since the TF could also bind to the complementary DNA strand. To test if these observed k-mers are among the top ranking k-mers according to our model, we assigned the corresponding ranks to them (as illustrated in Figure 4) and normalized the ranks by dividing by 4
k
in order to look at all k-mers simultaneously. The null hypothesis that the waiting times according to our model do not affect the appearance of real TF binding sites can then be formulated as

Example. Assuming that the SP1 motif is the set of 10-mers (and their reverse complements) with a score of at least 95% of the maximal score, we can derive the ranks for this 10-mer set, i.e., the ranks among all 10-mers in ascending order according to their waiting time until emergence and normalize them.
We performed Pearson's χ2-goodness-of-fit test yielding a p-value <2.2e − 16, i.e. one can reject the null hypothesis. The mean relative rank for the k-mers taken from JASPAR is 0.425 while the mean of the uniform distribution on [0, 1] is 0.5. Thus, k-mers with shorter waiting times are used more frequently as TF binding sites than other ones and as can be seen in Figure 5, a high proportion of existing TF binding sites belongs to the top ranking k-mers (around one quarter of them are among the top 10% ranks).
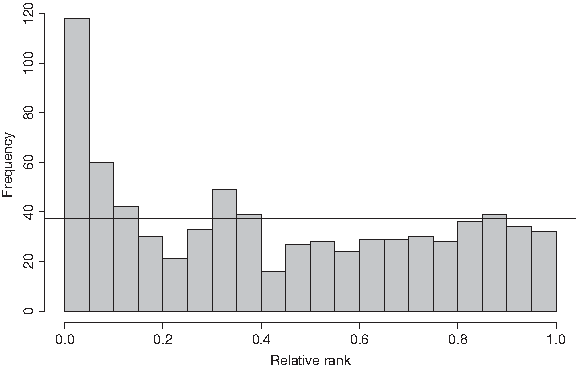
Histogram of the relative ranks of k-mers contained in JASPAR PCMs. For all JASPAR matrices of length k, 5 ≤ k ≤ 10, we assigned relative ranks to the k-mers with a relative score threshold of 0.95 (according to the procedure illustrated in Fig. 4). The horizontal line represents the uniform case, i.e., the case where the relative ranks would be distributed uniformly.
In this approach, for every TF, we have taken all of the possible k-mers (above a certain threshold) into account and thus, also included potentially slowly evolving k-mers. Hence, as a second step, we only looked at the rank of the fastest evolving binding site per TF. For every JASPAR TF we determined the k-mer with the smallest rank. Afterwards, we sorted all of the JASPAR TFs according to these minimal ranks. The results are depicted in Figure 6.
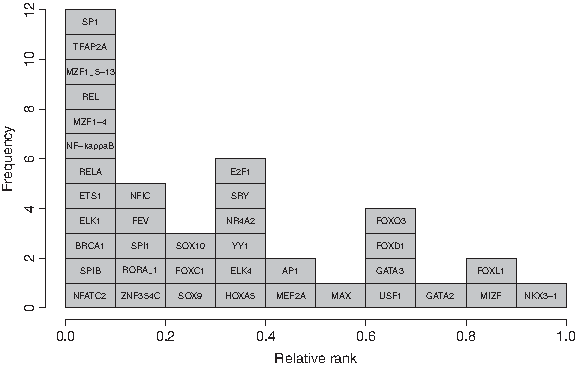
Histogram of the minimal relative ranks of JASPAR TFs. After having assigned relative ranks to the k-mers contained in JASPAR matrices (see Fig. 5), we determined the smallest relative rank for every TF. Thus, this figure depicts JASPAR TFs ranked according to their waiting time until appearance according to our model.
So under our model, the binding sites of the TFs SP1, TFAP2A, MZF1 5-13, REL, MZF1-4, NF-kappaB, RELA, ETS1, ELK1, BRCA1, SPIB and NFATC2 can be generated quickly while the appearance of binding sites like GATA2, FOXL1, MIZF and NKX3-1 binding sites requires long waiting times. Most of the TFs whose binding sites are predicted to be generated rapidly like BRCA1, NFKB, REL, RELA and SP1 are widely expressed and have been shown to interact with a lot of other proteins: e.g. BRCA1 has 225 interaction partners, REL 104, RELA 297, NFKB1 156, NFKB2 214 and SP1 has 156 interaction partners; numbers taken from the database UniHI (Chaurasia et al., 2007). In contrast, the binding sites of TFs which appear slowly according to our model are only expressed in certain tissues, e.g. the slowest evolving TF NKX3-1 is largely prostate and testis-specific, and have fewer interaction partners, e.g. NKX3-1 has 4, MIZF 11, FOXL1 30 and GATA2 21 interaction partners (numbers taken from UniHI).
4. Discussion
We have developed a probabilistic approach to study the evolution of regulatory regions allowing us to predict how long one has to wait for a given TF binding site of length k, k ranging from 5 to 10, to be created at random in the human species - either in one promoter of length 1 kb or in at least one of all the human promoters. Our results indicate that new TF binding sites can indeed appear on a small evolutionary time scale: for example, given that model M1 is an appropriate choice, on average around 7,500 years may be sufficient for a given 5-mer to emerge in at least one of all the human promoters, for 8-mers around 350,000 years and for 10-mers around 4.8 Myrs (model M1). But for some TF binding sites of length 10 like, for example, the SP1 binding site, a duration of 700,000 years may be enough. This reveals that new TF binding sites of length k, k ≤ 10, can easily appear in a time span significantly below or around e.g. the divergence time of human and chimp which is around 4 Myrs as stated by Hobolth et al. (2007).
According to our model, on average the expected waiting times increase exponentially with the length of the binding site. This suggests that in the evolution of primates, there should be a bias towards many short motifs instead of one long TF binding site in regulatory sequences. This is what one actually observes in eukaryotes; for example Wray et al. (2003) pointed out that promoters containing 10–50 binding sites for 5–15 different transcription factors are not uncommon. By computing the information content of eukaryotic TF binding sites, Wunderlich and Mirny (2009) found that in contrast to bacteria, single eukaryotic TF binding sites are too short and imprecise to guarantee specific binding which is compensated for by TF binding site clustering.
Furthermore, our results suggest that the composition of TF binding sites and not only their length play a crucial role concerning the waiting times for appearance: sometimes it is even more ”favorable” to wait for a particular (k + 1)-mer instead of waiting for another k-mer. For example, the waiting time for the 9-mer ACGTACGTA to appear in one of all promoters has been estimated to be around 1.3 Myrs and the one for the 10-mer CCCCCCCCCC to be only around 650,000 years. In consideration of the fastest and slowest emerging k-mers, one observes that k-mers containing products of the CpG methylation-deamination process (TG and CA) can rapidly appear in promoter sequences while TA- or CG-rich k-mers need a lot of time to be created at random. Hence, the CpG methylation-deamination process is probably a major determinant in generating new TF binding sites. It accelerates the emergence of some k-mers - which becomes obvious when comparing waiting times from the models M0 and M1. Simply assuming independently evolving nucleotides like Durrett and Schmidt (2007), Stone and Wray (2001), does not unveil the importance of this neighbor dependent substitution process for the creation of new TF binding sites. Thus, the more general model M1 should be preferred over the model M0.
We have tested whether our results are consistent with existing TF binding sites, i.e. if these TF binding sites are top ranking among all k-mers ranked in ascending order according to their waiting time till emergence. Based on PCMs from the database JASPAR (Portales-Casamar et al., 2010), we showed that this holds true for most of the cases. On the other hand, our model of predicting waiting times for the appearance of TF binding sites could be also used as a null model to detect TF binding sites which emerge slowly under the model but which are still observed. For example, the TATA-binding protein recognizes a motif containing TATA. But when looking at the waiting times in Table 2 (model M1), one surprisingly observes that k-mers containing TATA are among the slowest emerging k-mers. In this case, we speculate that due to the fact that the TATA-motif is probably one of the most crucial cis-regulatory elements, it ”has to” be quite rare and therefore ”should” not appear rapidly by the time passing to avoid drastic changes in gene regulation. Additionally, for future research it would be interesting to characterize the TFs with fast (resp. slowly) emerging binding sites with regard to biological properties (e.g. GO categories) similar to our approach in section 3.2.3. where we have examined the connection between the speed of binding site emergence and tissue-specificity/interaction partners. So far, we could observe that ubiquitous TFs are usually associated with fast emerging binding sites, while tissue-specific TFs are linked to slower emerging TF binding sites.
In summary, one can conclude that new TF binding sites are expected to emerge rapidly when taking all human promoter sequences as a basis. Apart from having computed the speed of de novo creation of k-mers, our approach now also reveals how the composition of a TF binding site as well as of the promoter sequence can influence the process of TF binding site emergence and therefore, extends the previous knowledge about the dynamics of promoter sequence evolution.
Footnotes
Acknowledgments
We would like to thank Peter Arndt, Hugues Richard, Szymon Kielbasa, and Tomasz Zemojtel for helpful discussions.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.