
Abstract
Objective:
Generalized anxiety disorder (GAD) in children and adolescents is associated with substantial morbidity and increases the risk of future psychopathology. However, relatively few psychopharmacologic studies have examined treatments for GAD in pediatric populations, especially in prepubertal youth.
Methods:
Children and adolescents aged 7–17 years of age with a primary diagnosis of GAD were treated with flexibly dosed escitalopram (10–20 mg daily, n = 138) or placebo (n = 137) for 8 weeks. Efficacy measures included the Pediatric Anxiety Rating Scale (PARS) for GAD, Clinical Global Impression of Severity (CGI-S) scale, Children's Global Assessment Scale (CGAS); safety measures included the Columbia-Suicide Severity Rating Scale (C-SSRS) as well as adverse events (AEs), vital signs, and electrocardiographic and laboratory monitoring.
Results:
Escitalopram was superior to placebo in reducing anxiety symptoms of GAD, as seen in the difference in mean change from baseline to week 8 on the PARS severity for GAD score (least squares mean difference = −1.42; p = 0.028). Functional improvement, as reflected by CGAS score, was numerically greater in escitalopram-treated patients compared with those receiving placebo (p = 0.286), and discontinuation owing to AEs did not differ between the two groups. Vital signs, weight, laboratory, and electrocardiographic results were consistent with previous pediatric studies of escitalopram.
Conclusions:
Escitalopram reduced anxiety symptoms and was well tolerated in pediatric patients with GAD. These findings confirm earlier reports of escitalopram efficacy in adolescents aged 12–17 years and extend the safety and tolerability data to children with GAD aged 7–11 years.
ClinicalTrials.gov Identifier:
NCT03924323.
Introduction
Decades of accumulating data from dozens of studies underscore the morbidity of anxiety disorders in children and adolescents (Asselmann et al., 2018; Crawley et al., 2014; Ramsawh et al., 2010; Strawn et al., 2021a). Anxiety disorders frequently emerge in childhood and adolescence and produce significant functional impairment and distress (Ramsawh et al., 2010). Furthermore, anxiety disorders, including generalized anxiety disorder (GAD), are common, chronic, and relapsing illnesses (Beesdo et al., 2010). Without effective treatment, anxious youth such as those with GAD experience a lifelong struggle; they are at increased risk of having difficulty transitioning to adulthood and developing depression (Bittner et al., 2007), substance abuse (Behrendt et al., 2011), and suicidality (Foley et al., 2006; Husky et al., 2012; O'Neil et al., 2012).
Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) reduce symptoms in pediatric patients with anxiety disorders (Dobson et al., 2019; Locher et al., 2017; Strawn et al., 2018) and are generally well tolerated (Mills and Strawn, 2020). However, the evidence base for most of these medications is established on relatively homogeneous populations that frequently have multiple anxiety diagnoses (e.g., separation, social, and GADs) (Walkup et al., 2008).
For youth with a primary diagnosis of GAD, four double-blind, placebo-controlled studies have evaluated SSRIs or SNRIs (Rynn et al., 2007, Rynn et al., 2001; Strawn et al., 2020, Strawn et al., 2015). In the first of these studies, (Rynn et al., 2001) found fixed-dose sertraline (50 mg/day) to be superior to placebo in youth aged 5–17 years (n = 22) and well tolerated. Later, venlafaxine was evaluated in children and adolescents aged 6–17 years with GAD (n = 320) and was superior to placebo in reducing anxiety symptoms (Rynn et al., 2007).
In a Food and Drug Administration (FDA)–requested registration trial, duloxetine was examined in a large, 10-week, multicenter trial of children and adolescents aged 7–17 years of age (n = 334) and it significantly decreased anxiety symptoms (Strawn et al., 2015). This resulted in the first medication to be FDA-approved for treating any pediatric anxiety disorder, in this case, GAD. Finally, in a small study of adolescents aged 12–17 years (n = 51), forced-flexible dosing of escitalopram significantly reduced anxiety compared with placebo (Strawn et al., 2020), and it normalized brain connectivity between the amygdala and prefrontal cortex (Lu et al., 2021; Lu et al., 2022). However, this escitalopram study, which was conducted under an investigational new drug exemption, did not examine efficacy in pediatric patients younger than 12 years of age.
Taken together, these studies reveal that SSRIs and SNRIs reduce anxiety symptoms for many youths; however, some patients fail to respond to an initial antidepressant treatment (Walkup et al., 2008), whereas others do not respond to one SSRI but may respond to a second SSRI trial (Walkup et al., 2002). Furthermore, anxious youth respond differently to SSRIs and SNRIs; SSRIs typically produce faster and larger improvements compared with SNRIs (Strawn et al., 2018). However, the only FDA-approved medication for pediatric patients with GAD is the SNRI duloxetine (Strawn et al., 2015).
With these considerations in mind, we sought to examine the efficacy of escitalopram in a large, multicenter trial of children and adolescents with a primary diagnosis of GAD. We hypothesized that escitalopram would reduce anxiety symptoms, based on results of the Pediatric Anxiety Rating Scale (PARS) severity score for GAD (PARS-GAD) (Strawn et al., 2015), and that it would be well tolerated, based on other studies of SSRIs in this population (Locher et al., 2017; Mills and Strawn, 2020).
Methods
This study (NCT03924323) was conducted at 33 sites in the United States from May 2019 to September 2021. The protocol was approved by an institutional review board (IRB) at each site and the study was conducted in compliance with ethical principles based on the Declaration of Helsinki, current good clinical practice guidelines, and International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines. Each participant's parent/legal representative provided written informed consent before participating and participants completed an informed assent.
Study design
This was a randomized, multicenter, double-blind, flexibly dosed, placebo-controlled, parallel-group trial of escitalopram in children and adolescents with GAD. The study consisted of an up to 3-week screening period, an 8-week double-blind acute treatment period, and a 1-week double-blind down-taper period; weekly study visits were conducted from screening through the end of the double-blind down-taper period, with follow-up telephone contact 1 week after the last visit
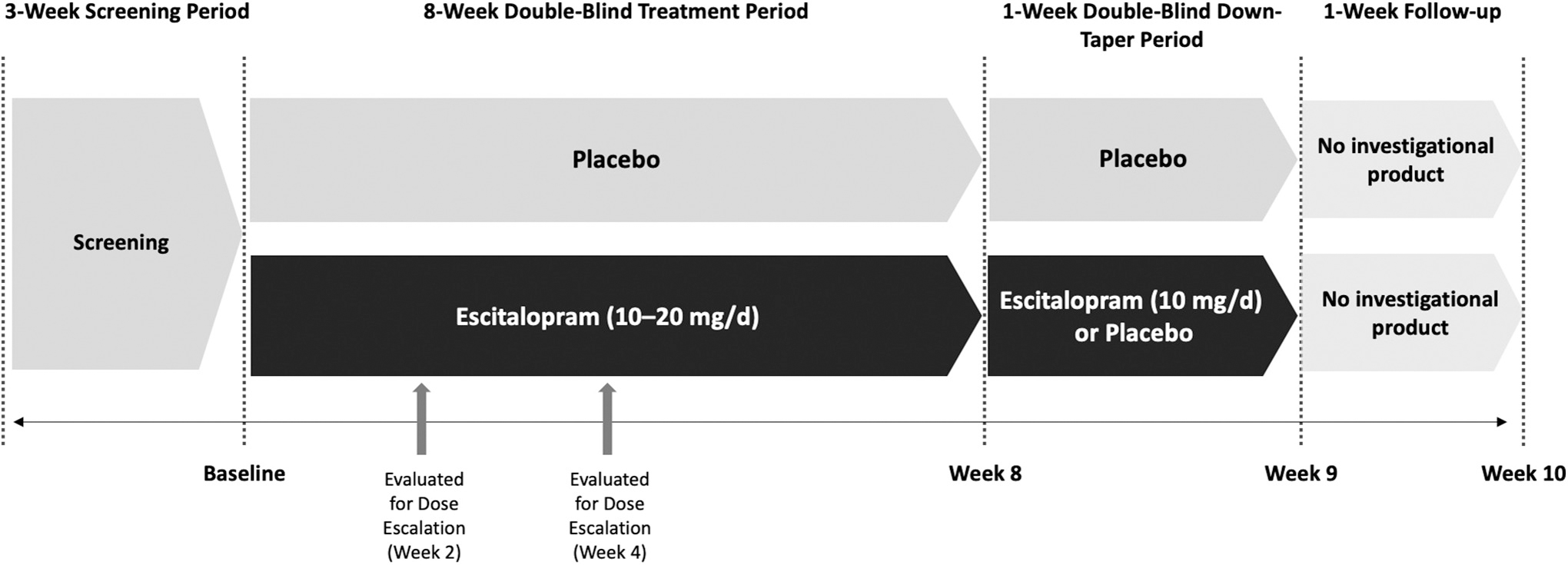
Study design. Patients randomized to escitalopram received 10 mg/day during weeks 1 and 2; patients with good tolerability were eligible for dose escalation to 20 mg/day at weeks 2 or 4 (Investigator judgment). Patients receiving 20 mg/day during double-blind treatment received 10 mg/day during down-taper; patients receiving 10 mg/day during double-blind treatment received placebo during down-taper.
An Interactive Response System was used to manage randomization, treatment assignment, and emergency unblinding, if necessary. At the end of week 2, patients with good tolerability could have their escitalopram titrated to 20 mg/day (clinician investigator's discretion); patients who remained at 10 mg/day were re-evaluated at week 4 and could titrate to 20 mg/day at that time. A subsequent decrease to 10 mg/day was allowed if escitalopram 20 mg/day was not well tolerated.
During double-blind discontinuation or at early withdrawal, patients receiving 20 mg/day at the end of the treatment period received 10 mg/day and patients receiving 10 mg/day at the end of treatment received placebo.
To ensure adherence to local safety guidelines and regulations after the COVID-19 pandemic began, protocol amendments were made to modify study procedures. Although in-clinic visits remained the preferred option, remote visits were allowed except for screening (all patients) and baseline for female patients of childbearing age. For the safety endpoints, assessments collected up to 1 week late were included in the safety analysis.
Patients
Randomized participants (n = 275) were outpatient children (7–11 years) and adolescents (12–17 years) who met Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) (American Psychiatric Association, 2013) criteria for GAD. Clinical inclusion criteria required that patients have a PARS-GAD ≥15 at the screening and baseline visits and a Clinical Global Impression of Severity (CGI-S) (Guy, 1976) score ≥4 at the two screening visits. The diagnosis of GAD was supported by the Mini International Neuropsychiatric Interview for children and adolescents (MINI-Kid) (Sheehan et al., 2010). At screening, patients were required to be medically stable based on physical examination, laboratory screening tests, and electrocardiograms (ECGs); girls of childbearing age were required to have a negative serum pregnancy test.
Patients with secondary diagnoses of separation anxiety or social anxiety disorder were enrolled if GAD was the primary diagnosis; however, patients with current major depressive disorder or any history of bipolar disorder, psychotic disorder, attention-deficit/hyperactivity disorder, eating disorder, obsessive compulsive disorder, or posttraumatic stress disorder were excluded. In addition, patients could not have a DSM-5 substance use disorder within the past 12 months and were required to have a negative urine drug screen at screening. Additional study inclusion and exclusion criteria are provided in Supplementary Table S1.
Efficacy assessments
The primary outcome was change from baseline to week 8 in PARS-GAD (assessed at screening, baseline, weeks 2 and 8). The PARS is a clinician-rated instrument for assessing the severity of anxiety symptoms associated with common anxiety disorders, including GAD (RUPP, 2002). Used in earlier registration trials for pharmacotherapy in pediatric patients with a primary diagnosis of GAD, the PARS-GAD is derived by summing 5 of 7 severity/impairment/interference items (items 2, 3, 5, 6, and 7) (Strawn et al., 2015). PARS raters were clinicians who participated in a standardized training program and met predefined criteria before conducting study ratings.
Secondary efficacy outcomes included change from baseline to week 8 on the Children's Global Assessment Scale (CGAS; screening, baseline, weeks 1, 2, 4, 6, 8) (Shaffer et al., 1983). The CGAS measures functional outcomes using a scale ranging from 1 to 100, with higher scores indicating better functioning (Shaffer et al., 1983). Change from baseline to week 8 on the CGI-S (a 7-point scale where 1 = Normal and 7 = Among the most extremely ill patients) was administered at screening, baseline, and weeks 2 and 8 (Guy, 1976). Other secondary outcomes included response and remission rates at week 8. PARS-GAD response and remission were defined by a 50% improvement from baseline score and score ≤8, respectively (Caporino et al., 2013); CGI-S and CGAS remission at week 8 were defined as a score ≤2 and a score >70, respectively.
A global COVID-19 impact assessment was also administered as the last assessment at every visit to evaluate the impact of the pandemic on the severity of the patient's GAD during the past week; patients with scores from 4 (moderate impact) to 7 (extreme impact) were considered to have been significantly impacted (either positively or negatively).
Safety assessments
Safety assessments included adverse event (AE) reporting (weeks 1–8, down-taper), vital signs (all weeks), physical examinations (screening and week 8), ECG (screening and week 8), and laboratory parameters (screening, week 8). In addition, suicidality was assessed at every visit via the Columbia-Suicide Severity Rating Scale (C-SSRS), an instrument designed to systematically assess and track suicidal ideation (category 1 [“Wish to be Dead”] to 5 [“Active Suicidal Ideation with Specific Plan and Intent and behavior”]) and behavior (category 6 [Preparatory Acts] to 10 [Completed Suicide]) (Posner et al., 2011).
Statistical methods
Efficacy analyses were based on the modified intent-to-treat (mITT) population, which consisted of all patients who received at least one dose of study drug and had a baseline and at least one postbaseline assessment of PARS-GAD. The primary efficacy parameter (change from baseline to week 8 in PARS-GAD) was analyzed using a mixed-effects model for repeated measures (MMRM) with treatment group, age group strata (7–11 years vs. 12–17 years), sex, pooled study center, visit, and treatment group-by-visit interaction as the fixed effects and the baseline value and baseline value-by-visit interaction as the covariates. An unstructured covariance matrix was used to model the covariance of within-subject PARS-GAD, and the Kenward–Roger approximation was used to estimate denominator degrees of freedom (Kenward and Roger, 1997). The analysis used observed cases of postbaseline scores without imputation of missing values.
Measurements after study withdrawal were treated as missing at random in the MMRM modeling, leading to an estimate of the treatment effect while on treatment. Last observation carried forward (LOCF), and pattern-mixture model sensitivity analyses were performed on the primary efficacy parameter to assess the impact of the missing at random assumption of the MMRM. To assess the impact of COVID-19 on the primary efficacy parameter, additional sensitivity analyses were conducted by (1) treating remote visits as missing and analyzing the remaining data using the primary MMRM model; (2) by repeating the primary analysis adding an indicator of when data were collected (i.e., before or on the start of the COVID 19 pandemic [March 17, 2020]) and treatment interaction to the model; and (3) by subgroup analysis repeating the primary analysis for subjects randomized before or on the start of the COVID-19 pandemic and after.
Changes from baseline to week 8 in CGAS and CGI-S (secondary endpoints) were determined using the same MMRM as in the primary analyses. Rates of PARS-GAD response and remission, and rates of CGAS and CGI-S remission were based on observed cases using a generalized linear mixed model, with logit link function, random intercept, and fixed terms of treatment, age group, sex, visit, treatment-by-age group interaction, treatment-by-sex interaction, treatment-by-visit interaction, treatment-by-age group-by-sex interaction, and baseline score using a Toeplitz covariance matrix. Inferential testing for the secondary endpoints was reported with nominal p-values; no adjustments for multiplicity were made. All statistical tests were two-sided hypothesis tests performed at the 5% significance level for main effects, and confidence intervals (CIs) were two-sided 95% CIs unless stated otherwise.
The study was powered to address the primary outcome (difference in PARS-GAD mean change from baseline to 8 weeks between escitalopram- and placebo-treated patients). To detect an effect size (treatment group difference of 2.30 U relative to pooled SD of 5.79) of 0.39, a sample size of 128 subjects in the escitalopram treatment group and 128 subjects in the placebo group would provide 85% power based on an MMRM model using a simulation method. The simulation assumed a correlation of 0.7 between the repeated measures and a common dropout rate of 14%, based on historical data of escitalopram in pediatric patients and prior randomized controlled trials of antidepressants in pediatric patients with anxiety disorders (Pine et al., 2001; Strawn et al., 2020; Walkup et al., 2008).
Safety analyses were based on the safety population, which consisted of all randomized patients who took at least one dose of study medication; baseline was the last nonmissing safety assessment before the first dose of double-blind treatment. Continuous variables were summarized by the number of subjects and mean, SD, median, minimum, and maximum values; categorical variables were summarized by the number and percentage of subjects.
Results
Patient characteristics and disposition
Of the 442 participants screened for participation, 275 were randomized to treatment (placebo = 137, escitalopram = 138); 273 patients received the study drug and were included in the safety population (placebo = 136; escitalopram = 137)
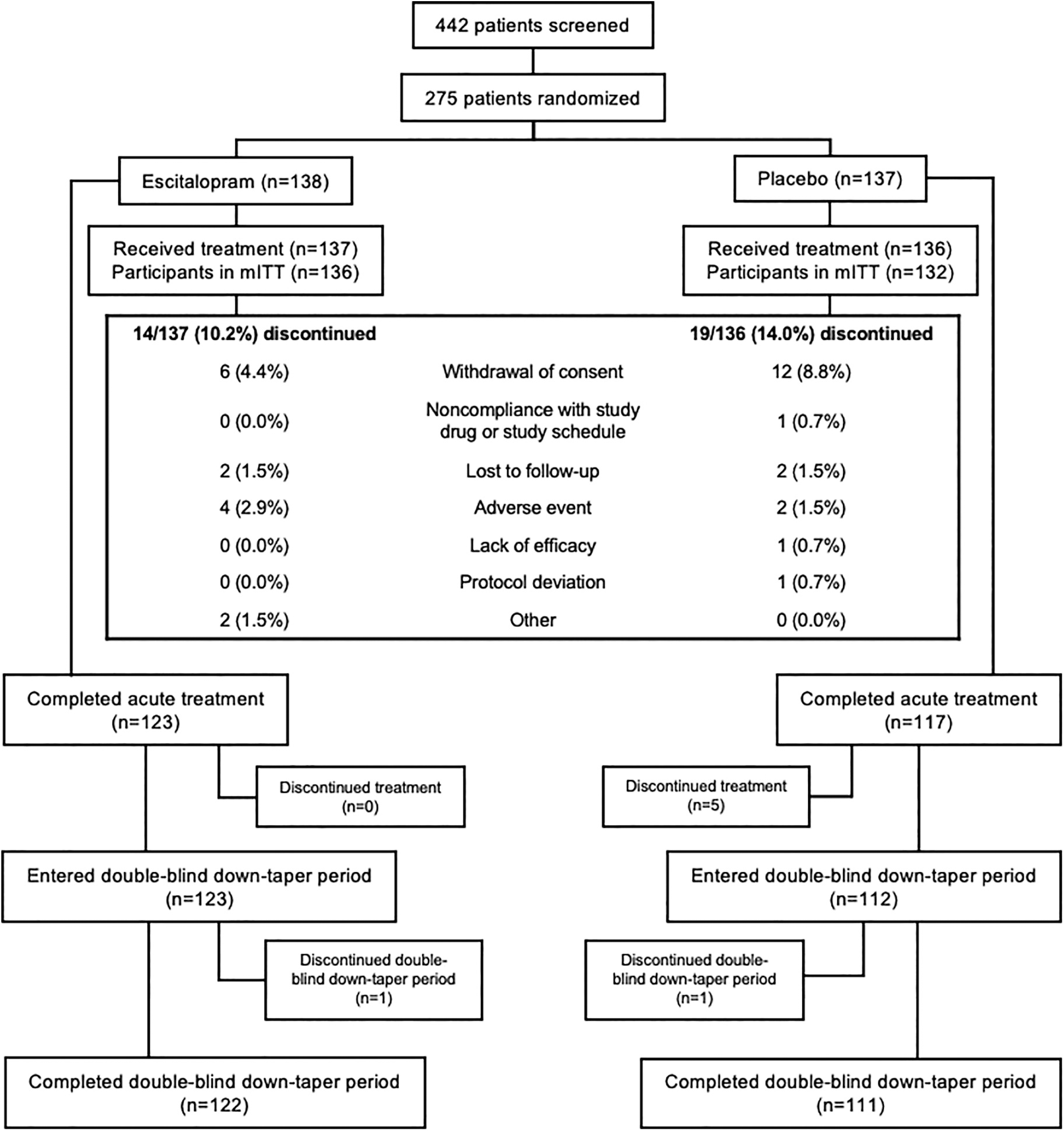
CONSORT flow diagram. mITT, modified intent-to-treat population.
Baseline Demographic Characteristics (Safety Population) and Efficacy Scores (Modified Intent-to-Treat Population)
PARS total score consists of the sum of general items 2, 3, 5, 6, and 7; PARS-GAD consists of the sum of GAD-specific items 2, 3, 5, 6, and 7.
Patients with a lifetime (current/past) diagnosis of ADHD were excluded from the study.
ADHD, attention-deficit/hyperactivity disorder; CGAS, Children's Global Assessment Scale; CGI-S, Clinical Global Impression of Severity-Severity; GAD, generalized anxiety disorder; MINI Kid, Mini-International Neuropsychiatric Interview for Children and Adolescents; mITT, modified intent-to-treat; PARS, Pediatric Anxiety Rating Scale; PARS-GAD, PARS severity score for GAD.
Extent of exposure
The mean (SD) double-blind treatment duration was 52.2 (14.3) days in the placebo group and 53.7 (10.4) days in the escitalopram group. The mean (SD) daily escitalopram dose was 13.1 (3.6) mg/day; the final daily dose was 20 mg/day for 94 (68.6%) patients and 10 mg/day for 43 (31.4%) patients. Most escalations to 20 mg/day occurred at week 2 (placebo = 78 [61.9%]; escitalopram = 81 [61.8%]); doses were escalated at week 4 for 11 (8.9%) placebo-treated and 15 (11.6%) escitalopram-treated patients. Two (2.2%) placebo-treated and three (3.1%) escitalopram-treated patients had dose reductions back to 10 mg/day; the median time to dose reduction was 15.5 days in the placebo group and 14.0 days in the escitalopram group.
Efficacy outcomes
Least squares (LS) mean (SE) change from baseline to week 8 in PARS-GAD was significantly different in favor of escitalopram versus placebo (−7.81 [0.484] vs. −6.38 [0.494]; LS mean difference [LSMD] = −1.42 [95% CI: 2.69 to −0.15]; p = 0.0281)
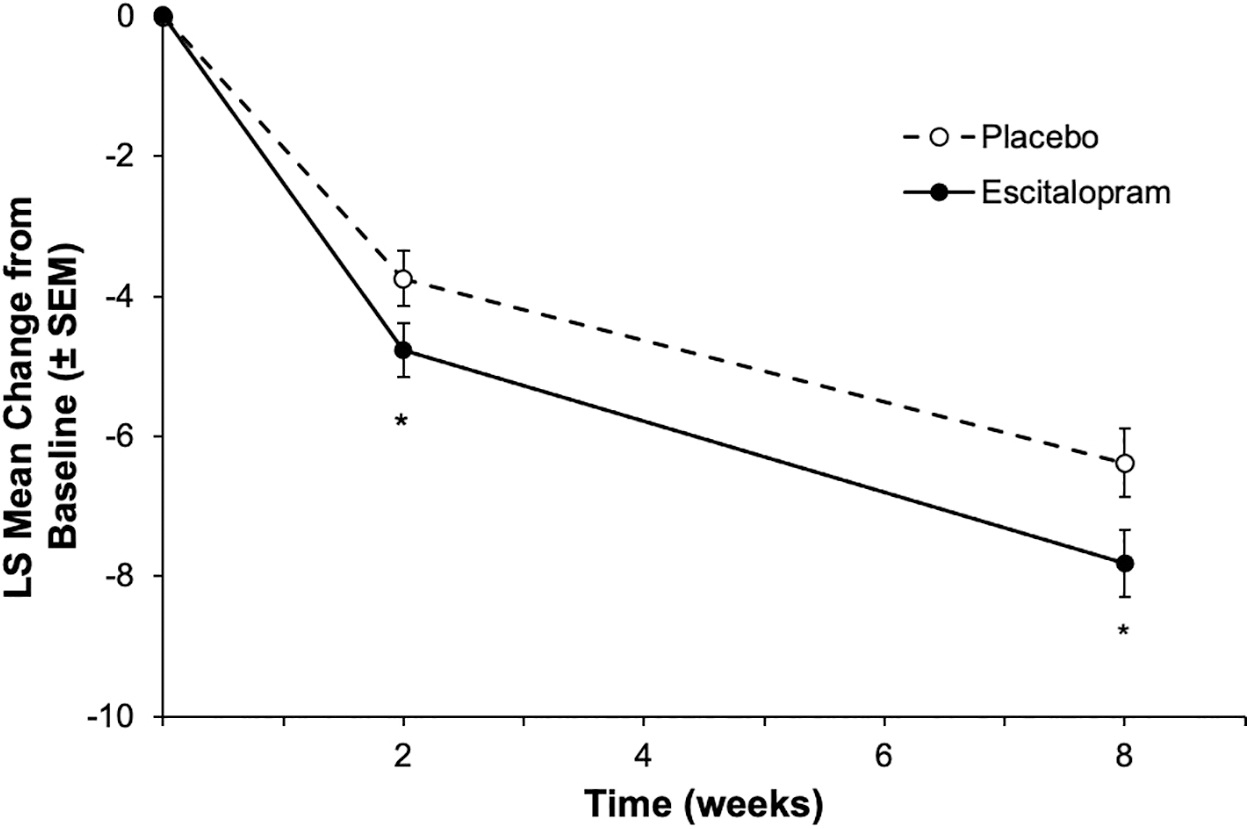
Primary outcome: change from baseline to week 8 in PARS severity score for GAD. LS mean change was significantly different in favor of patients treated with escitalopram versus placebo at week 8; at week 2, the difference between escitalopram and placebo was nominally significant (*p < 0.05). GAD, generalized anxiety disorder; LS, least squares; PARS, Pediatric Anxiety Rating Scale; SEM, standard error of the mean.
Results from additional sensitivity analyses assessing the impact of COVID-19 were in line with the primary analysis, showing statistically significant between-group differences in favor of escitalopram when remote visits were treated as missing, as well as when the date of data collection (before or after March 17, 2020) and treatment interaction were added as an indicator in the model. There was also no statistically significant between-group difference in the primary outcome for subjects randomized before or after the start of the pandemic.
LS mean changes from baseline to week 8 in CGAS and CGI-S (secondary efficacy endpoints) were numerically greater for escitalopram versus placebo; however, differences did not reach statistical significance
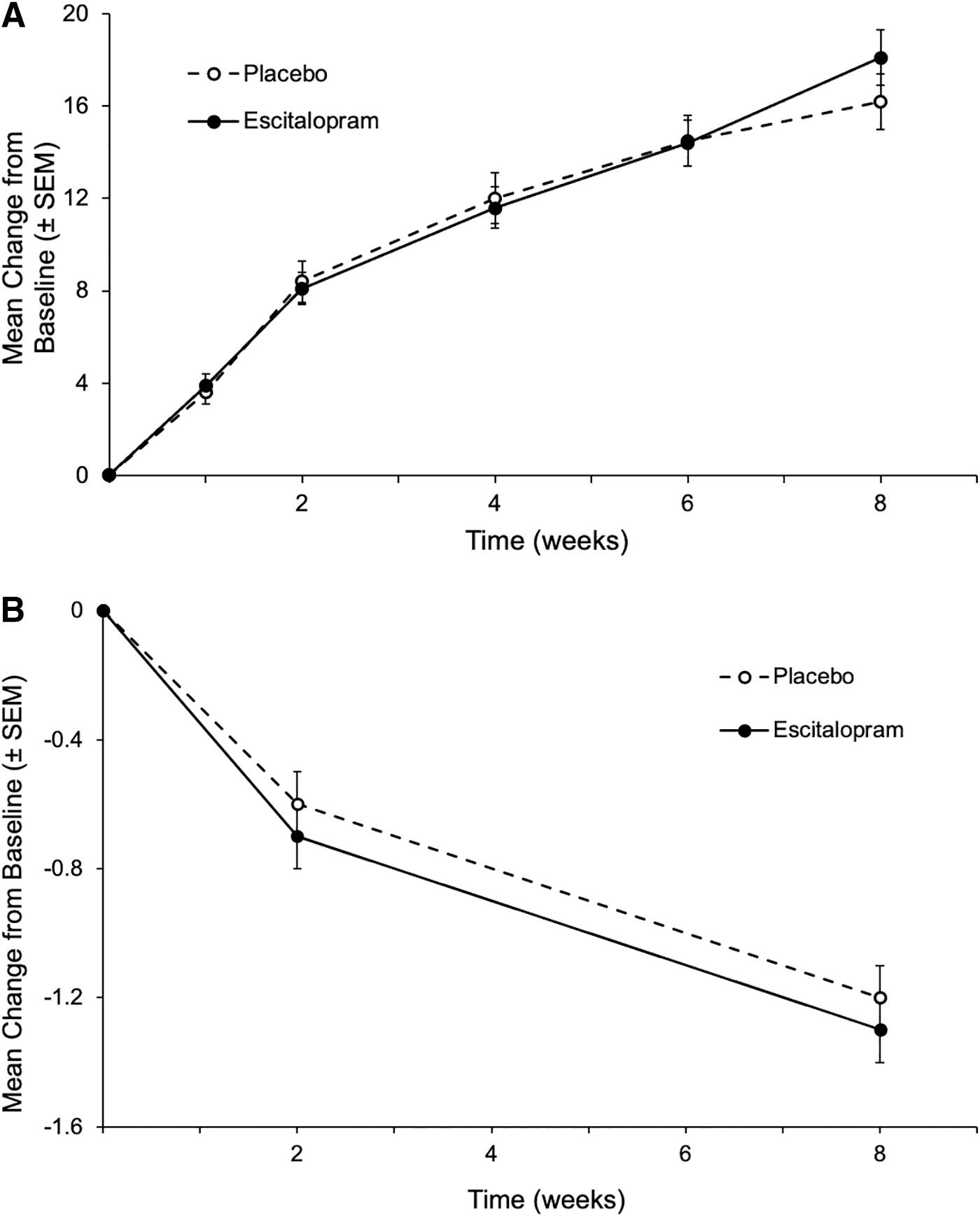
Secondary outcomes: mean change from baseline to week 8 in CGAS and CGI-S scores.
Secondary Outcomes: Change from Baseline, Response, and Remission at Week 8 (Modified Intent-to-Treat Population)
Estimates and p-values obtained from a mixed-effects model for repeated measures based on observed cases with treatment group, age group strata (7–11 years vs. 12–17 years), sex, pooled study center, visit, and treatment group-by-visit interaction as the fixed effects, and baseline and baseline-by-visit as covariates using an unstructured covariance matrix.
Analyses based on the observed cases using a generalized linear mixed model, with logit link function, random intercept, and fixed terms of treatment, age group, sex, visit, treatment-by-age group interaction, treatment-by-sex interaction, treatment-by-visit interaction, treatment-by-age group-by-sex interaction, and baseline score using a Toeplitz covariance matrix. For each outcome, n = number of patients in the modified intent-to-treat population; N1 = number of patients in the modified intent-to-treat population with a valid postbaseline visit score on the scale of interest.
c ≥50% reduction from baseline in the PARS severity score for GAD.
PARS-GAD score ≤8.
CGAS score >70.
CGI-S score ≤2.
CGAS, Children's Global Assessment Scale; CGI-S, Clinical Global Impression of Severity; CI, confidence interval; GAD, generalized anxiety disorder; LS, least squares; LSMD, least squares mean difference; PARS, Pediatric Anxiety Rating Scale; PARS-GAD, PARS severity score for GAD; SE, standard error.
Safety outcomes
Treatment-emergent AEs
The incidence of treatment-emergent AEs (TEAEs) was higher for escitalopram-treated (55.5%) than for placebo-treated patients (37.5%)
Treatment-Emergent Adverse Events Occurring at >2% in Escitalopram-Four Treatment-Emergent Adverse Event Were Observed with an Incidence Exceeding 5% and At Least Twice That of Placebo: Nausea, Decrease Appetite, Insomnia, and Somnolence
AE, adverse event; NEAE, newly emergent AE; SAE, serious AE; TEAE, treatment-emergent AE.
Premature discontinuation owing to an AE occurred in four (2.9%) escitalopram-treated patients (one patient each with activation syndrome, intentional self-injury, nausea, and epistaxis) and two (1.5%) placebo-treated patients (one patient with suicidal ideation and one patient with depression and fatigue); except for the event of intentional self-injury, all AEs associated with premature discontinuation were considered related to study drug.
Vital signs and clinical laboratory parameters
There were no clinically relevant differences between treatment groups for any hematology, chemistry, or urinalysis parameters during double-blind treatment. Furthermore, there were no differences between groups in potentially clinically relevant laboratory findings, vital signs, or ECG parameters. The only laboratory variables with potentially clinically significant results that were reported in >2 patients in either group were phosphate >1.1 × upper limit of normal (ULN) (placebo = 3/107 [2.8%]; escitalopram = 6/116 [5.2%]; normal range = 0.87–1.55 mmol/L [age 12–17 years] or 1–1.78 mmol/L [age 7–11 years]), potassium >1.0 × ULN (placebo = 4/101 [4.0%]; normal range = 3.5–5 mmol/L), and sodium <1.0 × lower limit of normal (escitalopram = 3/118 [2.5%]; normal range = 135–148 mmol/L).
ECG parameters
The only ECG variables that met potentially clinically significant criteria were PR interval of ≥250 ms (1 [0.8%] placebo patient) and corrected QT interval (Bazett) (QTcB) of >460 ms (1 [0.8%] placebo patient). QTcB increases >30 and ≤60 ms occurred in 8 of 118 (6.8%) patients receiving placebo and 6 of 125 (4.8%) patients receiving escitalopram; corrected QT interval (Fridericia) (QTcF) increases >30 and ≤60 ms were observed in 6 of 118 (5.1%) placebo patients and 3 of 125 (2.4%) escitalopram patients. No patient had QTcF increase >60 ms, and no patients had a QTcB- or QTcF-associated TEAE.
Suicidality
A higher percentage of escitalopram-treated (13 [9.5%]) than placebo-treated (2 [1.5%]) patients had suicidal ideation, with the most common ideation in the least severe category (“wish to be dead”; escitalopram = 9 [6.6%], placebo = 1 [0.7%]) (Supplementary Table S2). One (0.7%) patient receiving escitalopram had suicidal behavior, which was an actual attempt that was reported as an AE (intentional self-injury) and led to premature study discontinuation; this event was assessed as unrelated to the study drug. Suicidal ideation was reported as an AE by one (0.7%) placebo-treated and one (0.7%) escitalopram-treated patient; it was considered moderate and related to treatment in both patients and resulted in discontinuation for the placebo-treated patient. Additional details related to suicidality at baseline, during the double-blind portion of the study, and during discontinuation are given in Supplementary Table S2.
Discussion
This is the first multicenter trial and the largest prospective evaluation of the efficacy of escitalopram in pediatric patients with a primary diagnosis of GAD. The findings are encouraging; they replicate the findings from smaller, single-site studies of escitalopram (Strawn et al., 2020b) and extend efficacy and tolerability to younger patients. Several aspects of the trial warrant additional discussion.
As in two earlier studies of antidepressants in pediatric patients with GAD (Strawn et al., 2020b, Strawn et al., 2015), we included a week 2 efficacy time point and we observed improvement at week 2. This finding challenges conventional conceptions of SSRI response in youth that have encouraged clinicians to wait 4, 6, or even 8 weeks to see a response. Furthermore, this improvement at week 2 in escitalopram-treated youth is consistent with meta-analyses of SSRIs in pediatric patients with generalized, separation, and social anxiety disorders demonstrating early response (Strawn et al., 2018). Mechanistically, in adolescents with GAD, escitalopram (but not placebo) increases connectivity between the ventrolateral prefrontal cortex and amygdala during the first 2 weeks of treatment that predicts response at week 8 (Lu et al., 2021), suggesting that SSRIs produce early treatment-related changes in brain connectivity.
For clinicians, early improvement is important for two reasons. First, it frames patient and caregiver expectations and second, it predicts subsequent and continued response (Strawn et al., 2018). Knowing that medication may produce early improvement decreases uncertainty about the time course of treatment response and increases positive expectations about treatment. Beyond this, knowing that response tends to emerge earlier in treatment may help clinicians make the difficult decision to abandon an intervention—whether psychopharmacologic or psychotherapeutic. For example, in the largest trial of an SSRI in pediatric anxiety disorders, Child/Adolescent Anxiety Multimodal Study, patients who had no improvement by the eighth week of treatment had a 3:1 odds against additional improvement over the subsequent 4 weeks, suggesting that if there has been no response by 8 weeks, clinicians could consider switching treatments (Strawn et al., 2017).
However, clinicians must also keep in mind that the trajectory of improvement in individual trials and meta-analyses is an average treatment response, which may fail to capture variations that are attributed to clinical, demographic, pharmacodynamic, or pharmacokinetic factors (Strawn et al., 2020, Strawn et al., 2019).
Response and tolerability in pediatric anxiety disorders—including GAD—vary considerably in children and adolescents. Recently, several pediatric studies suggested that variation in CYP2C19 activity, the primary enzyme responsible for metabolizing escitalopram, produces substantial variation in escitalopram exposure (Strawn et al., 2021c; Vaughn et al., 2021). In fact, in adolescents, poor metabolizers have nearly three times the exposure seen in normal CYP2C19 metabolizers treated with either 10 or 20 mg/day, whereas ultrarapid metabolizers have approximately half of the exposure seen in normal metabolizers (Strawn et al., 2019). These differences in CYP2C19 metabolism are also associated with differences in escitalopram tolerability, including rates of activation and weight gain (Aldrich et al., 2019).
Indeed studies of the pharmacogenetic-related variation in pediatric escitalopram exposure and tolerability provide a lens through which to look at the results of our study. Taken together, they suggest substantial pharmacogenetically driven heterogeneity in response and tolerability potentially subtended by differences in escitalopram exposure. Certainly, future studies will be needed to refine escitalopram dosing approaches in pediatric patients to maximize response and tolerability (Strawn et al., 2021b) and to refine recommendations for considering CYP2C19 phenotype when using escitalopram in youth (Hicks et al., 2013).
Limitations
Although this is one of the largest prospective trials of an SSRI in pediatric patients with anxiety disorders and it demonstrates efficacy relative to placebo, several important limitations warrant additional discussion. First, compared with several earlier studies, the study only included two postbaseline efficacy measures. This limits our ability to examine the functional form of response. Second, we have a limited number of measures that would characterize family, environmental, and cognitive factors, as well as dimensional measures of comorbidity that could explain additional variation in either placebo or medication response (Compton et al., 2014; Strawn et al., 2017). Third, we did not include a dimensional measure of depressive symptoms that would have allowed us to better understand the relationship between changes in depressive symptoms and anxiety symptoms.
Fourth, the study examined currently used doses (i.e., 10 mg/day or 20 mg/day). Several pediatric studies and guidelines from the Clinical Pharmacogenetics Information Consortium (CPIC) suggest that for some youth with increased CYP2C19 metabolism, higher doses are required to generate similar exposure compared with normal metabolizers (Strawn et al., 2019). Thus, whether titration beyond the 20 mg/day dose would have boosted response rates remains to be determined, and patients were not genotyped for CYP2C19.
Conclusion
This large multicenter trial replicates earlier studies demonstrating the efficacy of escitalopram in adolescents with GAD and extends these findings to children aged 7–11 years. In addition, the study suggests that escitalopram is generally well tolerated in children and adolescents.
Clinical Significance
For clinicians, these findings buttress data that SSRIs effectively reduce anxiety severity in anxious youth and lend support for including escitalopram in the pediatric anxiety disorder armamentarium.
Footnotes
Acknowledgments
Writing and editorial assistance were provided by Carol Brown, MS, of Prescott Medical Communications Group (Chicago, IL), which was funded by AbbVie. Golda S. Ginsburg, PhD, served as the consultant for the PARS training. A poster of the results of this study was previously presented at the 2022 American Academy of Child and Adolescent Psychiatry Annual Meeting. AbbVie appreciates all the study sites, caregivers, and patients who participated in the study. The authors acknowledge Dr. Adena Rosenthal for her contributions to this study.
Authors' Contributions
All authors contributed to the interpretation of data and provided critical reviews of all article drafts. All authors approved the article for submission to this journal.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this article for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link:
Disclosures
J.R.S. has received research support from AbbVie, PCORI, the Yung Family Foundation and the National Institutes of Health. He has provided consultation to Cerevel, Intracellular Therapeutics and the FDA. He receives royalties from Springer Publishing and UpToDate and received material support from Myriad. He has also received honoraria from Medscape Live, Neuroscience Education Institute, the American Academy of Pediatrics, and the American Academy of Child and Adolescent Psychiatry. At the time of the study, L.M., R.D.H., A.W., K.B., and B.E. were employees of Syneos Health, which was funded by AbbVie to write the protocol and conduct the study. K.B. is currently an employee of Janssen Global Services and may hold stock. E.G., C.L., and M.G. are employees of AbbVie and may hold stock. M.M. reports royalties from American Psychiatric Publishing and grant funding from The Hartwell Foundation. J.A.K. is owner/employee of Core Clinical Research, which was funded by AbbVie to conduct the study.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.