
Abstract
Prebiotic synthesis of complex organic molecules in water-rich environments has been a long-standing challenge. In the modern deep sea, emission of liquid CO2 has been observed in multiple locations, which indicates the existence of benthic CO2 pools. Recently, a liquid/supercritical CO2 (ScCO2) hypothesis has been proposed that a two-phase ScCO2–water environment could lead to efficient dehydration and condensation of organics. To confirm this hypothesis, we conducted a nucleoside phosphorylation reaction in a hydrothermal reactor creating ScCO2–water two-phase environment. After 120 h of uridine, cytosine, guanosine, and adenosine phosphorylation at 68.9°C, various nucleoside monophosphates (NMPs), nucleotide diphosphates, and carbamoyl nucleosides were produced. The addition of urea enhanced the overall production of phosphorylated species with 5′-NMPs, the major products that reached over 10% yield. As predicted, phosphorylation did not proceed in the fully aqueous environment without ScCO2. Further, a glass window reactor was introduced for direct observation of the two-phase environment, where the escape of water into the ScCO2 phase was observed. These results are similar to those of a wet–dry cycle experiment simulating the terrestrial hot spring environment, indicating that the presence of ScCO2 can create a comparatively dry condition in the deep sea. In addition, the high acidity present in the aqueous phase further supports nucleotide synthesis by enabling the release of orthophosphate from the hydroxyapatite mineral solving the phosphate problem. Thus, the present study highlights the potential of the unique ScCO2–water two-phase environment to drive prebiotic nucleotide synthesis and likely induce condensation reactions of various organic and inorganic compounds in the deep-sea CO2 pool on Earth and potentially other ocean worlds.
Introduction
The origin of life is one of the most important questions to be addressed in the history of science. The challenge of creating complex biomolecules, such as nucleic acids, from basic chemical components has perplexed scientists for decades. To tackle this issue, researchers have utilized laboratory experimentation to recreate prebiotic conditions. The synthesis of nucleotides, which consist of nucleobases, ribose, and phosphate, can be primarily replicated through mimicking terrestrial hot springs found on Earth, wherein dehydration and condensation reactions prevail (Powner et al., 2009; Kim and Benner, 2017; Stairs et al., 2017; Becker et al., 2019). The concentration of these molecules has been proposed in the environment for the formation of the fundamental components of nucleobases and ribose (Benner et al., 2012; Yi et al., 2022). Studies have investigated the use of condensing agents or drying conditions to phosphorylate nucleosides via phosphorylation reactions (Lohrmann and Orgel, 1968; Gull, 2014; Burcar et al., 2016; Kim et al., 2016). Phosphate minerals in meteorites may provide the phosphorus needed for nucleotide synthesis, but the resulting geochemical cycle is unclear (Pasek, 2008; Pasek et al., 2013; Walton et al., 2023, 2021). Furthermore, phosphorus participates in various functions in biology, such as replication, genetic information, metabolism, and membranes in contemporary living systems. In addition, phosphate plays a significant role in the context of the origin of life due to its role as a high-energy molecule and thermodynamic instability outside of a biological context. However, it should be noted that any assessment of the importance of phosphate must be objective and supported by empirical evidence. There is currently a debate as to the supply of phosphate on early Earth due to its high reactivity with divalent cations such as calcium, which makes it a water-insoluble phosphate mineral. The benefit of a terrestrial lake or a hot spring is its dynamic behavior of dehydrating and condensing building blocks of life, such as amino acids and nucleotides, through wet–dry cycling (Deamer et al., 2006; Damer and Deamer, 2015; Damer and Deamer, 2020; Van Kranendonk et al., 2021). While the deep-sea environment is considered to be water rich, the introduction of temperature gradients and metal or mineral surfaces has been shown to enable the condensation of amino acids into peptides in a simulated hydrothermal system (Imai et al., 1999; Lemke et al., 2009; Takahagi et al., 2019). Similarly, RNA oligonucleotide synthesis has been demonstrated in the laboratory using iron- and sulfur-rich chimney structures, and nucleotide phosphorylation has been achieved using a flow reactor system with the support of metal catalysts (Ozawa et al., 2004; Burcar et al., 2015).
Given the importance of having a microenvironment with low water activity, might there be additional methods in the ocean depths that can allow for the formation of an interface between aqueous and nonaqueous phases? Here, natural condensed CO2 fluids have been recently identified in the deep-sea environment, which indicates the existence of a subsurface condensed CO2 fluid and hydrates (Sakai et al., 1990; Inagaki et al., 2006; Konno et al., 2006; Lupton et al., 2006; Kawagucci et al., 2011; Pedersen et al., 2010; Stensland et al., 2019; Takahashi et al., 2023). The critical point of CO2 is located at mild temperature and pressure conditions (T c = 31.1°C, P c = 7.38 MPa), which allows CO2 to exist in its liquid and supercritical states near the deep-sea hydrothermal system as a hydrophobic solvent (Shibuya and Takai, 2022). Supercritical fluids generally possess liquid-like solubility, gas-like diffusivity, and low viscosity, providing unique characteristics. The phase transition from liquid CO2 (LqCO2) to supercritical CO2 (ScCO2) changes the solubility, partition coefficient, dipole moment, and dielectric constant. Therefore, in industry, ScCO2 has been used as a green solvent for the extraction of various hydrophobic organic compounds as well as nonaqueous enzyme-catalyzed reactions in a wide variety of biotechnological applications (Beckman, 2004; Matsuda, 2013). A recent geochemical model suggests that the Lq/ScCO2 pool in the modern ocean is derived by phase separation from a mixed fluid of magma-derived CO2 gas and hydrothermal water caused by temperature and pressure changes during upwelling (Fig. 1a). This results in a two-phase interfacial environment of Lq/ScCO2 and water in the deep sea. Since ScCO2 has gaseous properties and can partly dissolve water molecules (Sabirzyanov et al., 2002), we hypothesize that the water escape leads to partial dehydration and condensation of organic molecules in the aqueous phase, similar to that of the wet–dry cycle simulating terrestrial hot springs (Deamer et al., 2006; Damer and Deamer, 2015; Damer and Deamer, 2020). Such an environment may provide another solution to the long-standing water problem in prebiotic chemical evolution in the ocean (Russell, 2021; Shibuya and Takai, 2022). Indeed, a similar environment has been shown to produce amino acids from hydroxylamine and keto acids in an ScCO2–water system (Fujioka et al., 2009). In addition, peptide synthesis through the amphiphilic vesicle formation process in the subcritical CO2–water environment, simulating the deep tectonic fault zone, has been reported (Mayer et al., 2018; Mayer et al., 2015). However, successful phosphorylation of organic compounds in the deep-sea environment has yet to be reported due to the nature of hydrolysis, which presents significant obstacles (Gerlt et al., 1975; Pasek, 2020).
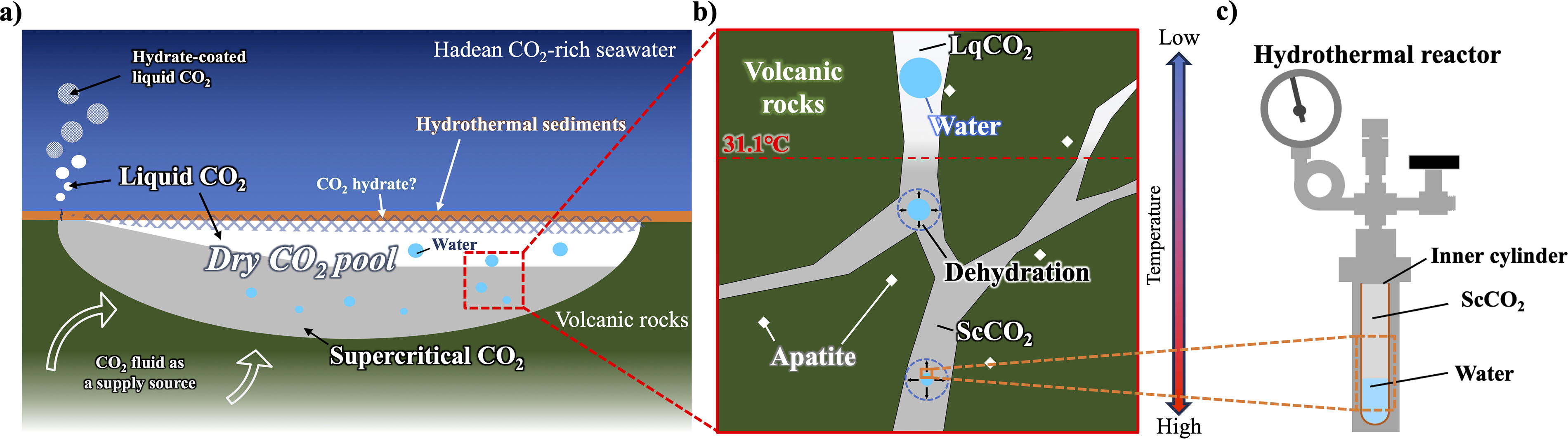
Simulated reaction environment in the CO2 pool using a hydrothermal reactor.
So far various inorganic phosphorus sources such as orthophosphate, phosphite, condensed phosphates, and diamidophosphate are considered potential agents for prebiotic phosphorylation (Gibard et al., 2018; Nicholls et al., 2023). Nevertheless, the major form of P on early Earth is considered to be orthophosphate minerals (Schwartz, 2006), and, thus, accessible dissolved P-sources on early Earth remain a challenging problem (Walton et al., 2023; Walton et al., 2021). One potential solution to achieving the formation of phosphorus-bearing organic compounds is to utilize highly soluble reduced oxidation state phosphorus compounds (Gull et al., 2023).
In the present study, we developed a hydrothermal reactor system to simulate the ScCO2/water bilayer in the deep sea. This environment provides an acidic water phase resulting from the high partial pressure of CO2 (Craig, 2014), which leads to the dissolution of phosphate minerals (Lundager Madsen, 1975; Walton et al., 2021). Therefore, we examined the phosphorylation of nucleosides, as one of the commonly used model organic compounds for prebiotic phosphorylation reaction (Gull, 2014). While it is likely that nucleosides are less abundant in the deep sea than on terrestrial surfaces (Orgel, 2004; Damer and Deamer, 2020), it is important to investigate the unique properties of the ScCO2/water environment in comparison to the well-studied terrestrial wet–dry conditions that lead to nucleotide synthesis.
Simulated CO2 fluid/water two-phase environment
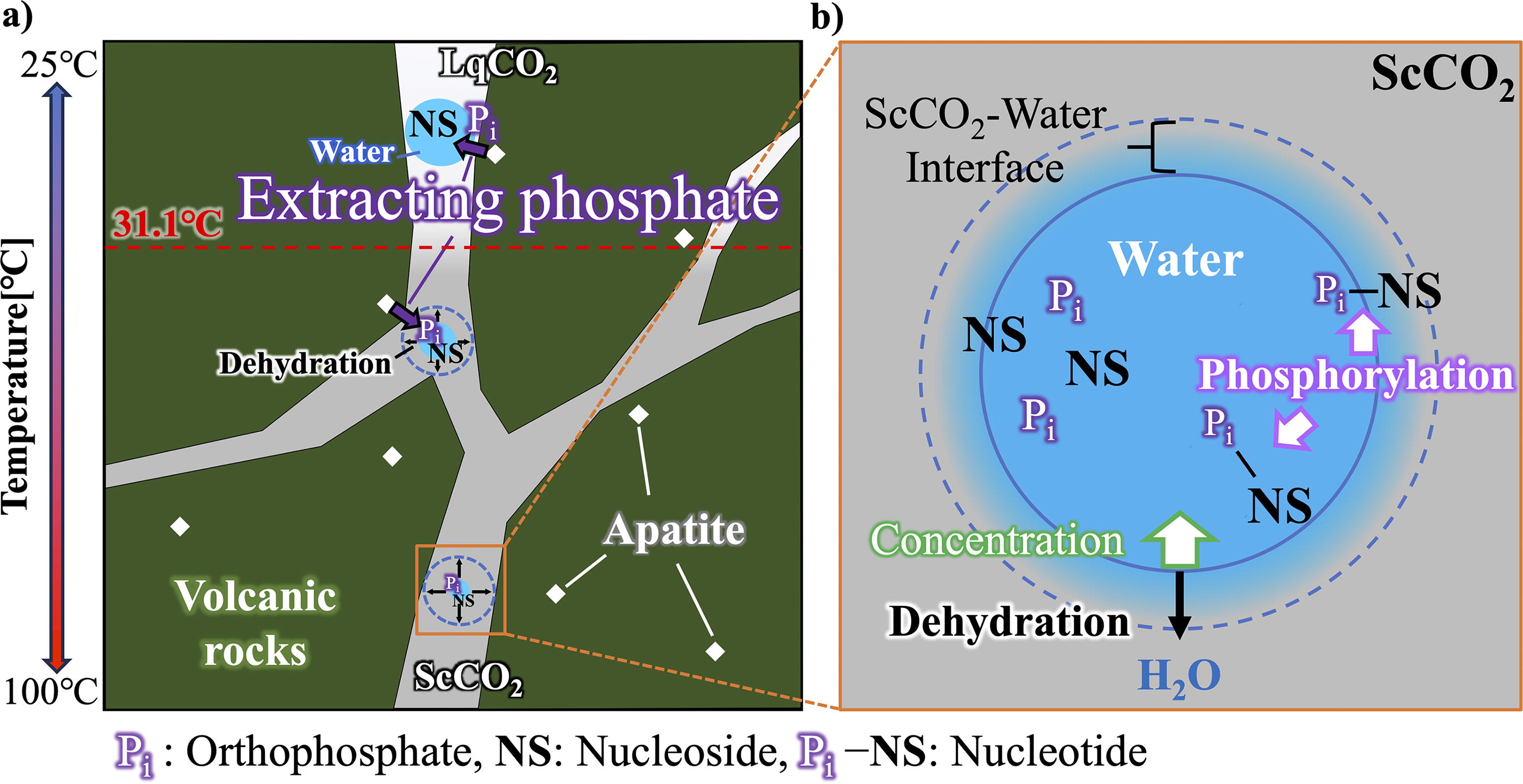
A detailed model of the formation of a CO2 pool is described by Shibuya and Takai (2022). In brief, Hadean Earth presumably had atmospheric and oceanic CO2 levels as high as the early Archean (Sleep and Zahnle, 2001), and therefore, CO2 fluid was continuously supplied to the subseafloor through phase separation of CO2-rich seawater and magmatic gases. Near the low-temperature seafloor, a CO2 pool is trapped by hydrothermal sediments capped with CO2 hydrate. If the surrounding temperature exceeds 10°C, LqCO2 will leak and rise from the seafloor and lead to the formation of a hydrate-coated CO2 bubble (Fig. 1a).
Whereas in the subseafloor, CO2 fluid and the precipitated water droplet (due to low solubility of H2O) coexist within the pore space of the permeable volcanic rocks (Fig. 1b). Due to the geothermal gradient, LqCO2 will become ScCO2 above 31.1°C causing higher miscibility of water in the CO2 phase that leads to the dehydration of water droplets. In this study, we used the hydrothermal reactor to simulate the two-phase environment of water and CO2 inside the hydrothermal reactor system (Fig. 1c).
Hydrothermal reactor system
The reactor system comprises an autoclave reactor fabricated from SUS316 stainless steel (TVS-N2-30, Taiatsu Techno Co., Japan) linked to an LqCO2 inlet/outlet line with SUS316, along with a pressure gauge and a valve (Supplementary Figure S1a). A heater (RSH-1DN, AS ONE Co., Japan) was connected to a thermocouple to monitor the temperature of the silicone oil in a beaker with the stirrer bar (oil bath). A PEEK (Poly Ether Ether Ketone) inner cylinder was utilized to prevent any reaction with the stainless steel. A stirrer bar inside the inner cylinder was used to directly mix the reaction solution. To simulate the water-rich environment, a gold tube was used (Supplementary Figure S1b). Because the oil temperature does not directly reflect the internal temperature of the reactor, we obtained a correlation plot of the oil bath temperature and the internal temperature prior to the experiment using organic and chemical compounds (Supplementary Figure S1c). Hence, the temperatures described in this article reflect the internal temperatures converted from the isothermal oil bath temperatures (Supplementary Table S1).
Hydrothermal reactor system with glass window
The glass ceramic cylinder-based autoclave system (HPG-30-3GC, Taiatsu Techno Co., Japan) was used to visualize the interface of the CO2 fluid and water (Supplementary Figure S2). The reaction was conducted for 24 h using 1.2 mL water containing uridine 10 mmol/kg, monosodium phosphate (NaH2PO4) 30 mmol/kg, and urea 200 mmol/kg. Oil bath temperature was set to 140°C (internal temperature 94.1°C), and pressure was set at 10 MPa using a digital pressure gauge (NAGANO KEIKI CO., LTD., Japan). The reactor was video recorded for 24 h to observe the LqCO2 to ScCO2 phase transition as well as the partial dehydration from the aqueous phase leading to the change of the water level. Fast forward video of the first 3 h (reaching equilibrium) were edited as Supplementary Video S1.
Reagents and gases
Nucleobases (adenine, guanine, cytosine), nucleosides (uridine, adenosine, guanosine, cytidine), and nucleotides (5′-UMP, 3′-UMP, 5′-AMP, 2’-AMP, and 3′-AMP in mixed powder, 5′-GMP, 5′-CMP), as well as hydroxyapatite (Hap) and C13-urea, were procured from Sigma-Aldrich Co. LLC. Uracil, urea, and sodium dihydrogen (NaH2PO4) were procured from FUJIFILM Wako Pure Chemical Co., Japan. Other reagents were specified in each section. LqCO2 was purchased from KAYAMA SANSO Co., Japan.
Nucleoside phosphorylation experiment in an ScCO2–water two-phase environment
We prepared a 1.5 mL mixture composed of a nucleoside mix (including uridine, adenosine, guanosine, and cytidine) at a concentration of 10 mmol/kg, a phosphate source (either 30 mmol/kg NaH2PO4 or 50 mg Hap), and other chemicals (at a concentration of 200 mmol/kg urea). The pH of the solution was analyzed using a 0.5 mL aliquot of the mixture. The remaining 1 mL of the mixture was introduced into the inner cylinder with a volume of 24.5 cm3 (Supplementary Figure S1a), which was then inserted into the hydrothermal reactor, capped, and purged with LqCO2. The hydrothermal reactor was positioned within an oil bath and heated to reach an internal temperature ranging from 25°C to 94.1°C at 11–12 MPa for a duration of 24–240 h. The oil bath situated on a heater facilitated temperature regulation. The heater temperature was set to meet the target internal temperature of the reactor. Subsequent to the nucleoside phosphorylation experiment, the oil bath was no longer heated, and the reactor was removed from it. The reaction vessel was rapidly cooled using liquid nitrogen to freeze the samples, and CO2 fluid was subsequently released to decrease pressure. After the ice was thawed from the solution, it was transferred into 1.5 mL glass vials and quickly frozen with liquid nitrogen. The samples were then stored at −30°C until analysis. The internal pressure was adjusted by releasing the CO2 fluid after reaching the initial temperature by monitoring the pressure gauge.
Nucleoside phosphorylation conditions in a water-rich environment using a gold tube
A gold tube (with an outer diameter of 5 mm and inner diameter of 4 mm) was cut into ∼3.7 cm length. One end was arc welded using a Lampert PUK U5 welding machine. Then, the tubes were filled with a reaction solution consisting of nucleoside, urea, and NaH2PO4. The other end was also arc welded (refer to Supplementary Figure S1b) to fully enclose the inner solution. Subsequently, the gold tube was placed inside an inner PEEK cylinder of the reactor. After capping the reactor, LqCO2 was injected from the top. The procedures that followed those outlined earlier were identical until the conclusion of the experiment. During sample collection, a section of the gold tube was cut, and the solution within was extracted using a pipette and placed into a glass vial of 1.5 mL capacity. The vial was then stored at a temperature of −30°C.
Ultrahigh-performance liquid chromatography analysis
Ultrahigh-performance liquid chromatography (UPLC) analysis was carried out on a Waters Acquity UPLC H-Class PLUS system (Waters Corp., MA, USA) using an Atlantis Premier BEH C18 AX 1.7 µm column (2.1 × 100 mm), which is known for its mixed-mode reversed-phase/weak anion exchange stationary phase for clear separation of various anions including nucleotides. Running buffers included 10 mM ammonium acetate pH 9.2 (A) and 90:10 acetonitrile/H2O (B). The flow rate was 0.3 mL/min, the injection volume was 3 µL, and the column temperature was maintained at 25°C throughout the analysis. The reaction sample was separated using a linear gradient elution program. This program began at 100% A and transitioned to 90% A between 0 and 4 min. The elution then progressed from 90% A to 0% A between 4 and 6 min. Subsequently, it transitioned to 0% A between 6 and 7 min. The elution then progressed to 100% A between 7 and 8 min before returning to 100% A between 8 and 10 min. The detection wavelengths ranged from 210 to 300 nm, monitoring nucleobases, nucleosides, and nucleotides. The data were processed, and the peak area was calculated using MassLynx 4.2 software. The samples were diluted from 20 to 100 μL with ultrapure water. To analyze the samples using UPLC, the solutions were filtered through a 0.2 μm filter. However, if NaH2PO4 was used in the samples, the filter could not be used. The diluted sample solutions were then transferred to 0.3 mL glass vials for analysis by UPLC. Peak identification involved comparing a standard peak to determine peak attribution and using time of flight-mass spectrometry (TOF-MS) analysis to identify nucleotides through nucleoside phosphorylation. Prior to analyzing the reaction sample, reference standards were employed, which included nucleobases (uracil, adenine, guanine, cytosine), nucleosides (uridine, adenosine, guanosine, cytidine), and nucleotides (5′-UMP, 3′-UMP, 5′-AMP, 2’-AMP, and 3′-AMP in mixed powder, 5′-GMP, 5′-CMP).
Mass spectrometry
Mass spectrometry was performed using a Waters H-Class/Xevo G2-XS QTof system attached to a Waters Acquity UPLC H-Class PLUS system (Waters Corp., MA, USA) with data acquisition in sensitivity mode under positive electrospray ionization. The range of acquisition was 100–1200 m/z with a capillary voltage of 5.0 kV and a cone voltage of 10 V. The column temperature was maintained at 25°C, whereas the sample manager temperature was at 4°C. The spray chamber temperature was set to 30°C. Cone gas flow was maintained at 50 L/min. MassLynx 4.2 software facilitated data acquisition and processing. The same buffer as that used in the previously described UPLC method was used for the running buffers.
Ammonia assay
Dissolved ammonia was quantified using an ammonia assay kit (Cell Biolabs, Inc., CA). The assay kit was employed following the instructions provided by the company. Calibration curves were established by preparing 1/2 dilutions of the standard solution (80 mM ammonium chloride) from 12.5 to 800 μM, a total of six times, and a blank solution (Milli-Q water). The solution collected after conducting the phosphorylation reaction under ScCO2–water conditions (solution containing uridine, NaH2PO4, and urea heated to 68.9°C at 11–12 MPa for 24 h) was diluted 1000-fold. The diluted solution was then subjected to analysis with an ammonia assay kit using a 96-well microtiter plate and EnSpire Multimode Plate Reader® (PerkinElmer) by reading 630 nm.
Phosphate assay
Phosphate concentration extracted from Hap in the ScCO2–water two-phase environment was measured with the QuantiChromTM Phosphate Assay Kit (DIPI-500) (BioAssaySystems, CA). Procedures for analysis were performed according to the protocol provided in the kit. Using the standard reagents supplied with the kit, a calibration curve was prepared, ranging from 0 to 40 μM. Reaction samples were diluted 1000–2000 times so that the concentrations fell within the calibration curve. Fifty microliters of standards or samples were transferred to a 96-well microtiter plate, and then 100 μL of reagent was added to each well. After 30 min of incubation at room temperature, the absorbance of the solutions in the plate was measured at 620 nm using an EnSpire Multimode Plate Reader (PerkinElmer).
Results
Nucleoside phosphorylation in the ScCO2–water environment
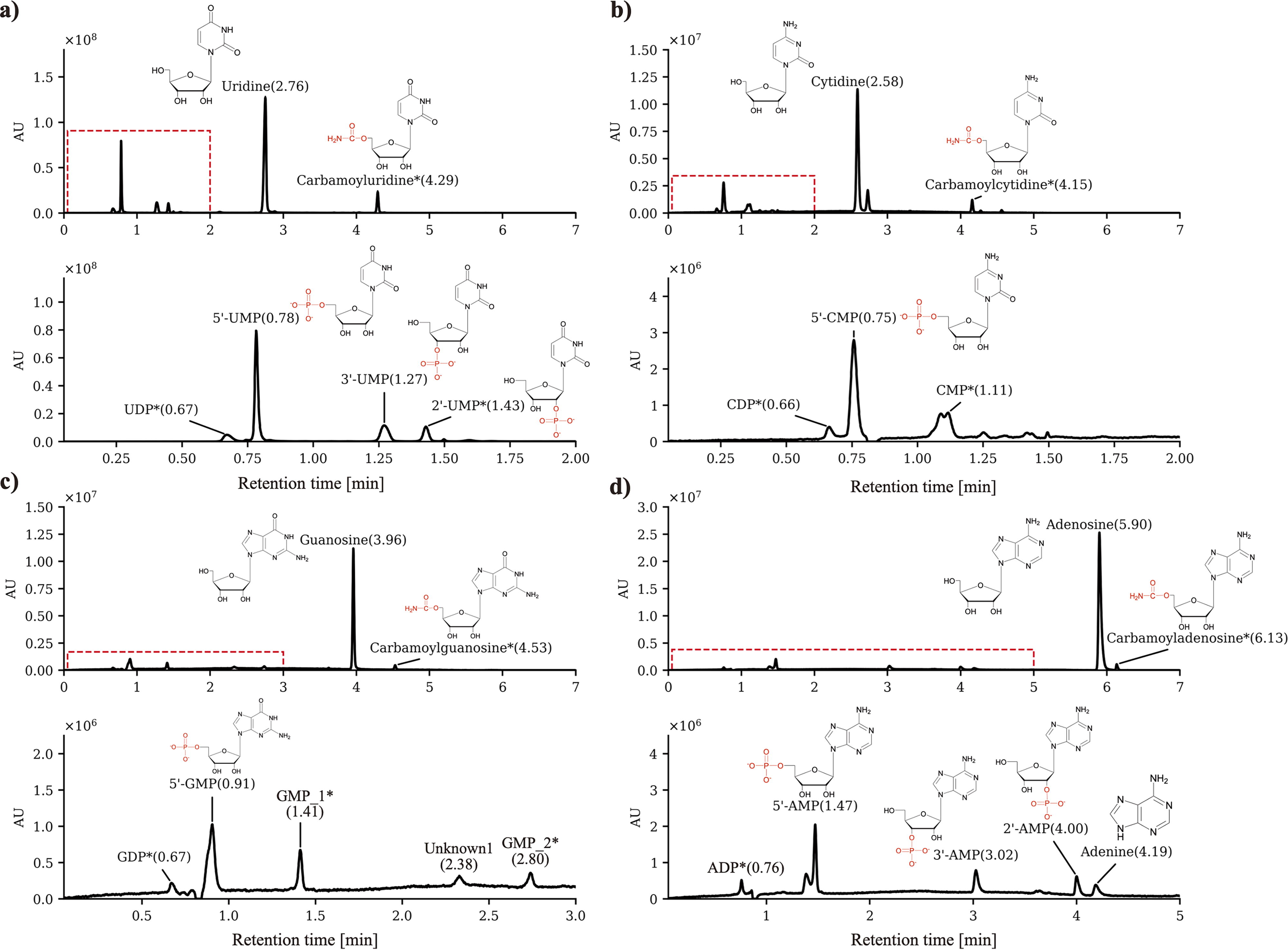
We first performed a nucleoside phosphorylation experiment using four different nucleosides (uridine, cytosine, guanosine, and adenosine), each mixed with NaH2PO4 and urea in a hydrothermal reactor. The reactor was pressurized with LqCO2 to create a CO2/water volume ratio of 23.5:1 (Supplementary Figure S1a). After incubation for 120 h at 68.9°C, reactants were analyzed using UPLC. As a result, we were able to identify the production of various nucleoside monophosphates (NMPs), nucleoside diphosphate (NDP), and carbamoyl nucleosides (Fig. 2, Supplementary Figures S3, S4, S5, and S6). Each peak was further identified and annotated using downstream TOF-MS (Supplementary Figures S7 and Figure S8). We have listed all detected peaks and their mass-to-charge ratio (m/z) in Supplementary Table S2. For carbamoyl nucleosides, TOF-MS data were obtained for all four nucleotides. Overall, 5′-NMPs showed the highest yield independent of the type of nucleoside. This trend is consistent with previous wet–dry cycle and dry-up experiments simulating a land-based hot spring environment (Costanzo et al., 2007; Burcar et al., 2016; Kim et al., 2016). Among the four nucleotides, uridine showed the highest yield of 5′-NMP, reaching 15.4% yield (Supplementary Table S3). Hence, we further carried out various experimental conditions using uridine as a model compound for nucleoside phosphorylation. UMP isomers were identified with reference to the standards (5′-UMP and 3′-UMP), whereas additional peaks with a mass of UMP (most likely 2’-UMP), uridine diphosphate (UDP), and carbamoyluridine were detected using TOF-MS (Supplementary Figures S7 and Figure S8a). The synthesis of 2’,3′-cyclic mononucleosides (cNMPs) in aqueous conditions (100°C, 2–24 h) was previously reported (Lohrmann and Orgel, 1971). However, no cNMPs, such as 2’,3′-cNMPs, were identified under our experimental conditions. To compare the nucleotide yields from the negative control of ScCO2, we performed a similar experiment using gold tubes (Supplementary Figure S1b) to simulate a water-only environment (Table 1). Under this condition, UMP could not be detected even after 120 h. Next, to confirm the effect of urea as a catalyst, we also performed an experiment without adding urea and observed that only a small amount (<0.5% yield) of 5′-UMP was detected via UPLC. Since we were not able to observe carbamoyl nucleosides without urea, we speculated that the carbamoyl group is likely derived from urea. To confirm our prediction, we used four different nucleosides along with isotope-labeled 13C urea and performed ultrahigh-performance liquid chromatography-time of flight-mass spectrometry (UPLC-TOF-MS) analysis. Indeed, it showed that this carbamoyl group was from urea (Supplementary Figure S8a, S8e). During this reaction, it is predicted that urea decomposes into isocyanate and ammonia (Katsuura and Inagaki, 1966). To check this prediction, we checked the amount of ammonia in the resultant experimental sample using ammonia assay. Approximately 382.9 mM ammonia was present in the sample (Supplementary Figure S9). Carbamoyluridine have been reported as the result of heating a mixture including uridine, ammonium phosphate, ammonium chloride, ammonium bicarbonate, and 14C urea at 100°C (Lohrmann and Orgel, 1971). It is predicted that carbamoyl nucleosides were also synthesized under our reaction conditions.
Products Obtained After Uridine Phosphorylation With and Without Supercritical CO2 Phase and Urea
Products Obtained After Uridine Phosphorylation With and Without Supercritical CO2 Phase and Urea
Species predicted based on their mass. All experiments were conducted at 68.9 °C.
Gold tube.
N.D., none detected; ScCO2, supercritical CO2; UDP, uridine diphosphate.
Hap is regarded as a significant source of phosphate during the early stages of Earth’s formation. However, its low solubility in neutral-to-alkaline aqueous solutions makes it more difficult to release phosphoric acid from the mineral than in acidic solutions (Pasek et al., 2017). However, regardless of pH, an increase in pCO2 has been shown to contribute to the increased solubility of apatite due to the complexation of calcium by carbonate (Pan and Darvell, 2010; Pan and Darvell, 2009). The aqueous solution in contact with the ScCO2 fluid possesses a highly acidic pH (∼pH 3) due to high pCO2 (Craig, 2014). This high acidity likely results in the extraction of phosphate from Hap minerals. For this reason, we performed the phosphorylation of uridine by replacing NaH2PO4 with 50 mg Hap mineral with urea (200 mmol/kg) at 68.9°C. Initially, no production of UMPs was detected within the first 24 h. However, after 120 h of incubation, final values of 0.9% (5′-UMP), 0.6% (3′-UMP + 2’-UMP*), and 6.1% (carbamoyluridine) were verified (Table 2). Assuming that the low yield is due to the limitation of accessible phosphate, we next carried out a phosphate quantification assay to determine the amount of orthophosphate present in the solution. Fifty milligrams of Hap was incubated at 25°C and 68.9°C in an ScCO2–water environment with and without urea (200 mmol/kg). Samples were collected after 24 and 120 h of incubation, and orthophosphate was quantified using the QuantiChrom Phosphate Assay Kit. As shown in Figure 3, the orthophosphate concentration ranged between 1.9 and 4.7 mM, in which the presence of urea had an effect of lowering the concentration. Higher orthophosphate concentration was observed at 25°C (4.7 mM without urea, 3.6 mM with urea) than at 68.9°C (2.8 mM without urea, 1.9 mM with urea). Further elongation of incubation time to 120 h did not affect the final concentration, which suggests that dissolved orthophosphate concentration has reached an equilibrium after 24 h.
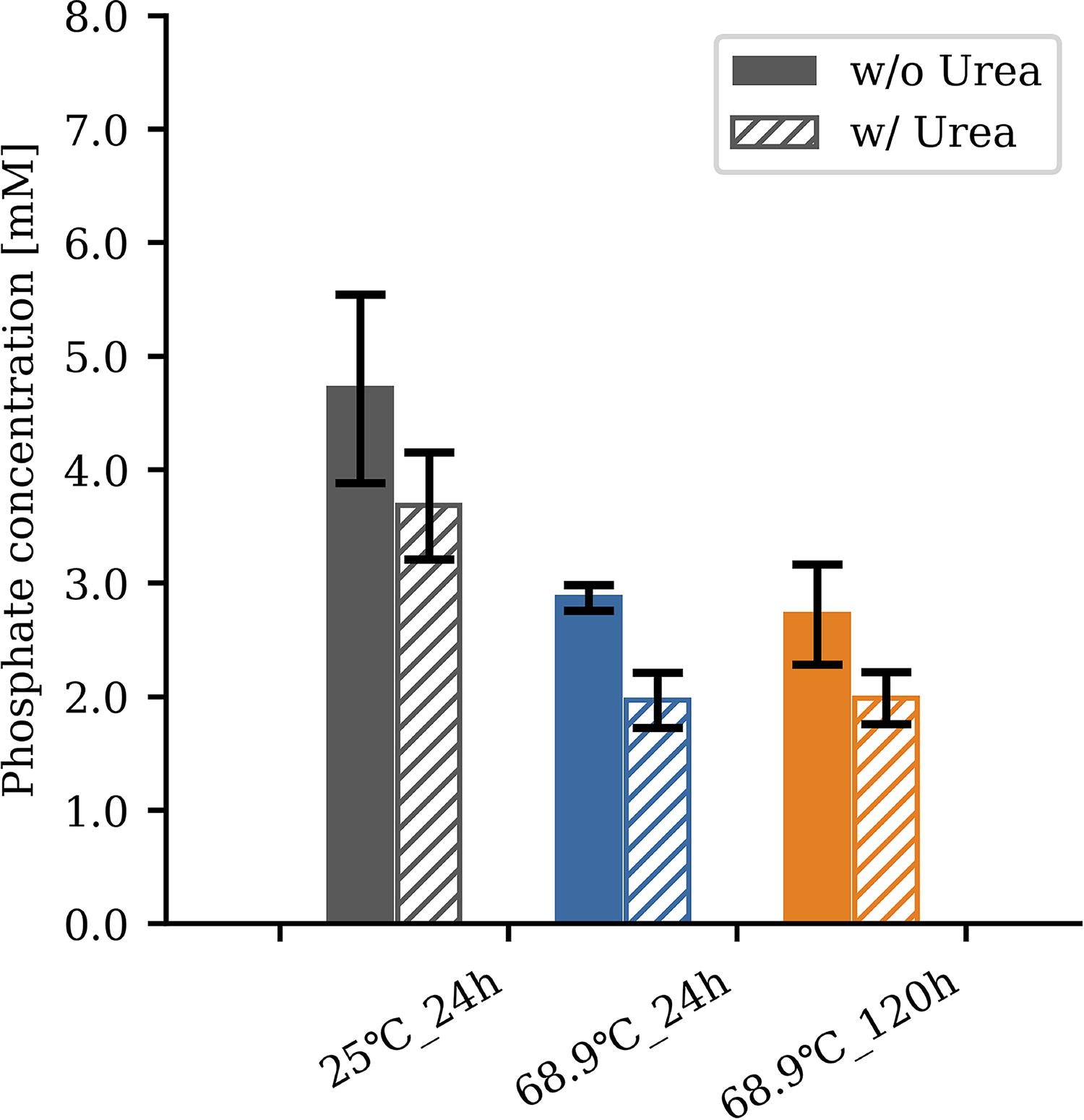
Quantification of orthophosphate released from Hap. The graph shows the estimated concentration of dissolved orthophosphate released from Hap minerals in an ScCO2–water environment. The experiment was conducted with 50 mg Hap with/without 200 mmol/kg urea at 25 °C for 24 h (gray), at 68.9 °C for 24 h (blue), and at 68.9°C for 120 h (orange). Hap = hydroxyapatite. Error bar represents the standard error obtained from a sample size of n = 3.
Products Obtained After Uridine Phosphorylation Using Hydroxyapatite (Hap) as a Phosphorus Source
Species predicted based on their mass. All experiments were conducted at 68.9 °C.
N.D., none detected.
Previous studies have shown a clear correlation between increasing temperature and the yield of phosphorylated products (Burcar et al., 2016; Lohrmann and Orgel, 1968; Schoffstall, 1976; Schoffstall et al., 1982). To investigate the effect of temperature on nucleoside phosphorylation, we performed a temperature gradient experiment ranging from 25°C to 94.1°C in an ScCO2–water environment for 24 h using 10 mmol/kg uridine, 30 mmol/kg sodium dihydrogen phosphate, and 200 mmol/kg urea. As a result, uridine phosphorylation was only detectable above 60.4°C, with the yield proportional to temperature (Fig. 4a). This trend was also consistent with other nucleotide isomers and carbamoyluridine (Fig. 4b). The pH of the aqueous phase was higher, after the experiments conducted under high-temperature conditions, compared with those conducted under low-temperature conditions, which indicates the degradation of urea (Supplementary Table S4). To verify whether phosphorylation of nucleosides in this reaction environment can be achieved even with a low initial concentration, an additional experiment was conducted at 94.1°C for 24 h using the starting materials at a 1/100 dilution (uridine 0.1 mmol/kg, NaH2PO4 0.3 mmol/kg, urea 2 mmol/kg). As a result, overall product yields decreased but remained within one order of magnitude (Supplementary Table S5).
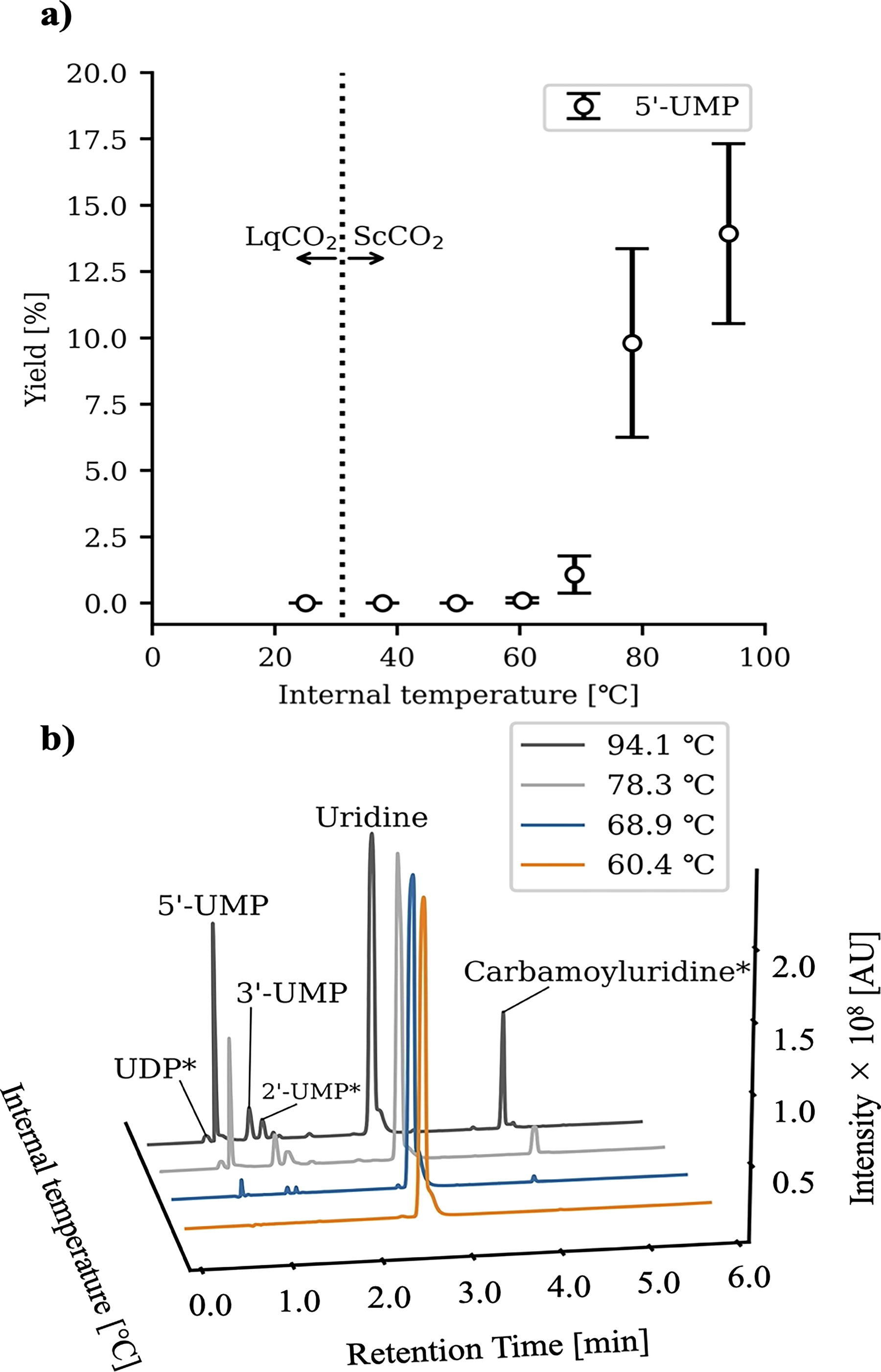
Temperature dependence of uridine phosphorylation.
To understand the duration needed to reach equilibrium, time course experiments up to 10 days (240 h) were conducted using uridine as a substrate, and the products were quantified by UPLC-TOF-MS. The yield of the four major compounds (5′-NMP, 3′-NMP, 2′-NMP, and carbamoyluridine) reached a maximum at ∼120 h and remained almost consistent after 240 h with a sign of slight degradation, which suggests that the reaction had reached equilibrium within 120 h (Fig. 5, Supplementary Table S6). 5′-NMP appeared to be the most abundant product, exceeding 2’-NMP and 3′-NMP, which is consistent with previous studies. However, we did not observe the formation of 2′,3′-cNMP even after 240 h. This feature was also consistent even in the high-temperature regime (78.3–94.1°C). Another outstanding feature was the production of carbamoyluridine, which constantly existed as the second most abundant product during the time course reaction (Fig. 5). As shown in Figure 2, carbamoyl nucleoside peaks were detected in all four nucleoside experiments. The urea origin of the carbamoyl moiety was further determined by isotope-labeled UPLC-TOF-MS analysis (Supplementary Figure S8a, S8e), suggesting that urea is first decomposed to isocyanate and NH3 and that isocyanate reacts with the nucleoside to form carbamoyl nucleosides.
UPLC chromatogram of nucleoside phosphorylation in the ScCO2–water two-phase environment. A concentration of 10 mmol/kg nucleosides (uridine, cytosine, guanosine, and adenosine) mixed with 30 mmol/kg NaH2PO4 and 200 mmol/kg urea showed uncharacterized species. The top graph shows the full UPLC chromatogram with an RT from 0 to 7 min, while the bottom graph shows an enlargement of the highlighted area shown with a red dotted line.
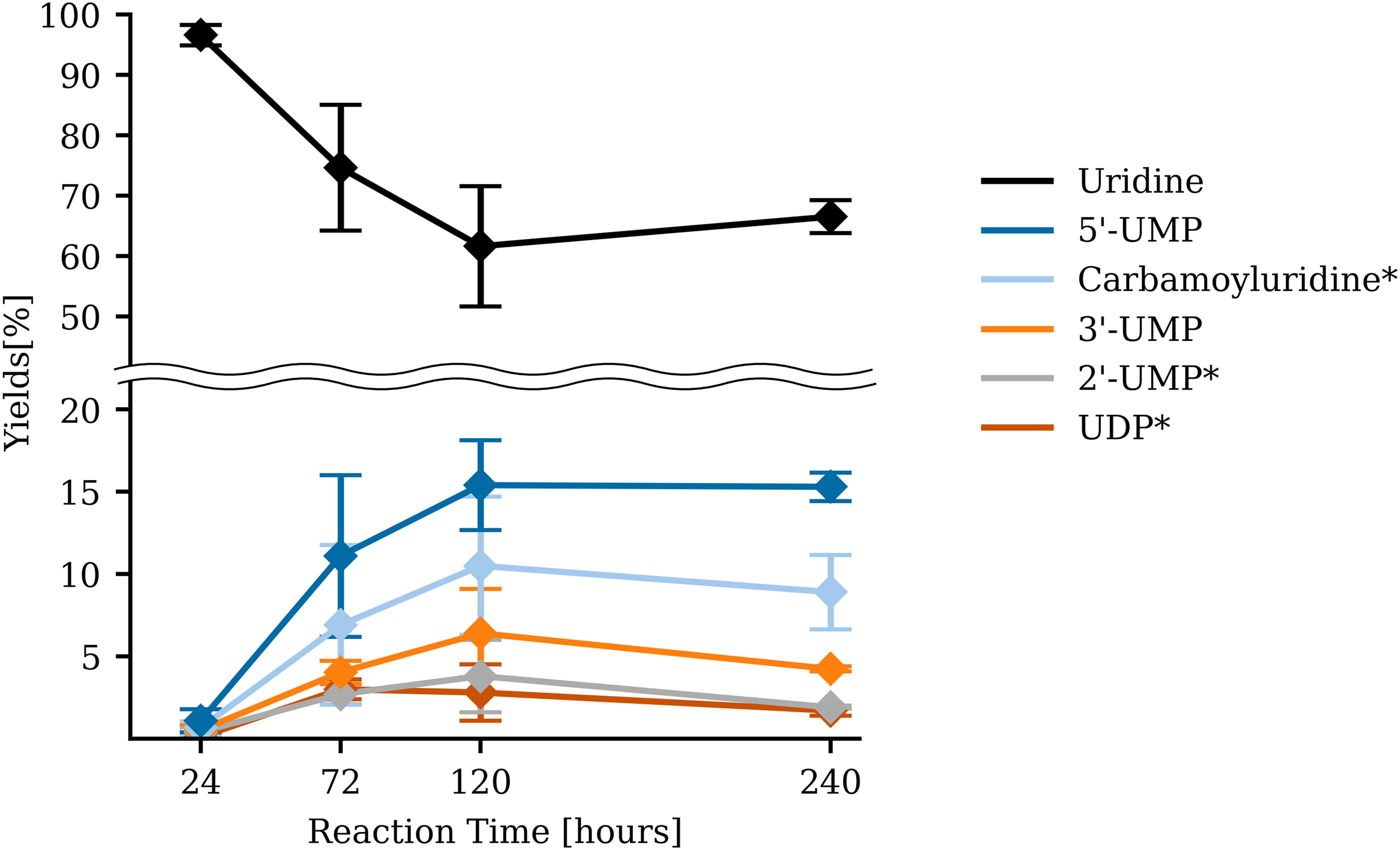
Time-series data of the synthesized products during uridine phosphorylation reaction. The sample was analyzed after 24, 72, 120, and 240 h. The initial substrate is uridine (black), 5′-UMP (dark blue), carbamoyluridine* (light blue), 3′-UMP (orange), 2’-UMP* (gray), and UDP* (brown). Asterisk (*) indicates the predicted compound based on the m/z determined by UPLC-TOF-MS. UDP = uridine diphosphate. Error bar represents the standard error obtained from a sample size of n = 3.
To understand the behavior of water and CO2 fluid during the incubation, we performed 24-h recording of two-phase using the glass window hydrothermal reactor. As presented in the Supplementary Video S1, we were able to confirm the CO2 fluid interface inside the reactor. Beyond the critical point, constant water vapor created an opaque cloud in the upper zone of the reactor filled with ScCO2. As the temperature increased, condensed water droplets started to form on the inner glass wall. Simultaneously, the water level decreased to approximately half of its original volume, and afterward, constant small fluctuation of water level were observed due to the continuous influx and efflux of water by evaporation and condensation. A recondensed droplet is most likely pure water (Bischoff and Pitzer, 1989; Takenouchi and Kennedy, 1965), and it serves as an ideal analog of the water droplet that can be formed in the low-temperature range of a CO2 pool within the cavity of volcanic rock (Fig. 1b).
Discussion
Similarity of phosphorylation reaction between the ScCO2–water environment and the terrestrial environment
Previously, phosphorylation in the deep-sea environment has not been considered fully, aside from the recent insight on serpentinization of ultramafic rock as a source of reduced phosphate (Pasek et al., 2022). Here, we have experimentally demonstrated that the ScCO2–water two-phase environment in the deep sea can promote nucleoside phosphorylation. The type of phosphorylated products and their relative yields have shown features of similarity to that of previous studies simulating the nucleoside phosphorylation under a wet–dry environment or in nonaqueous solvents (Costanzo et al., 2007; Burcar et al., 2016; Furukawa et al., 2015). For example, in Figure 2, we found a similar trend in the product yield following the order of (5′-NMP > 3′-NMP > other isomers and NDP). Also, the overall yields were proportional to the temperature increase (Fig. 4). It has been reported that the water miscibility in CO2-rich phase increases with the increasing temperatures (Sabirzyanov et al., 2002; Spycher et al., 2003; Takenouchi and Kennedy, 1964; Wiebe and Gaddy, 1941). As expected, continuous escape of water into the ScCO2 phase was observed using the transparent glass window reactor (see Supplementary Video S1). The constant semiwet condition with the presence of more than half of the aqueous solution remaining inside the reactor indicates that a major driving force for nucleoside phosphorylation in our reactor system is not due to the increase in solute concentration. Rather, we consider that the ScCO2–water interface is creating a unique microenvironment for the reaction to proceed. This could explain why a water-only gold tube experiment did not yield any detectable reactants, while 1/100-diluted starting material in a two-phase environment can still provide sufficient yield of nucleotide isomers (Supplementary Table S5). A study based on interfacial tension measurements and molecular dynamics simulation indicated that water protrudes into the ScCO2 layer and vice versa, resulting in a less well-defined interface with a thickness of ∼0.8 nm providing a gradient of water density (Da Rocha et al., 2001; Tewes and Boury, 2005). Hence, organic compounds such as nuclesides and nucleotides with an affinity to both water and ScCO2 might be attracted to the interface (Fig. 6b). A low number of water molecules at the interface could be considered as semidry, a condition driving condensation and simultaneously preventing newly formed compounds from hydrolysis, which is similar to the environment with eutectic (Burcar et al., 2016; Gállego et al., 2015; Gull et al., 2014) and nonaqueous solvents (Furukawa et al., 2015; Schoffstall, 1976). It is important to note that, in the natural environment (an open system), the constant escape of water from the droplet volume will greatly increase the solute concentration, resembling that of a dry-down experiment on land.
Characteristics of a water droplet in the geological setting.
Another observed similarity to the previous wet–dry or dry-down experiment is the positive effect of urea for the reaction. Urea has been previously used as an organocatalyst to enhance prebiotic phosphorylation (Burcar et al., 2016; Lohrmann and Orgel, 1968, 1971; Powner et al., 2009). As shown in Table 1, the yield of phosphorylated products was relatively higher compared with that of urea (-) experiments, which indicates that urea is indeed capable of serving as a catalyst in the ScCO2–water two-phase environment. While urea supply in the deep sea is a separate topic to be considered, it has been shown that nitric (HNO3) or nitrous (HNO2) acid can be reduced to NH3 near hydrothermal vents by molybdenum sulfide (Li et al., 2017; Nishizawa et al., 2021), and further urea can be synthesized from NH3 and CO using sulfide reduction in aqueous solution (Kitadai et al., 2022).
As a unique feature of the ScCO2–water two-phase environment, we could not detect any cyclic nucleotides under any of our experimental conditions. Previously, it has been shown that prolonged incubation at high temperature (95°C) significantly increased the formation of 2′,3′-cNMP (Reimann and Zubay, 1999). Furthermore, nucleoside phosphorylation in formamide has also been shown to lead to the formation of various cNMP species (Costanzo et al., 2007; Schoffstall, 1976; Schoffstall et al., 1982; Schoffstall and Laing, 1985), but it was later shown to be strictly prohibited with regard to borate minerals (Furukawa et al., 2015). Because cNMP can be considered as dehydrated 2′- and 3′-NMPs, we assume that the presence of water prohibits NMPs to further convert into cNMPs. cNMPs were not detected even under our two most expected low water activity conditions Figure 4 (94.1°C, 24 h) and Figure 5 (68.9°C, 240 h). It would seem that an ScCO2–water environment does not exhibit an extremely “dry” condition compared with that of dry heating and nonaqueous solvents. No significant difference in total yield was observed with or without stirring the aqueous solution (Supplementary Table S7).
Presence of carbamoyl nucleosides and the possible reaction mechanisms
Throughout our experiment, carbamoyl nucleosides were constantly observed as the second most present nucleoside-derived compounds (Supplementary Tables S2 and S3). To verify the source of carbamoyl moiety, we performed UPLC-TOF-MS along with isotope-labeled 13C urea and confirmed that carbamoyl moiety of carbamoyluridine is derived from urea (Supplementary Figure S8e), although the exact position of carbamoyl moiety on uridine is yet to be solved. Urea decomposition leads to the formation of isocyanate and ammonia, and isocyanate further undergoes hydrolysis to ammonia and carbon dioxide (Chin and Kroontje, 1963; Randall et al., 2022; Shaw and Bordeaux, 1955; Zhu et al., 2021). It is known that the decomposition of urea is facilitated by an increase in temperature (Randall et al., 2022; Shaw and Bordeaux, 1955), and a lower pH is considered to have a faster kinetic rate with respect to the hydrolysis of isocyanate (Borduas et al., 2016). Since we also observed pH becoming less acidic along with an increase of the temperature (Supplementary Table S4), we expect that urea decomposition has proceeded under the condition that we observed the carbamoylnucleosides (>60.4°C, >15 h). Interestingly, Lohrmann and Orgel (1971) have also reported the formartion of carbamoylnucleoside during the urea-catalyzed phosphorylation reaction, which they concluded as a side product. Nevertheless, the presence of carbamoylnucleosides suggests that urea-driven carbamoylation of organics also proceeds in the ScCO2–water environment.
Previously, two different phosphorylation reaction mechanisms that involve urea have been proposed as shown in Figure 7. The first model suggests that urea undergoes condensation to form a reactive urea-phosphate intermediate (Burcar et al., 2016; Lohrmann and Orgel, 1968; Powner et al., 2009). Another model assumes that isocyanate derived from urea reacts with phosphate to form a reactive carbamoyl phosphate intermediate (Katsuura and Inagaki, 1966). While both reaction paths are possible in our system, we propose a third scenario where carbamoyl nucleosides could also potentially serve as an intermediate (Fig. 7). The fact that products are more efficiently formed between 24 and 72 h than the first 24 h (Supplementary Table S6) had us speculating that isocyanate formed by urea decomposition may play a major role in nucleoside phosphorylation in our system (Model 2 or 3 in Fig. 7). However, characterizing the thermodynamics of carbamoyl nucleosides as well as exploring the exact reaction sequence is beyond the scope of this study due to the complexity of the two-phase environment. Hence, approaches such as quantum chemical calculations would be of interest for future studies.
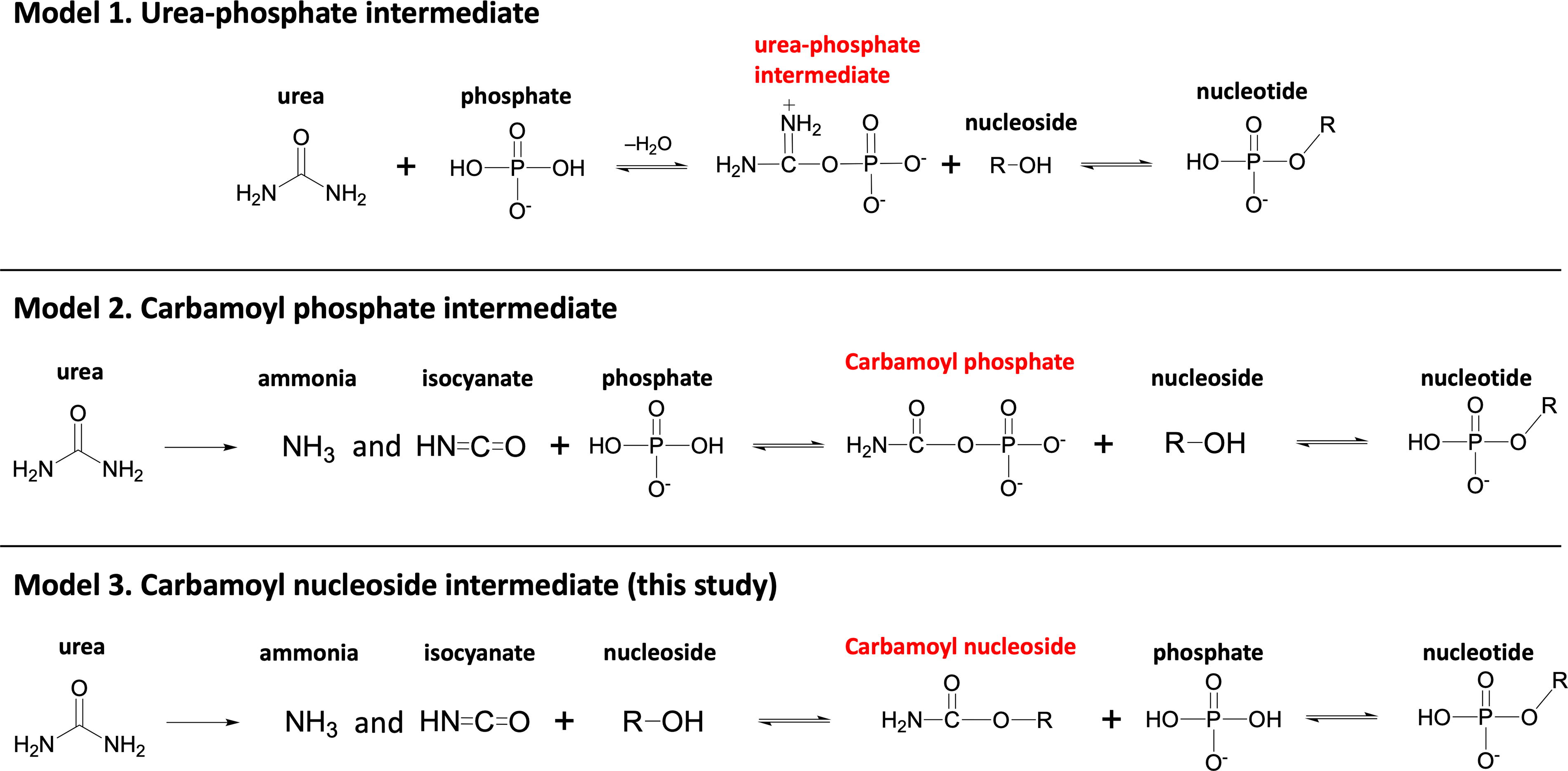
Possible reaction models for nucleoside phosphorylation using urea as a catalyst or reactant.
We also conducted uridine phosphorylation using Hap as a main P-source (Table 2). The most abundant naturally occurring P-containing mineral in Earth’s crust is apatite [Ca5(PO4)3(F, Cl, OH)] (Paytan and McLaughlin, 2007), and Hap, which is also considered to have been one of the most common phosphate minerals during the Hadean (Hazen, 2013). Some predicted that the pH of an aqueous solution coexisting with ScCO2 fluid becomes highly acidic. The pH in this condition is predicted to be ∼3.2–3.4 (Craig, 2014). In this acidic pH range, Hap dissolves in water (Lundager Madsen, 1975; Pasek et al., 2017; Walton et al., 2021). Although the reaction field in this study is expected to be under acidic conditions, the high pCO2 suggests that the solubility of apatite can be affected by the presence of carbonate ions (Kakegawa et al., 2002; Walton et al., 2021). The solubility effect of carbonate and bicarbonate ions on the calcium phosphate mineral (apatite) is expected to be greater in an alkaline hydrothermal environment (Toner and Catling, 2020), which is indicative of the high orthophosphate concentration in Enceladus’s ocean (Postberg et al., 2023). However, because the hydrolysis rate of phosphoester bonds is higher at alkaline pH (Kluger et al., 1969), unless an environment with low water activity exists, a constant yield of phosphorylated compounds would be difficult. In fact, ribose phosphorylation with apatite in a carbonate- or formate-rich environment has tended to show lower yields of ribose phosphate at high pH (Takabayashi et al., 2023). Thus, it is important to understand whether an ScCO2–water environment can dissolve orthophosphate from Hap and drive a phosphorylation reaction. At the very least, under our experimental conditions with excess amounts of Hap, we were able to observe few milimolar level orthophosphate existing in an aqueous solution after 24 h (Fig. 3), which is sufficient to produce a detectable amount of UMP isomers after 120 h (Table 2). While yields were significantly lower than that when using NaH2PO4, as a P-source, the trend was consistent with previous studies showing that, when Hap was used, the overall yield of nucleotide isomers decreased compared with the inorganic phosphate, while several mineral phosphates tended to show better yield than Hap (Burcar et al., 2019; Costanzo et al., 2007). We also found that the amount of phosphate dissolved from Hap at 25°C was higher than at 68.9°C (Fig. 3). This result is in line with previous results attained when simulating the equilibrium solubility of Hap in the presence of a calcite buffer, considering the effects of temperature and CO2 pressure (Toner and Catling, 2020). No difference was observed in free phosphate concentration from apatite dissolution experiments at 68.9°C for 24 and 120 h (Fig. 3). Although we were not able to perform apatite dissolution experiments at longer times, we consider that the amount of apatite dissolved had approximately reached equilibrium. Dissolved phosphate was slightly affected by the presence of urea shifting the pH higher, but the change in the overall concentration was <1 mM (Fig. 3).
Geological location and microscopic feature of the water droplets within ScCO2 fluid
According to a previously proposed model (Shibuya and Takai, 2022), we infer that the concentration and condensation of solutes in acidic water droplets can occur due to the water that escapes into the ScCO2 phase in subseafloor environments (Figs. 1 and 6). In the seafloor, temperature gradients exist as a function of the distance from the hydrothermal vents (Miyoshi et al., 2019), while the amount of water that dissolves into the Lq/ScCO2 pool increases along with the temperature (Sabirzyanov et al., 2002; Spycher et al., 2003; Takenouchi and Kennedy, 1964; Wiebe and Gaddy, 1941). Hence, phosphorus fluxes in the prebiotic ocean could have been regulated by hydrothermal activity associated with carbonated oceanic crust, which might have enriched phosphorus in volcanic rocks (Kakegawa et al., 2002; Rasmussen et al., 2021). CO2-saturated acidic water droplets have the potential to dissolve these phosphate minerals due to their acidity in the Lq/ScCO2 pools. According to the results in Figure 3, the amount of dissolved apatite was higher in the lower temperature environment, which suggests that the phosphate concentration could be different in each temperature range due to the geothermal gradient under the seafloor (Fig. 6a).
While synthesis of nucleosides in the deep sea has not yet been reported, syntheses of simple organic compounds and biomolecules in submarine hydrothermal environments have been reported (Huber and Wächtershäuser, 1997; Imai et al., 1999; Keller et al., 2014; Kitadai et al., 2019; Lemke et al., 2009; McDermott et al., 2015; Simoneit, 2004). Thus, we can expect that the phosphorylation of relatively simple organic compounds is likely to proceed in the ScCO2–water environment, that is, given the fact that sub-milimolar concentration of nucleoside, orthophosphate, and urea is sufficient to drive phosphorylation (Supplementary Table S5), a possible reaction field in which a high concentration of phosphate is acquired in the lower temperature zone and increasing temperature (Fig. 7).
Solubility and localization of organics in ScCO2–water environment
ScCO2 has been known as a green solvent for a wide range of hydrophobic compounds, including biological, pharmaceutical, and noncharged organometallic compounds (Škerget et al., 2011). The solubility of uracil has been reported with a mole fraction solubility of 0.878 × 10−8 at 15 MPa and 65°C (Zhou et al., 2008). However, no solubility data in ScCO2 are available for nucleotides, nucleosides, ribose, or phosphate. Some of these compounds could be attracted to the ScCO2–water interface but require spectroscopic analysis using the glass window reactor to provide further evidence. Chemical modification such as acetylation has been shown to enhance sugar solubility due to Lewis acid–Lewis base interactions between CO2 and the acetyl group (Ma et al., 2010). Hence, if such reaction proceeds at the ScCO2–water interface, it will dynamically change the solubility as well as partitioning of the organic molecules across different phases.
Observation of the Lq/ScCO2–water two-phase system
During the initial incubation below the critical point (31.1°C, 7.38 MPa), small bubbles were produced from both the LqCO2 and water. This behavior is likely due to the phase transition of LqCO2 (Meyers and Van Dusen, 1933) and dissolved CO2 in the aqueous solution (Spycher et al., 2003; Wiebe and Gaddy, 1939). After surpassing the critical point, opaque cloud started to form in the cooler upper zone of the reactor, given the vertical temperature gradient caused by the oil bath heating (Supplementary Figure S1). This phenomenon can be explained by the dissolution of water vapor into the ScCO2 phase (Sabirzyanov et al., 2002; Spycher et al., 2003; Takenouchi and Kennedy, 1964; Wiebe and Gaddy, 1941). Hence, the continuous cycle of water droplet formation and evaporation across the temperature gradient clearly indicates that the dynamic wet–dry process is a fundamental feature that occurs in the deep-sea CO2 fluid pool, both in the past and present Earth.
Conclusion
We investigated the effects of the ScCO2–water two-phase environment on the phosphorylation reaction using nucleoside as a model organic compound and by simulating the deep-sea pressure and temperature using a hydrothermal reactor system. Two vital functions were discovered in this environment. First, the presence of ScCO2 results in an effect on concentration and low water activity. Second, nucleoside phosphorylation was solely verified in the ScCO2–water two-phase environment, not in the water-only environment. Our speculation is that an interface between ScCO2 and water serves as a water-scarce microenvironment that is further enhanced by the continuous water evaporation observed when using the glass window reactor, thereby altering the thermodynamic equilibrium in favor of the production of phosphorylated organic molecules. Moreover, the pH of the aqueous solution coexisting with ScCO2 fluid becomes acidic with an increase in pCO2, which results in the release of metal cations and anions from the contact minerals. In this study, we simulated the ScCO2–water fluid within Earth’s crust by using an excess amount of Hap. We confirmed that orthophosphate can reach concentration in milimolar order at 68.9°C and 11–12 MPa. This dissolved phosphate was successfully employed for nucleoside phosphorylation. Finally, an increased yield of products was observed as the temperature increased to 94.1°C. 5′-NMP was consistently overproduced compared with 3′-NMP and 2’-NMP, while no cyclic forms were observed, in contrast to the previous wet–dry cycle experiments simulating terrestrial hot springs/warm little ponds. Finally, the production of carbamoyl nucleoside highlights the potential of isocyanate chemistry. To conclude, this work provides clear experimental evidence that wet–dry cycling-driven chemistry can also occur within the ScCO2–water environment in the deep sea. From an astrobiology perspective, it is therefore interesting to consider the possibility of nonequilibrium wet–dry cycling on ocean world planets that are completely covered by ocean.
Footnotes
Acknowledgments
The authors thank Dr. Ken Takai for discussion about phase separation in the ScCO2–water system and geological environments, Dr. Shawn McGlynn and Dr. Tomoaki Matsuura and Dr. Ryuhei Nakamura for discussions about nucleoside phosphorylation under the ScCO2–water two-phase environment, Dr. Matthew A. Pasek for discussing the phosphate minerals, and Dr. Shuya Tan for discussing the thermodynamics of nucleoside phosphorylation under the ScCO2–water two-phase environment. The authors also thank Mr. Mikhail Makarov for support with the phosphate assay, Dr. Kesuke Aratsu and Ms. Kumiko Nishiuchi, and Dr. Tien-Tzu Liang for support with UPLC-TOF-MS analysis, and Ms. Nozomi Adachi, Mr. Shota Suzuki, and Ms. Nazugumu Maimaiti for support with experiments.
Authors’ Contributions
K.F. conceptualized this research. S.T. performed the experiments mainly, in which R.H. and K.M. provided support. All authors participated in data analysis and discussion about the results. S.T. and K.F. wrote this paper with assistance from all authors.
Author Disclosure Statement
The authors do not have any competing interest statements.
Funding Information
This work was supported by
Supplementary Material
Supplementary Data S1
Supplementary Video S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Associate Editor: Nita Sahai
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.