
Abstract
Life on Earth relies on mechanisms to store heritable information and translate this information into cellular machinery required for biological activity. In all known life, storage, regulation, and translation are provided by DNA, RNA, and ribosomes. Life beyond Earth, even if ancestrally or chemically distinct from life as we know it, may utilize similar structures: it has been proposed that charged linear polymers analogous to nucleic acids may be responsible for storage and regulation of genetic information in nonterran biochemical systems. We further propose that a ribosome-like structure may also exist in such a system, due to the evolutionary advantages of separating heritability from cellular machinery. In this study, we use a solid-state nanopore to detect DNA, RNA, and ribosomes, and we demonstrate that machine learning can distinguish between biomolecule samples and accurately classify new data. This work is intended to serve as a proof of principal that such biosignatures (i.e., informational polymers or translation apparatuses) could be detected, for example, as part of future missions targeting extant life on Ocean Worlds. A negative detection does not imply the absence of life; however, the detection of ribosome-like structures could provide a robust and sensitive method to seek extant life in combination with other methods. Key Words
Introduction
Life is commonly defined as a “self-sustaining chemical system capable of Darwinian evolution” (Benner, 2010; Joyce, 1994; Luisi, 1998; Neveu et al., 2018). In all known forms of life, Darwinian evolution occurs via heritable changes encoded into molecular features (i.e., nucleic acids), which are then translated into cellular machinery and other proteins. These products of translation are responsible for a variety of biological functions within and outside the cell, such as maintenance, growth, reproduction, regulation, structure, storage, and transport.
In all known life, the ribosome is integral to the “central dogma of biology,” which traces the flow of genetic information into organismal structure and function: genetic information is stored in DNA, which is transcribed into RNA, which is then translated into protein. Specifically, the ribosome is responsible for the process of translation, using mRNA-encoded information to synthesize proteins. All known life is descended from a common ancestor (Pace, 2001), and the structure and genetic code of the ribosome are highly conserved across evolutionary time and the tree of life. Indeed, the most highly conserved sequences across biological systems correspond to 16S and 23S ribosomal subunits and transfer RNAs, and these sequences have experienced little change in the ∼3.5–4 billion years since life evolved (Bray et al., 2018; Isenbarger et al., 2008). Ribosomes are found within all cells regardless of place in the tree of life or level of activity, and they are instrumental for a cell’s ability to carry out life-sustaining processes such as growth, reproduction, and metabolism (Bernstein and Baserga, 2004; Bowman et al., 2020).
This conservation of ribosome-associated genomes has been posited as evidence for the existence of an RNA-protein world before DNA became the dominant form of nucleic acid-based information storage (Fournier et al., 2010; Goldman et al., 2010; Harish and Caetano-Anollés, 2012; Hsiao et al., 2009; Jerome et al., 2022). This “RNA world” model for the origin of life posits that RNAs may have acted as both heritable material and cellular machinery before the evolution of the ribosome or other more sophisticated biological structures. This feature persists today, with RNA molecules being capable of both storing genetic information and catalyzing biochemical reactions. However, there is good reason to assume that life beyond Earth or an alternate origin of life could utilize a heritable storage system that is physically distinct from translation-performing cellular machinery, as discussed in Section 2.1.
Translation-performing molecules similar to ribosomes could function as an agnostic biosignature that could provide evidence for “life as we don’t know it” that may not share a common origin or physiochemical basis with life on Earth. Given the uncertain range of possibilities for alien biochemistry, morphology, and other characteristics, various methodologies to search for nonterran life-forms have been proposed that are not specific to known biochemistry. Such strategies can identify potential agnostic biosignatures via direct detection, such as using DNA probes (aptamers) to bind to repeating patterns within compounds (Johnson et al., 2018). They can also be elucidated by analyzing observed molecules: for example, the probability that a molecule of a certain complexity will form abiotically can be quantified (Marshall et al., 2021), and identification of such molecules can be accomplished via mass spectrometry (Chou et al., 2021). Such methods generally rely on the assumption that observation of abundant molecules with sufficiently high complexity can indicate a biotic origin.
Molecules with specific structural or functional features have been proposed as potential agnostic biosignatures. Such features are generally analogous to what is known about terran life without presupposing any origin or biochemical basis. For example, information-encoding charged linear polymers could serve as a mechanism to store, regulate, and transmit genomic information, analogous to nucleic acids on Earth (Benner, 2017). We extend this point and argue that the “Darwinian evolution” definition of life not only implies a mechanism to store heritable information but further necessitates a mechanism to translate this information into material that is useful for the biological system. In terran life, this need is satisfied by the ribosome, a molecule that is composed of RNA and proteins and is responsible for using genetic information to synthesize polypeptides and proteins through a process known as translation. The ribosome has a very specific structure and size (although it is important to note that eukaryotic ribosomes are 25–50% larger in diameter), and we similarly argue that nonterran life may use a molecule of similar structure and size. In our work, we recapitulate the structure of the central dogma of biology by examining DNA, RNA, and ribosomes via solid-state nanopore (SSN) instrumentation. We also describe the case for targeting the translational apparatus as a biosignature, and we demonstrate the ability to target our only known example, the ribosome, using SSNs. Finally, we show that machine learning (ML) can discriminate between different classes of biomolecules (DNA, RNA, and ribosomes) based on the features associated with their SSN detections.
The Case for the Translational Apparatus as a Biosignature
What would life look like without translation?
Any life with separate heritable material and cellular machinery must have a translation system to convert instructions into machinery. If these functions are not separate, then heritable material must also act as machinery, to include both self-copying and carrying out all other life functions (metabolism, maintenance, growth, evolution). While ribozymes have been discovered and engineered that are capable of copying short RNA sequences (Been and Cech, 1988; Ekland and Bartel, 1996; Horning and Joyce, 2016; Sczepanski and Joyce, 2014), and RNA sequences can be inherited via viral replication (Rampersad and Tennant, 2018), we are not aware of any truly self-replicating informational polymer system of nontrivial complexity (i.e., capable of replicating nucleic acid strands longer than tens of bases). Furthermore, such systems would be unlikely to competitively support the chemical richness required to subsume all the roles of cellular machinery (e.g., an RNA-only RNA world in the absence of proteins). In other words, if such a system evolved, it would likely be displaced by a system with separate coding and machinery due to selective pressures, just as may have happened with life as we know it (Gilbert, 1986; Orgel, 1968).
Separation of coding and machinery would decouple conflicting functions. To perform a wide range of complex functions, cellular machinery requires chemical diversity, whereas enabling copying requires chemical simplicity to facilitate copying robustness. Even while RNA can serve as both as an informational storage molecule and as cellular machinery (as evidenced by ribosomes, tRNAs, and other ribozymes), a physically separate translation apparatus might be expected to arise because decoupling these two functions offers significant benefits, as observed by these functions being decoupled in all known life.
Furthermore, separation among classes of functional biomolecules allows for their maintenance and enables structural differences that align with function. For example, linear charged polymers are considered a potential universal heritable information storage format for aqueous life (Benner, 2017). In terran life, such polymers exist as nucleic acids, which store genetic information in linear base sequences. Their topology contributes to copying fidelity: branched or otherwise more complicated structures would pose difficulties for effective copying. In addition, the charge of nucleic acids separates the properties of the molecule from its information content, so that the molecule can be effectively copied regardless of information content. Conversely, the ribosome has a complicated and specific structure that aids in its preservation and enables its highly specialized role in translation. The assembly of two types of biopolymers (polynucleotides and polypeptides) yields a functional and stable ribosome capable of conducting its important catalytic role, leveraging the qualities of both nucleic acid and protein to enable translation (Runnels et al., 2018). The differing charges across the ribosome similarly allow it to function by enabling compatibility with the biomolecules that interact with various parts of the ribosome (Leininger et al., 2021). Furthermore, self-complementarity (the propensity for a biomolecule to bind with itself) among biopolymers enables self-preservation and functional separation of different types of biomolecules. Meanwhile, heterogenous assemblages of different types of biopolymers can beget more complex functionality and “partner-preservation,” where mutualistic assemblies of different biomolecule types can confer protective effects on each other (Runnels et al., 2018). All known life requires a translation apparatus. However, let us consider life that does not have this layer of abstraction, such that hereditary molecules also serve as cellular machinery. As alluded to above, this introduces complexity into copying: changes in information content would result in changes in machinery, which could overly constrain such life’s exploration of evolutionary space. In short, life without translation would be limited in complexity, capabilities, and evolvability (Cuevas-Zuviría et al., 2023), the latter of which is central to our definition of life.
Detection approaches
A classical approach for the detection and identification of bacteria and archaea involves using polymerase chain reaction to amplify and sequence the gene encoding for 16S ribosomal RNA (rRNA), which is highly conserved across prokaryotes (Isenbarger et al., 2008). This genomic approach is integral for the study of microbial communities on Earth (Kim and Chun, 2014), since it targets a genomic sequence that exists in nearly all bacteria and archaea, or for some choices of primers, certain large clades of organisms. Similarly, the eukaryotic 18S rRNA gene can be used to identify eukaryotic community members. Ribosome profiling techniques can be used to sequence mRNA fragments that are being actively translated, which enables the study of translation with single-cell resolution (Heiman et al., 2008; VanInsberghe et al., 2021). However, targeting specific ribosome-associated RNA sequences is only suitable for seeking life-forms that are ancestrally related to known life. One instance where this may be relevant would be in the search for martian microbial communities that share an origin with life on Earth, for example, transferred between the planets via lithopanspermia (Carr, 2022; Mileikowsky et al., 2000).
Analysis of rRNA can be used to identify active taxa and assess activity in environmental microbial communities, although caveats exist for these use cases. The relationship between rRNA concentrations and microbial activity is complex; therefore, simple correlations do not often exist between the two, and any existing relationships are not consistent across taxa (Blazewicz et al., 2013). Furthermore, in certain environmental conditions (e.g., low temperature, low water activity, high osmolarity), the rRNA itself may persist after environmental conditions have changed, influencing conclusions about which specific microbes are actually currently active in an environment (Schostag et al., 2020). Although drawing explicit conclusions about which microbial taxa are currently active is difficult from rRNA analysis alone, detection of ribosomal material can serve as an indicator that microbial activity has occurred recently or is actively underway in a habitat.
Content-agnostic approaches can be used to detect ribosomes without making assumptions about their genetic associations. Microscopy techniques, including fluorescence microscopy and cryoelectron microscopy, can provide information about the ribosomal structure and morphology, location and organization within cells, and study of translation (Bai et al., 2013; Bakshi et al., 2012; Fu et al., 2011; Murphy et al., 2020). However, such technologies are destructive, require extensive sample preparation, or are susceptible to image artifacts. In addition, the size, sensitivity, and complexity of these instruments present difficulties for adapting these types of microscopes for in situ space exploration applications.
Dynamic light scattering (DLS) can be used to characterize molecules in an aqueous sample by measuring their diffusion coefficient, which in turn can be used to infer molecule size and shape (Stetefeld et al., 2016). For example, DLS can be used to interrogate ribosomal shape (Bruining and Fijnaut, 1979), track structural changes (Patel and Cunningham, 2002), and make comparisons between ribosomal types (Müller et al., 1986). DLS is a good strategy for making accurate size and shape particle measurements in minutes with a small amount of sample (<3 µL). In addition, sample preparation is minimal, and this technique is compatible with high-concentration and turbid samples. However, measurements are sensitive to temperature and solvent viscosity, and DLS is not capable of achieving sufficiently high resolution to distinguish between closely related molecules, such as monomers versus dimers. Finally, DLS is sensitive to changes in size and concentration (Jose et al., 2019). Overall, DLS is a good technique for interrogating pure ribosomal samples at high concentration but is not viable for identifying or characterizing ribosomes within a heterogenous sample or when ribosomes are present at low abundance.
SSNs can provide a useful method for ribosomal detection with single-molecule or single-particle resolution. This instrumentation detects individual molecules or particles (hereafter referred to in the generic sense as particles) in an aqueous sample of known conductivity by applying an ionic current to a nanometer-scale pore in a synthetic membrane, using a voltage or pressure differential to pass particles through the pore, then measuring how the particles disrupt the ionic current (Fig. 1A). The magnitude of the change in conductance and duration of the blockage can then be used to determine the relative size of each particle. SSN instrumentation is distinct from biological nanopore sequencing (e.g., the MinION from Oxford Nanopore Technologies), which uses controlled translocation of nucleic acids regulated by biological components with specific base sequences inferred from changes in ionic current flow. Instead, due to its larger, nonspecific pore, SSN tools can analyze a wide size range of particles and are not constrained to a specific class of molecules. This benefit is also a challenge to achieving molecular specificity and to achieving single-base resolution, as discussed below.
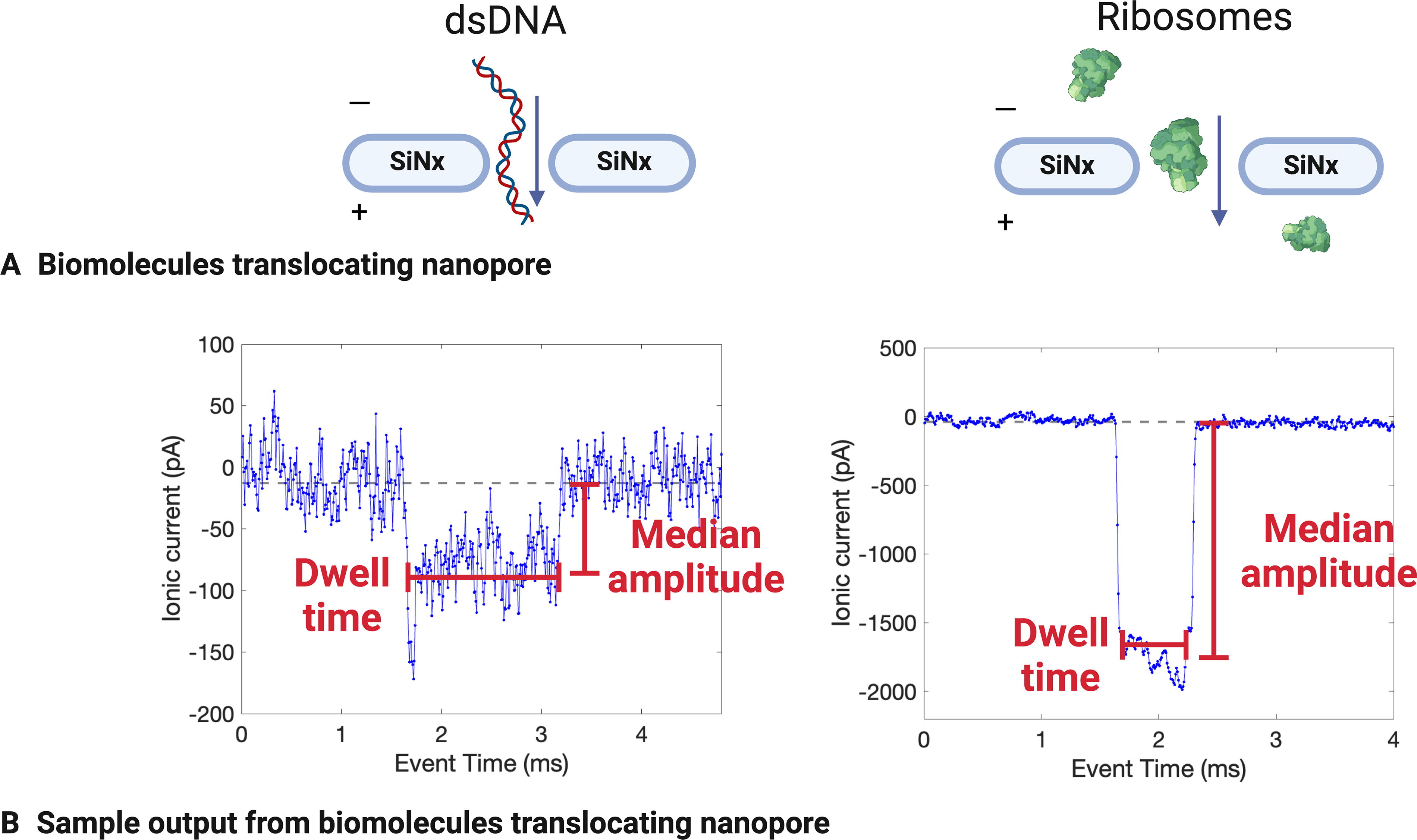
Examples of DNA and ribosome detection using a solid-state nanopore (SSN) in 2M LiCl. Molecules passing through the nanopore
In recent years, major advancements have been made in SSN technologies to improve their sensitivity, selectivity, and throughput capacity, which have enabled development of applications in biology, chemistry, and medicine (Xue et al., 2020). Particularly of interest to this work and other astrobiological applications, SSN-based methodologies have been used to detect biomolecules such as nucleic acids and ribosomes in laboratory conditions. Raveendran et al. (2020) used nanopipettes fitted with ∼60 nm nanopores on the tips to detect eukaryotic 80S ribosomes (derived from human and Drosophila melanogaster cell cultures) and distinguish ribosomes from polysomes (groups of ribosomes bound together by mRNA strands). Rudenko et al. (2011) used a nanopore-microfluidic chip to detect individual prokaryotic 50S Escherichia coli ribosomes and control their rate of nanopore translocation. Similarly, Rahman et al. (2019) used an optofluidic chip to control the flow of ribosomes, DNA, and proteins through a nanopore and measure these biomolecules as they translocated the nanopore. Xia et al. (2022) used a nanopore drilled into a silicon nitride membrane on a glass chip to detect and characterize DNA that was extracted from Mars analog soils, directly demonstrating SSN applications for astrobiological life detection.
ML can be applied to solid-state pore measurements to classify and identify biological particles, given the appropriate training data. Xia et al. (2021) applied multiple ML models to their SSN data sets to differentiate signals of monosaccharides and disaccharides, which enabled them to determine the composition of polysaccharide compounds. Arima et al. (2020) achieved over 99% accurate classification of viruses using ML. Using a molybdenum disulfide membrane, instead of the commonly used silicon nitride membrane, Farimani et al. used ML to identify individual amino acids within polypeptide chains (Farimani et al., 2018). On the micropore scale, ML can be similarly used to identify and classify cells. Using solid-state micropore data, Hattori et al. applied ML to discern between five clinically relevant bacterial species based on physical features, including cell shape, surface charge density, mass, surface proteins, and motility (Hattori et al., 2020). Tsutsui et al. tagged bacterial walls with synthetic peptides and analyzed them with a solid-state micropore, then used ML to identify bacterial features and discriminate between strains (Tsutsui et al., 2018).
SSNs are nonspecific and can be used to detect not just biomolecules such as nucleic acids and ribosomes but also nonbiological particles. Due to this nonspecificity, it has been proposed that SSNs may be useful for detecting agnostic biosignatures (Bywaters et al., 2017). It has also been proposed that alien life-forms with a separate origin and alternative biochemistry from terran life may store genetic information in charged linear polymers that are structurally and functionally analogous, but not necessarily chemically similar, to nucleic acids (Benner, 2017). We argue here that translational machinery analogous to ribosomes may be present and could additionally serve as an agnostic biosignature. SSN instrumentation can detect and characterize both nucleic acids and ribosomes and holds potential for detecting similar structures produced through alternate biochemistries.
In this study, we explore the utility of ribosomes (and agnostic translational machinery) as a biosignature, and we demonstrate nucleic acid and ribosomal detection using SSN instrumentation. In addition, we propose that detecting one or more peaks indicative of translational machinery, in a sample that has been agitated to sufficiently lyse any existing cells, could indicate the presence of life.
Life beyond Earth, even if ancestrally or chemically distinct from life as we know it, may utilize a structure similar in size to the ribosome, balancing the energetic costs of assembling translation machinery with the minimum complexity required for such a machine. In short, the translation machinery must have some minimum size related to the complexity of its translation function. In addition, there is likely a selective advantage to the translation machinery not being far larger than the minimum size, due to the energetic and time cost of synthesis and assembly (Li et al., 2014; Shore and Albert, 2022). In addition, different life-forms within the descendants of a particular origin of life may have a translation apparatus that differs in size, similar to how prokaryotic and eukaryotic ribosomes have different sizes on Earth. The ancient ribosomal core, which was capable of translation and has been evolutionarily conserved since the last universal common ancestor is about 2 MDa in size. Contemporary prokaryotic ribosomes are about 2.5 MDa, with a slightly larger size due to additional proteins and a slightly longer rRNA sequence (about 100 nucleotides larger than the core) (Bowman et al., 2020). Smaller bacterial ribosomes also exist, with certain ribosomal proteins being absent in bacteria with exceptionally small genomes (Nikolaeva et al., 2021). Eukaryotic ribosomes range in size from about 3.5 to 4 MDa, similarly attributable to longer rRNA sequences (hundreds to thousands of nucleotides larger than the core) and additional proteins (Bowman et al., 2020). It has been proposed that the size difference between prokaryotes and eukaryotes reflects additional complexity in the eukaryotic regulation of translation (Lafontaine and Tollervey, 2001). Comparisons of eukaryotic and prokaryotic ribosomes can allow for identification of the common ancestral core, and molecular models can be used to infer how ribosomes may have evolved (Petrov et al., 2014). Modeling suggests that various ribosomal capabilities for interpreting genetic encoding and creating polypeptides in response were sequentially added, resulting in a molecule composed of functional proteins and nucleic acids (Petrov et al., 2015). The tight size distribution and number of ribosomal proteins (∼50 in prokaryotes, ∼80 in eukaryotes) may reflect selection for optimal duplication efficiency (Shore and Albert, 2022). We do not presume any chemical specificity for ribosome-like machinery; rather, we argue that function and approximate size may be conserved across multiple origins of life with molecules that may differ in structure or composition.
Methods
Reagents and reagent preparation
Aliquots of thawed biomolecule samples (E. coli ribosome [NEB P0763S], 1 kilobase [kb] double-stranded DNA [dsDNA] ladder [Thermo Scientific #SM0311], high-range dsDNA ladder [Thermo Scientific # SM1351], DNA plasmid pUC19 [NEB #N3041], single-stranded RNA [ssRNA] ladder [NEB #N0362], 1 kb double-stranded RNA [dsRNA ladder [NEB #N0363]) were added to Ontera Start-Up Buffer (2M LiCl, conductivity 11.96 S/m). The E. coli ribosome was selected as a well-characterized example of a prokaryotic ribosome. DNA and RNA ladders were used to encompass a broad range of possible lengths and to explore how the same pore can differentiate various molecule sizes in the same run. The sample mass loaded ranged from 2 to 114 ng, and 10 µL of each sample was loaded, resulting in sample concentrations ranging from 0.2 to 11.4 ng/µL (Supplementary Table S1).
SSN operation
The Ontera NanoCounter was operated using the associated software (NanoCounter v0.19.1). The flow cells contained prefabricated nanopores in 30 nm silicon nitride membranes, connected to dual fluid channels with an internal capacity of 8 µL each. Nanopores arrived in a desiccated state. To hydrate and prepare the nanopores for experimentation, the Ontera Start-Up Buffer (2M LiCl, conductivity 11.96 S/m) was added to the flow cells. Nanopores were conditioned using a software-embedded electrochemical approach, which widened and then stabilized the pore within the target diameter of 20–45 nm. Postconditioning pore size was reported; then measurements could proceed. Biomolecule samples and negative controls were loaded into nanopores and analyzed following the manufacturer’s instructions. Ten microliter aliquots of diluted biomolecule samples were loaded, and volumes and dilution factors were used to calculate loaded mass (Supplementary Table S1). Two hundred microliters aliquots of buffer were used for flow cell flushes and subsequent recording. The applied voltage was 100 mV, the bandwidth was 30 kHz, and the sampling rate was 125 kHz. Flow cell configuration and operation were similar to methods reported in previous studies using Ontera systems (Morin et al., 2018; Pearson et al., 2019).
To independently validate that the nanopores were behaving ohmically, the Elements Data Analyzer application (Elements s.r.l., version 1.4.6) was used to generate current-voltage characteristic curves (hereafter known as I/V curves) after all analyses. For each nanopore, voltage protocol 3 was used to apply a series of voltages ranging from −50 to 50 mV, and the associated current was recorded. To assess nanopore noise, Welch Power Spectral Density (PSD) estimates were generated using the “pwelch” function on MATLAB with a window size of 216 and otherwise default parameters.
Detection of dsDNA, ssRNA, dsRNA, and ribosomes
For a given experiment, the same pore was loaded with a sequence of samples after conditioning, using an alternating pattern of negative control (0.2-µm-filtered buffers without added biomolecules) (run time 3–5 min), followed by a sample (run time 6–10 min), followed by the next negative control. This was done to assess buffer cleanliness, flush out and assess any carryover from prior samples, and quantify the risk of false positives. For the purposes of this study, an “event” is defined as a single translocation of a molecule through the nanopore. The NanoCounter software used a proprietary automatic event extraction program to identify events with a signal-to-noise ratio (SNR) high enough to consider the event to be a particle translocating the nanopore (Supplementary Fig. S1). The software also recorded event features, including dwell time, SNR, mean/maximum/median current change, standard deviation of the current change, and mean/maximum conductance change. Data were collected using two nanopores. The first nanopore was used for DNA samples (Supplementary Table S2, Session ID 1), and the second nanopore was used for RNA and ribosomal samples (Supplementary Table S2, Session ID 2) as well as one DNA sample to validate nanopore similarity.
Application of ML to differentiate biomolecule types
To prepare the SSN data for ML classifier training, event metadata was compiled for all biomolecule events with an SNR greater than 5. The MATLAB Classification Learner app (with MATLAB_R2023a) was used to train several ML algorithms on the data. Thirty-three ML algorithms were tested, including neural networks, support vector machines (SVM), k-nearest neighbors (KNN), kernel approximation, naive Bayes, discriminant, logistic regression, and tree-based models (Table 1). Data were partitioned by the toolbox, with 25% used for holdout validation, 10% used for testing, and the remaining 65% used for training. The response variable was the sample identifier (“sample_name”), and the predictor variables were dwell time (“dwell_sec”), signal-to-noise ratio (“SNR”), mean current amplitude during event (“mean_amp_pA”), maximum current amplitude during event (“max_amp_pA”), median current amplitude during event (“med_amp_pA”), standard deviation of amplitude (“std_amp_pA”), and area of the event disruption (“area_pA_sec”). The Ontera NanoCounter software recorded current values in picoamps and the associated conductivity values in nanosiemens; since the applied voltage was constant across experiments and thus conductivity always scaled with current, only current values were used for the ML training to avoid redundancy.
Summary of Various Machine Learning Algorithms on Solid-State Nanopore Data
Summary of Various Machine Learning Algorithms on Solid-State Nanopore Data
Table is sorted by testing accuracy. Model numbers are as indicated by the MATLAB Classification Learner app (MATLAB_R2023a).
Two chips were measured, which resulted in 28,419 total events (26,6667 biomolecule events and 1752 buffer events) across all sample types (Supplementary Table S2). A table with dwell time, mean current, and other data features was generated, where each row represents a single extracted event (e.g., Fig. 1B). The table can be regenerated using the data uploaded to OSF and GitHub code (see the “Data Availability” section). Nanopores passed quality metrics within the Ontera NanoCounter software before measurements, and I/V curves of both nanopores indicated ohmic responses (Supplementary Fig. S4). PSD plots generated before sample addition indicated that noise was low and consistent across nanopores, although Nanopore 2 had slightly more low-frequency noise (Supplementary Fig. S5).
Different biomolecule classes and sizes have distinct signatures
DNA and RNA samples were suspended in 2M LiCl and detected using the Ontera NanoCounter under an applied voltage of 100 mV, bandwidth of 30 kHz, and sampling rate of 125 kHz. The DNA samples were plasmid pUC19 (a circular fragment with a length of 2676 base pairs), 1 kb dsDNA ladder (linear DNA fragments of 14 different sizes ranging from 250 to 10,000 base pairs), and high-range dsDNA ladder (linear DNA fragments of eight different sizes ranging from 10,171 to 48,502 base pairs). The RNA samples were ssRNA ladder (linear ssRNA fragments of seven different sizes ranging from 500 to 9000 bases) and dsRNA ladder (linear dsRNA fragments of seven different sizes ranging from 21 to 500 base pairs). Events were automatically extracted from these runs using the NanoCounter software, totaling 2284 events over 6.62 min for plasmid pUC19, 919 events over 5.93 min for 1 kb dsDNA ladder, 3115 events over 5.99 min for high-range dsDNA ladder, 1874 events over 10.03 min for ssRNA, and 1398 events over 11.01 min for dsRNA (Supplementary Table S2). Raw data were baseline-adjusted, and the noise was calculated for each biomolecule sample (Supplementary Fig. S1, Supplementary Table S2). Key features were also identified, most importantly dwell time (the amount of time that the molecule spent translocating the nanopore) and change in conductance (which resulted from the current disruption of the molecule translocating the nanopore) (Fig. 2). Given the low number of events when running biomolecule-free buffer solutions (Section 4.2), we assume that all events from these runs are indeed the target biomolecule translocating the nanopore.
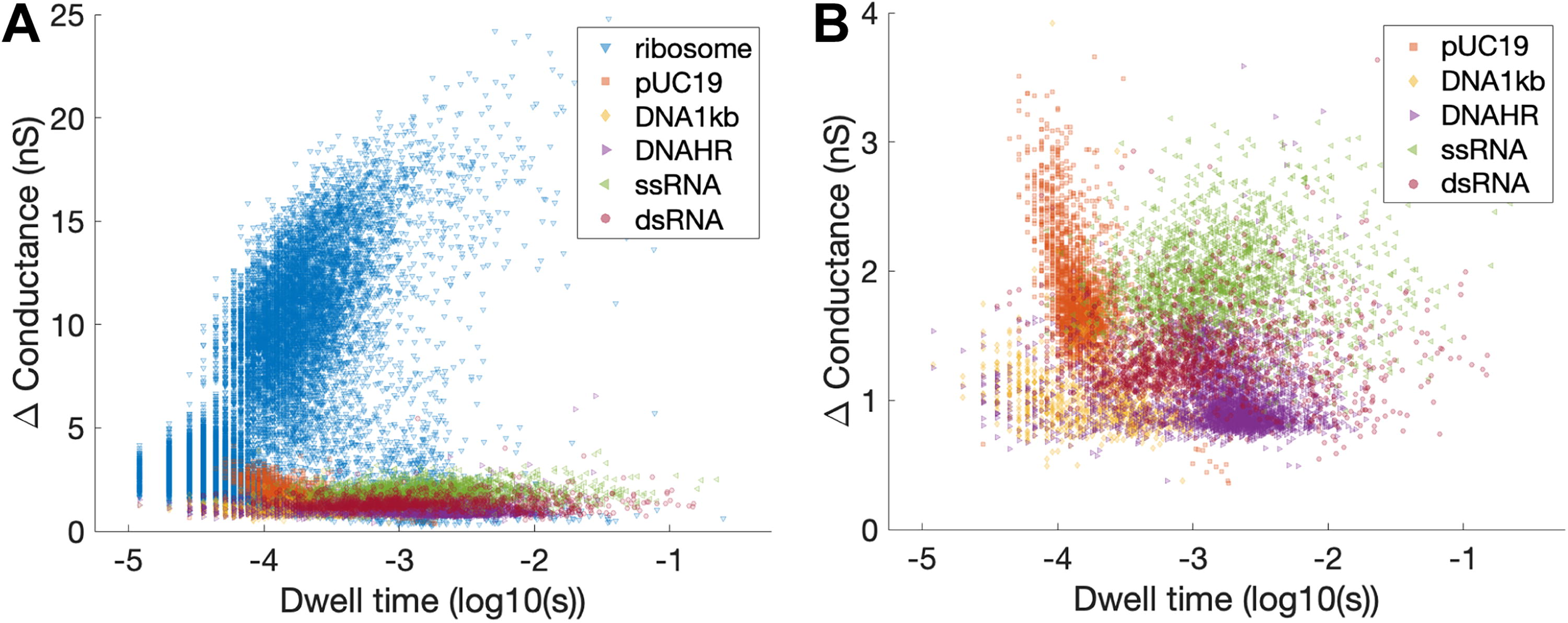
Scatter plots of conductance versus dwell time for various biomolecule types, sizes, and conformations. Panel
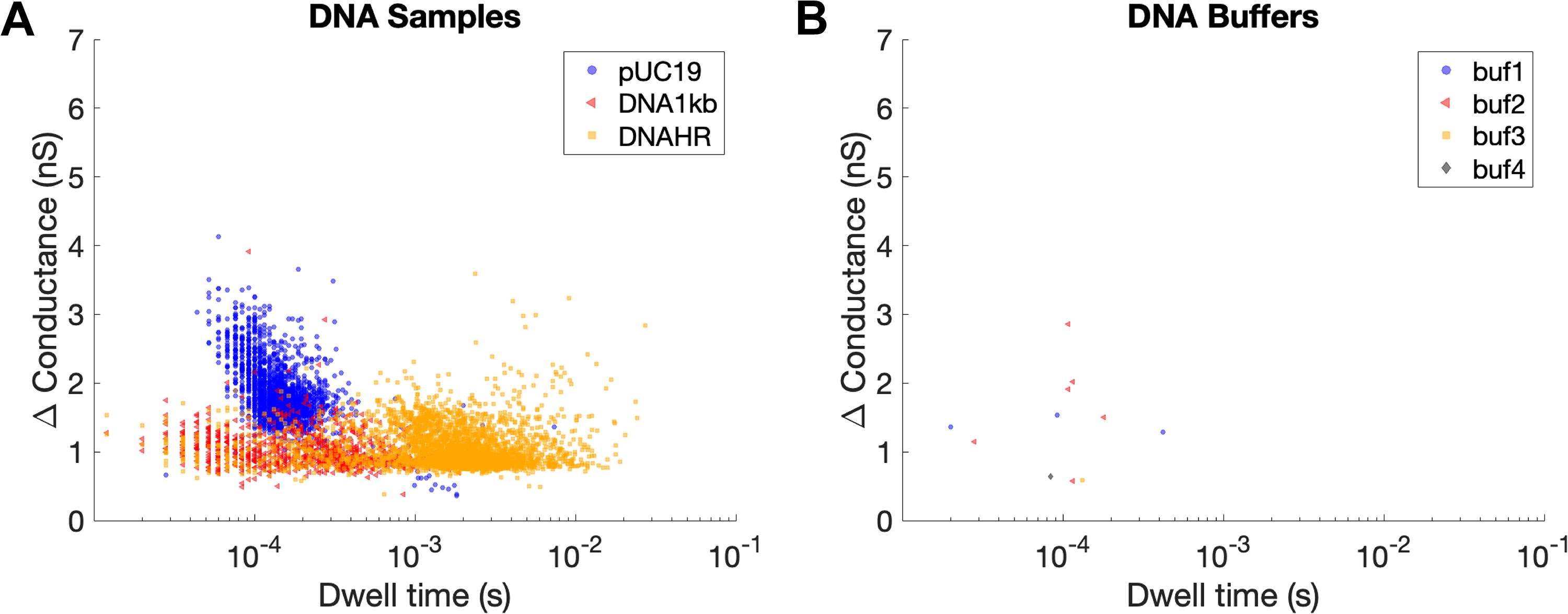
DNA events displayed specific features that were indicative of biomolecule size and structure (Fig. 3). Examples of individual events, heat maps of change in conductance versus dwell time, and plots of overlaid sample events are provided to show what individual translocations look like, display the distribution of event type within each sample, and visualize high-level emerging patterns in translocation features for each sample type. The change in conductance for pUC19 was generally approximately double that of the high-range and 1 kb ladders. This is thought to be attributed to four strands (two dsDNA strands) passing through the nanopore at once for a circular DNA plasmid, compared with the two strands (one dsDNA strand) that would pass through at once with a linear ladder. Individual strands of dsDNA that are folded or exhibit other complex structures would similarly have a larger change in conductance, which may explain the events with larger changes in conductance for the DNA 1 kb ladder and HR ladder samples (Fig. 3E, H). The intact pUC19 plasmid translocation has a comparable amplitude to what has been previously reported for circular dsDNA fragments analyzed by the Ontera NanoCounter (Rheaume and Klotz, 2023). The lower range ladder (1 kb) had a shorter range of dwell times compared with the upper range ladder (HR), which is indicative of the respective ladder fragment lengths.
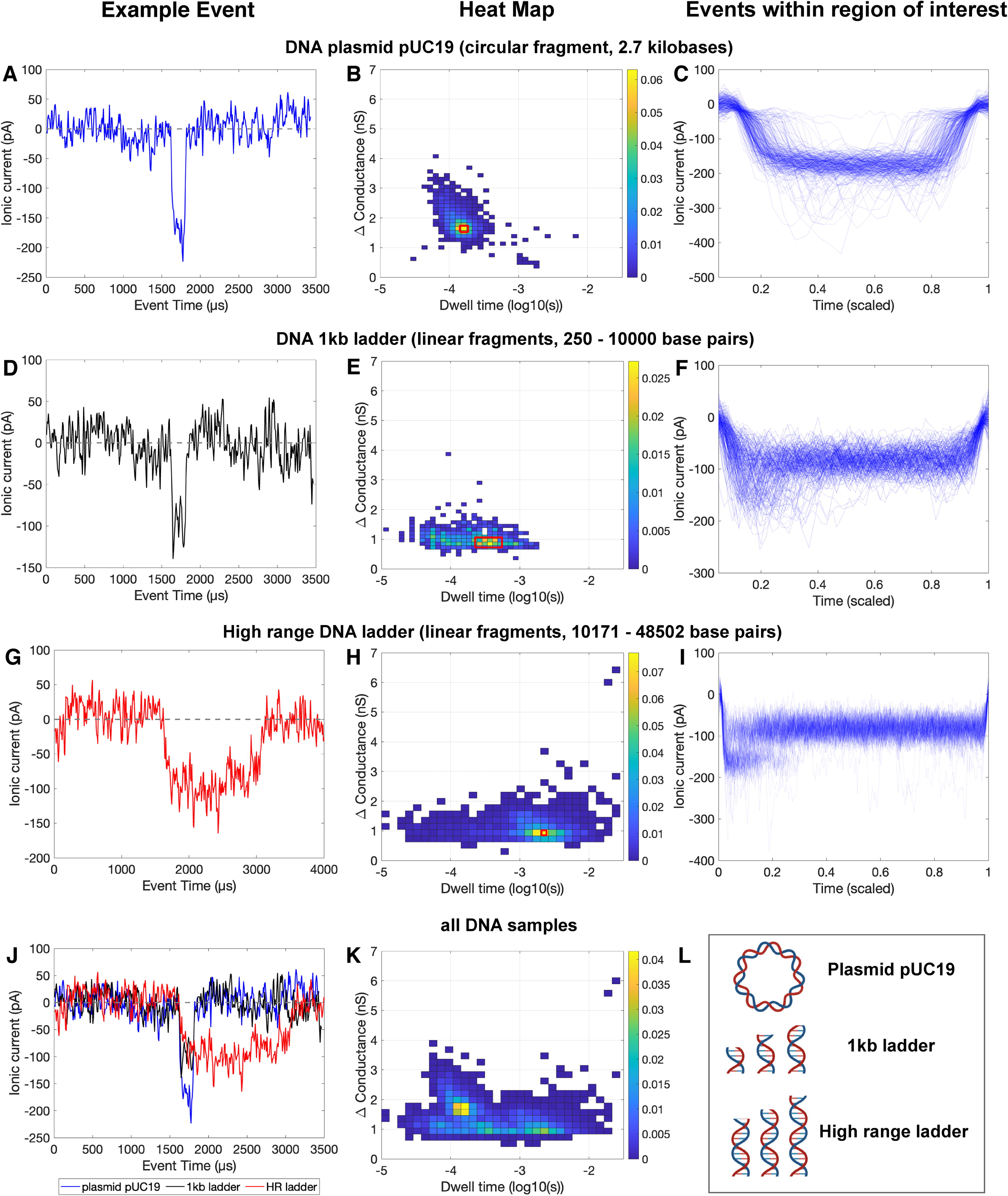
Event data for DNA plasmid pUC19
The ssRNA and dsRNA events also differed in their change in conductance and dwell time, with ssRNA samples generally having a slightly larger change in conductance and slightly smaller dwell time than the dsRNA samples (Fig. 4). Examples of individual events, heat maps of change in conductance versus dwell time, and plots of overlaid sample events are provided to show what individual translocations look like, display the distribution of event type within each sample, and visualize high-level emerging patterns in translocation features for each sample type. However, these differences are less discernible for the RNA samples than the DNA samples. This may be attributable to several factors. The RNA samples and following buffer runs were much noisier than the preceding biomolecule and buffer runs, which made it difficult to directly visualize molecular translocations (Supplementary Fig. S2). High numbers of events in the negative controls (buffer only) immediately after running RNA suggest that precipitation and resolubilization may have occurred for these samples, which could similarly result in low-quality events due to interactions with the nanopore. This is further supported by changes in the measured nanopore diameter throughout these runs (Supplementary Table S2, Supplementary Fig. S2)—after adding RNA, the measured nanopore diameter decreased, which suggests that molecules may have coated the interior of the nanopore. This reduced diameter was only partially alleviated throughout the negative control runs, during which the flow cell was flushed by a clean buffer. This clogging and resolubilization would be in line with expected RNA-LiCl interactions, indicating that a different buffer would be needed to optimize SSN RNA detection. In addition, the two types of DNA and RNA differ in molecule size and structure: the two DNA samples differed in conformation (circular plasmid of a specific length vs. linear fragments of varying lengths), while both RNA samples consisted of linear fragments spanning similar ranges of sizes but in double- or single-stranded form. Strand number or topology can be expected to influence persistence length and thus secondary structure (e.g., ssRNAs are highly flexible). These factors combine to indicate that although RNA was detectable, the detection was nonideal and would greatly benefit from further optimization.
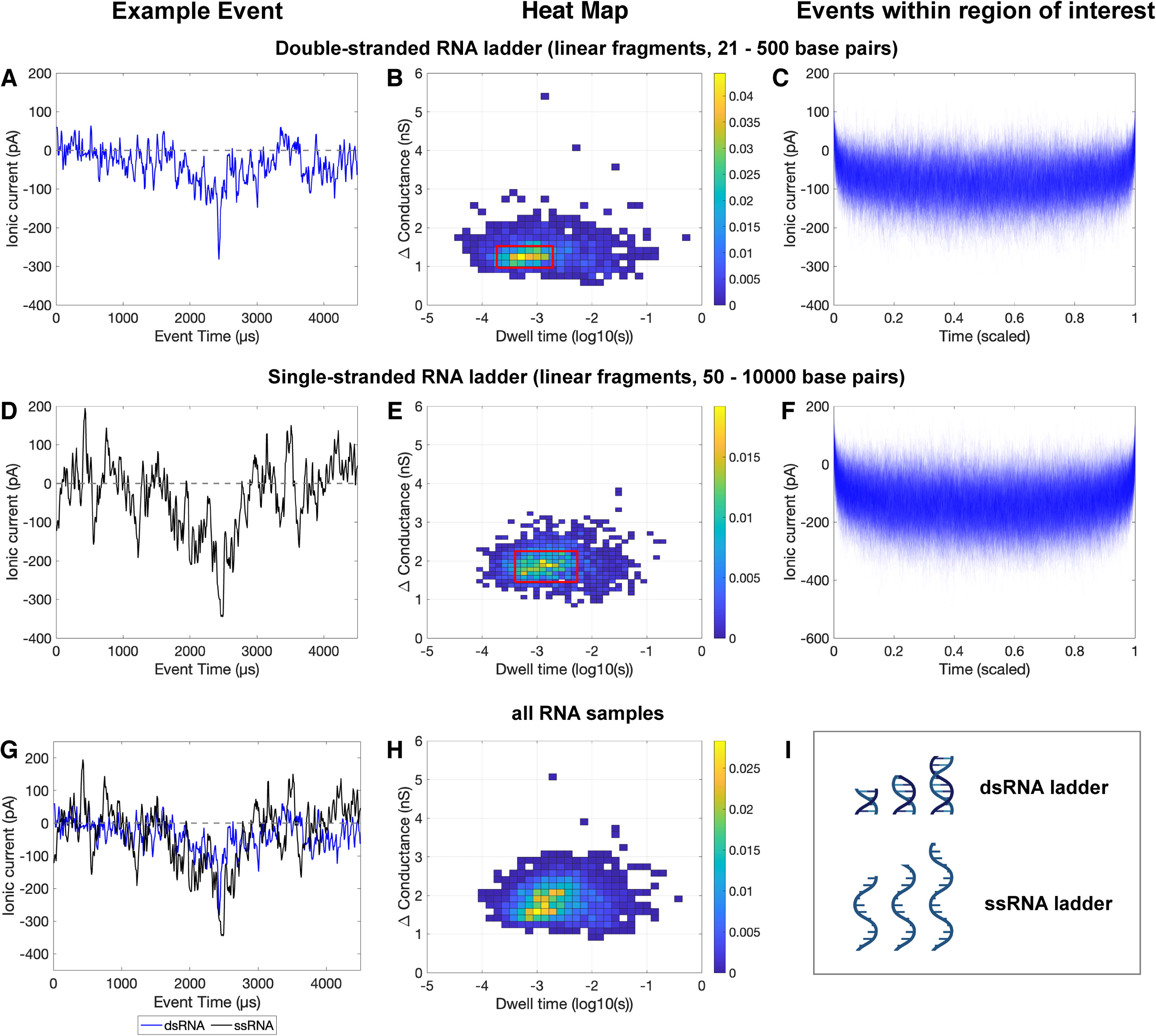
Event data for RNA samples: double-stranded RNA
Similarly, E. coli ribosomes were suspended in 2M LiCl and measured with the Ontera NanoCounter under an applied voltage of 100 mV, bandwidth of 30 kHz, and sampling rate of 125 kHz. From 10.03 min of run time, 17,077 events were extracted (Supplementary Table S2). Figure 5 contains a heat map of events from this run (Panel A), individual examples of events interpreted to be an intact ribosome and a ribosomal fragment (Panel B), and overlaid events from the regions of the heat map interpreted to be intact ribosomes (Panel C) and ribosomal fragments (Panel D). Examples of individual events, heat maps of change in conductance versus dwell time, and plots of overlaid sample events are provided to show what individual translocations look like, display the distribution of event type within each sample, and visualize high-level emerging patterns in translocation features for each sample type. Of the 17,077 events, 6885 are thought to represent intact ribosomes translocating the nanopore (defined as events with a change in conductance greater than 8 nS). Most of these intact ribosome events have a dwell time between 0.0001 and 0.001 s, with some longer dwell times. This is consistent with previous SSN measurements of prokaryotic ribosomes by Rudenko et al. (2011), who reported most dwell times being approximately 0.0001 s with a spread of longer events. The magnitude of translocations reported by Rudenko et al. differs from the magnitude observed in this study, which may be attributable to different applied voltages and different nanopore geometries. The large number of events with changes in conductance smaller than 5 nS is thought to be ribosomal fragments. Within this results’ space, there is a high concentration of events with a change in conductance between 1 and 5 nS and a dwell time between 0.00001 and 0.0001 s. These are interpreted as protein fragments. Some events have a larger dwell time (0.0001–0.1 s), which closely aligns with the reign occupied by RNA events (Fig. 4). Thus, it is thought that these fragments may be RNA products of ribosome fragmentation.
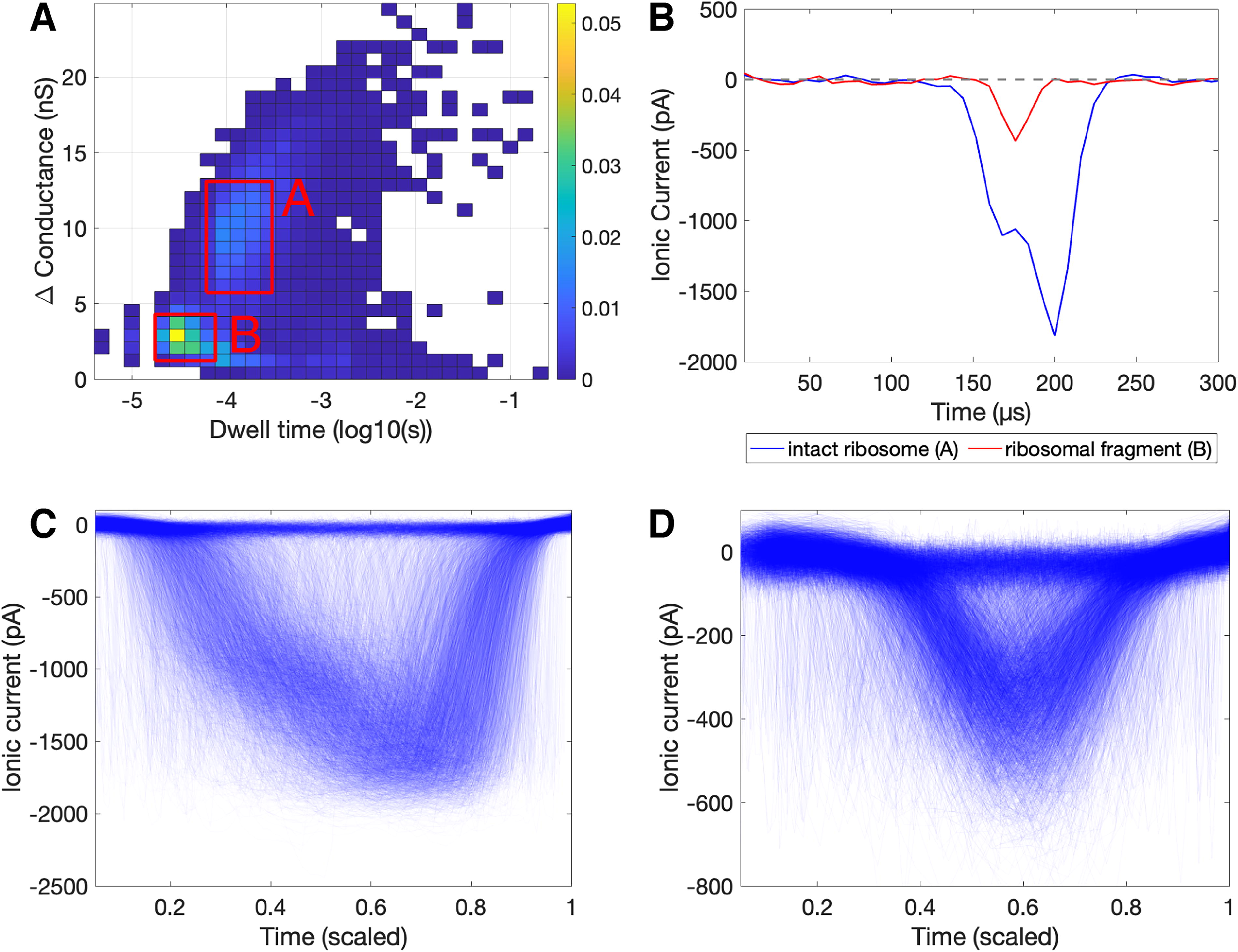
Event data for Escherichia coli ribosome in 2M LiCl. Panel
Overall, the signals of ribosomal events were much stronger than those of nucleic acids. An example of this is given in Figure 1B, where the change in conductance for a ribosomal translocation is larger than that of a DNA strand by about an order of magnitude. This can be primarily attributed to the relative size of the molecules passing through similarly sized pores. As shown in Section 4.4, ribosomes are also more predictable than nucleic acids when using a selected ML algorithm. That being said, signals from nucleic acids are also discernible, predictable, and differentiable. Based on dwell time and change in conductance, signals from each biomolecule class cluster together, although there is some overlap (Fig. 2). The cluster of ribosomal events was markedly different from nucleic acids (Fig. 2A), which demonstrates the high utility of SSN to differentiate between different classes of molecules. A scatter plot of event change in conductance versus dwell time is also shown for just nucleic acids (Fig. 2B) so that differences between the various DNA and RNA ladders could be more visible. ML was implemented in attempts to better differentiate between sample types.
Overlaid event data (conductance vs. dwell time) for biomolecule (
Negative controls were run before every test sample to assess background and carryover from previous runs. When testing sterilized and filtered brines on new nanopores, event abundance was low (<10 events per minute). Events detected in this case are likely particle contaminants introduced during buffer preparation, sample loading, or present within the flow channels of the nanopore. Event abundance in the negative controls when run between DNA samples was similarly low, as shown in Figure 6. A higher abundance of events than usual was sometimes detected in pores when running negative controls between biomolecule samples (Supplementary Table S2). This was especially prominent in cases where clogging (e.g., high-concentration ribosomal samples) and biomolecule precipitation (e.g., RNA in lithium chloride) were suspected to occur.
DNA detection yields similar results across nanopores
To demonstrate that measurements are replicable between the two nanopores used in this study, the DNA 1 kb sample was run on the nanopore that was previously used for RNA and ribosomal samples (as indicated in Supplementary Table S2 as Session ID 2). Events were automatically extracted, and heat maps of change in conductance versus dwell time for both pores were plotted on heat maps (Supplementary Fig. S3). Both heat maps had prominent peaks within the same region of the graph, particularly around a change of conductance of 1 nS and a dwell time of 10−3.5, indicating that the two nanopores were behaving similarly. The second nanopore had a wider distribution of events, with individual events scattered further across the data space. These events that majorly deviate from the general peak may be attributable to nanopore age, collection of more events with the second nanopore (919 events with the first nanopore, compared with 4449 events with the second nanopore), or residual clogging from the RNA and ribosomal samples that were previously measured with the second nanopore.
ML algorithms can differentiate biomolecule types and structures
The Classification Learner app identified the validation and testing accuracy of a variety of ML classifier methods, including neural networks, SVM, KNN, kernel approximation, naive Bayes, discriminant, logistic regression, and tree-based models (Table 1). To provide an illustrative example, detailed results from the medium tree model are shown. This specific model was selected for its high interpretability, as other top-scoring algorithms (e.g., neural networks, SVM, KNN) can be more difficult to interpret. The medium tree was specifically selected from the applied decision tree models (fine tree, medium tree, coarse tree) as it balances ease of interpretation with accuracy—the fine tree model had higher accuracy levels (94.41% validation accuracy and 95.02% testing accuracy, compared with the medium tree’s 92.89% validation accuracy and 93.34% testing accuracy), but the fine tree involved a much more complex array of decisions. In addition, with limited training data, it is difficult to preclude overfitting, which is more likely to occur for finer trees. The medium tree (Supplementary Fig. S6) contained 19 splits to classify biomolecules based on event features (Supplementary Table S3). It is possible to reach the same biomolecule classification by following different paths throughout the tree, which may be attributable to the presence of different molecule morphologies within a single sample (e.g., intact vs. fragmented ribosome, supercoiled vs. unsupercoiled plasmid, bent vs. straight DNA/RNA fragment). The medium tree’s confusion matrix (Fig. 7) demonstrates validation performance and shows how biomolecules were generally classified correctly by this model. This algorithm was particularly powerful for discerning ribosomes from nucleic acids, with the positive predictive value (PPV, the percentage of correctly classified events within all events of that class) being 98.5% and the true-positive rate (TPR, the percentage of correctly classified events within all events assigned to that class) being 98.2%. The medium tree model is shown here due to its interpretability, but other models achieved even higher performance and warrant further exploration.
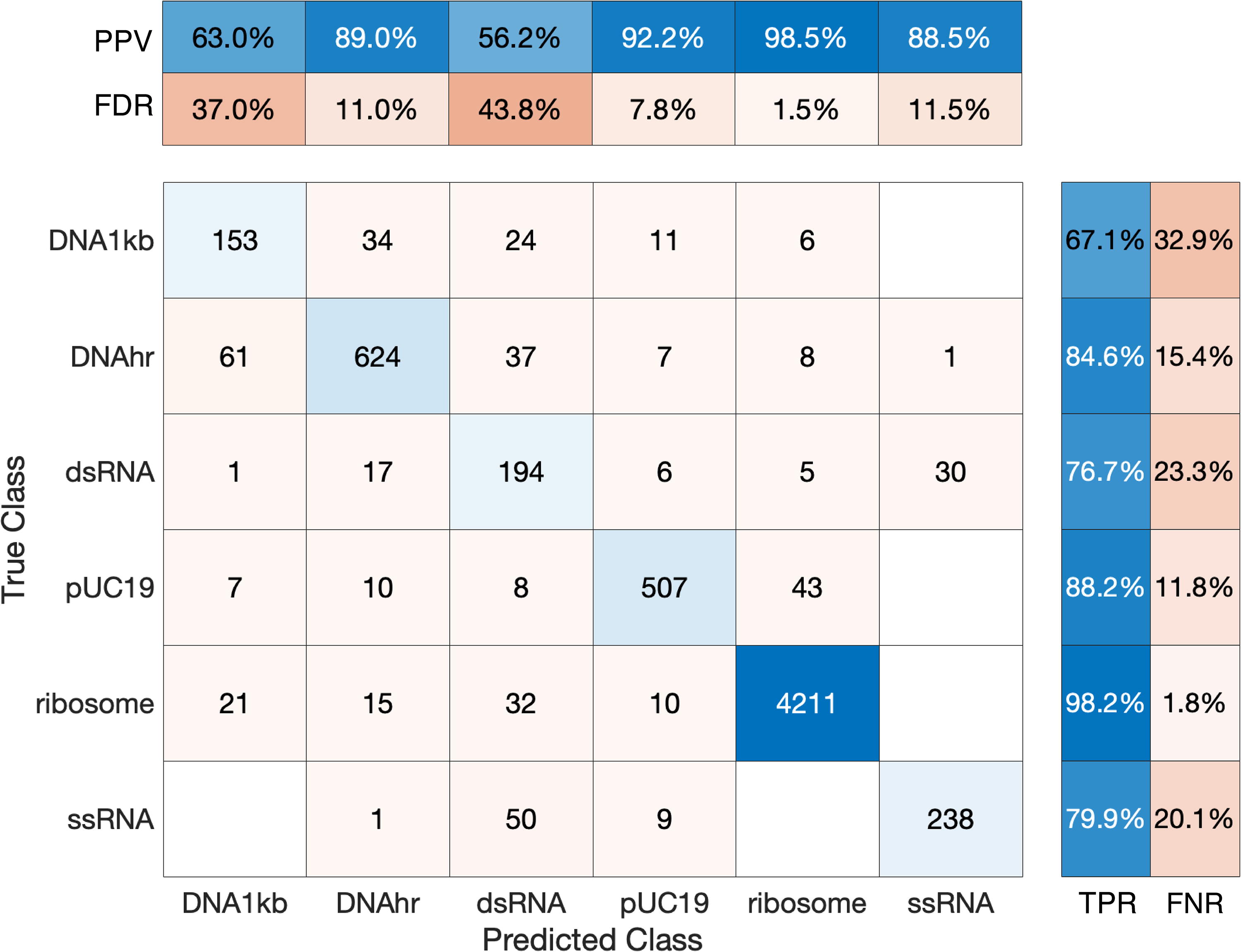
Confusion matrix for medium tree model. This matrix was generated by the medium tree model by the MATLAB Classification Learner app (MATLAB_R2023a). True class versus predicted class matrix shows the raw numbers of events for each true/predicted class combination. The upper matrix shows the likelihood that an event is correctly classified given the true class by showing the positive predictive value (PPV), the percentage of correctly classified events within all events of that class, and the false discovery rate (FDR), the percentage of incorrectly classified events within all events of that class. The right matrix shows how many of the events were correctly predicted within a predicted class by showing the true-positive rate (TPR), the percentage of correctly classified events within all events assigned to that class, and the false-negative rate (FNR), the percentage of incorrectly classified events within all events assigned to that class.
In this study, we argue that ribosomes can serve as a biosignature indicative of terran-like life, and another instance of cellular translational machinery of similar size and structure could serve as an agnostic biosignature. In the case of “life as we don’t know it,” this putative structure could be distinguished as a higher abundance of particles within a certain size range compared with the rest of the sample. Using SSN instrumentation, we demonstrate that such cellular machinery can be detected and distinguished from other biomolecules in aqueous samples.
Such measurement is sensitive (capable of correctly identifying target molecules in the sample), as evidenced by successful detection of various biomolecules, and specific (able to discern whether a sample does not contain target molecules), as evidenced by the low abundance of events within the negative controls. When measuring negative controls, no special precautions were taken (e.g., filtration of buffers), which suggests the potential for lowering the false-positive event rate further. Target biomolecules were evident within heat map distributions as specific peaks corresponding to different molecule sizes and conformations. This is especially true for ribosomes, which exist in very specific quantized sizes for prokaryotes and eukaryotes and are often highly abundant, for example, 25,000–31,000 per µm3 for active E. coli cells, as reported by Bakshi et al. (2012). High abundance of ribosomes is not limited to active cells. Slow-growing ultrasmall archaea of volume 0.03 µm6 (effective spherical diameter of 0.4 µm) have an average of 92 ribosomes/cell (Comolli et al., 2009), or 3000 per µm3. Bacterial endospores contain ribosomes at a similar abundance to active cells, even when mRNA has been degraded (Chambon et al., 1968). This high abundance of ribosomes across different types and activity levels of cells may allow for the distinguishing of ribosomes within cellular lysate or environmental samples treated to lyse cells. Conversely, abiotic material that was similarly treated would be expected to have a more even particle size distribution.
Certain features could confound a search for translational machinery as a biosignature, including other cellular components and abiotic features. The severity and implications of such mistaken identities vary. If another cellular component (e.g., genetic information storage molecules, functional proteins, other cellular organelles) is mistaken as a translational apparatus, then life has still been detected. In addition, structures associated with biological systems (e.g., bacteriophages or other viral agents) could be detected, which could similarly indicate the presence of life. However, if a particle of abiotic origin is mistaken for a ribosome-like molecule, this could cause a false-positive life detection result. For example, in Earth’s atmosphere, mechanically generated particles tend to be in the coarse-mode (>1 µm) size range, with particle sizes in the nanometer to tens of nanometer range forming from nucleation and thus likely to exhibit a continuous size range rather than high abundance at a specific size. Accumulation of nucleated particles can result in larger sizes (typically hundreds of nanometers). However, in astrobiological contexts, additional caution is warranted. While 99% of dust in the Enceladus plume is expected to be below 10 nm with a peak at 2 nm (Meier et al., 2014), the presence of hydrothermal activities at Enceladus was subsequently inferred from Cassini-detected silica nanoparticles in the 4–16 nm size range (Hsu et al., 2015). One mitigation in this case would be to filter a sample to retain cells and reject free silica nanoparticles before lysis. An additional approach would be to heat-treat a sample and observe if the particles are heat tolerant or exhibit denaturation or degradation. Further and varied analyses of any detected molecules would be necessary to discern their composition, function, and potential biogenicity. One such analysis could be employment of assembly theory, which involves quantifying the complexity of the formation of a molecule and making predictions about how likely such a molecule is to have arisen from a biotic process. Identification of a high-assembly index molecule such as a ribosome in high abundance could indicate that evolutionary selection has favored its existence (Sharma et al., 2023). While outside the focus of this article, future work should evaluate the potential abiotic production of particles in different settings to assess the risk of false positives.
The results presented here are intended to serve as a proof-of-concept study that lays the foundation for more extensive and robust work, with multiple avenues identified for further experimentation. One factor that warrants further study is buffer-biomolecule compatibility. In this study, RNA was particularly difficult. The RNA samples and following buffer runs were much noisier than the preceding runs, and it was particularly difficult to achieve low event abundance in the negative control buffer samples run between RNA and ribosomal samples. This is likely due to the effects of lithium chloride buffer on these biomolecules, which is thought to have caused molecular precipitation that hindered flow cell flushing and subsequent molecule detection. Similarly, it was difficult to discern ssRNA and dsRNA events by looking at current disruptions, possibly due to the precipitation of RNA fragments and/or interaction with the nanopore. It seems that most of the detected events were attributable to true translocations of RNA strands through the nanopore, since the event rate was much higher for the RNA samples than the blank runs immediately before and after (Supplementary Table S2). However, many of these events had a relatively low SNR compared with what was observed for other samples, and all RNA samples and following buffers were very noisy, which further obscured the influences of any molecular translocations. Refining buffer and pH compatibility with RNA (and other target biomolecules) is a future target for optimization.
Pore size constrained the size of biomolecules that can be analyzed with this system, and it may have played a role in the ribosomal fragmentation observed. This study used nanopores embedded within prefabricated flow cells that arrived in a desiccated state, which we then hydrated and subjected to an electrochemical conditioning process that widened and stabilized the pore. This conditioning process would result in a pore with a diameter between 20 and 45 nm, which could not be significantly altered. This was sufficient to allow the passage of single strands of nucleic acids (∼2 nm diameter) and prokaryotic ribosomes (21 nm diameter). It is important to match pore and biomolecule size as closely as possible: if the nanopore is too small, it inhibits free flow of molecules through the nanopore. Conversely, if the nanopore is too large, signals from translocating molecules may be obscured by noise—the closer the size of the biomolecule is to the size of the nanopore (without being large enough to clog the nanopore), the higher the SNR (Xia et al., 2022). With the nanopores used in this study, we were able to detect nucleic acids but may be able to achieve stronger signals with a smaller nanopore. In addition, we were able to detect ribosomes but also observed the presence of smaller particles thought to be ribosomal fragments. Such fragmentation is hypothesized to have been caused by buffer chemistry and exacerbated by RNA precipitation and clogging of the nanopore. Using a larger nanopore in future studies may help alleviate this issue, especially if coupled with a buffer that is more compatible with ribosomes. Overall, having access to a wide variety of nanopore sizes will be useful for future work with molecules of various sizes, structures, and biochemistry.
Future analysis of biomolecules with SSN should also include varying salt composition, salt type, pH, and mineralogy. Nucleic acids and ribosomes are both structurally sensitive to these conditions (Klein et al., 2004; Shukla and Mikkola, 2020; Spitnik-Elson and Atsmon, 1969; Sturtevant et al., 1958; Swadling et al., 2012; Tian et al., 2023), as well as biomolecule translocation itself (Bell et al., 2016; Firnkes et al., 2010; Kowalczyk et al., 2011; Roozbahani et al., 2020). SSNs themselves have been shown to be impacted by variations in pH and salinity (Firnkes et al., 2010; Wadsworth and Cockell, 2017; Wanunu and Meller, 2007) and, as demonstrated by these experiments, can be susceptible to clogging in cases of analyte and solution incompatibility. More extensive study of SSN performance under various chemical conditions will be necessary to achieve better understanding of how solution composition influences results.
In addition, it is important to consider the potential for internanopore variation when comparing measurements that are made with different nanopores. No two nanopores are unique, as physical and chemical variations can impact measurements, even if the nanopores are manufactured under identical conditions and have the same pore size. In this study, one DNA sample was run in both flow cells, and the results were compared. The two distributions of events had similar delta conductance and dwell times (Supplementary Fig. S3), which indicates that the pores yielded similar measurements. We use this observation as justification that comparisons can be made between the other measurements made on these two nanopores. Future studies of interchip and intersystem variability can be used to develop normalization approaches to train and validate ML models that can be applied across many devices, despite device variability. In addition to blanks, a positive control can be used to validate in situ spaceflight measurements, such as the use of a standard DNA sample (if storage conditions permit) or a synthetic polymer (which may be more robust to the physical and temporal conditions of long-duration spaceflight). Nanopore 2 was originally used on May 28, 2021, and used again on April 30, 2024; this demonstrates that SSNs can be stored and reused on mission-relevant timescales. If SSNs were to be integrated into a spacecraft that is traveling several years to its destination, the likelihood of having several usable pores would be maximized by fabricating and conditioning pores at the destination. However, the successful storage and reuse of old and previously used nanopores indicate that long-term storage and in situ conditioning may be a viable option. The Ontera system is no longer available, but alternative commercial solutions do exist. We compare our results with studies using different SSN systems, demonstrating consistency. Understanding differences in SSN setup and experimental conditions is crucial for accounting for internanopore and intersystem variations when analyzing SSN data.
Furthermore, we explore the utility of ML for processing of SSN data. We demonstrate that different biomolecule classes and structures can be differentiated and predicted by decision tree ML algorithms with over 90% accuracy on both validation and test data. Both a fine and a medium tree were generated, with the medium tree giving a more comprehensible assessment of how biomolecules could be discerned at the cost of less decisions and lower accuracy, and the fine tree yielding higher accuracy classification by making more decisions, at the cost of a very lengthy list of nodes and the potential for overfitting. A confusion matrix was generated for the medium tree (Fig. 7) to demonstrate the predictive power of the model. This model was particularly skilled in discerning ribosomes from nucleic acids, with ribosomal samples scoring highest for PPV and TPR. The sizes of the nanopores were closer to those of ribosomes than nucleic acids, which may help explain the high metrics for classifying ribosomal events. This demonstrates the benefit of closely matching pore size to target analyte size for maximal SNR, while it also shows that smaller analytes can also be detected with reasonable accuracy even when the nanopore has not specifically been optimized to its size range. Other less-interpretable ML models (e.g., neural networks) yielded even higher accuracy overall, further indicating the potential that ML classification holds for SSN data. This is useful for summarizing and transmitting data during future missions with highly constrained data budgets, such as at or within Ocean Worlds.
These ML results are intended to justify more rigorous future application of ML-based classification of SSN data, including exploring the utility of various algorithms, hyperparameter tuning, and feature engineering. In addition to this computational work, it will be critical to test these algorithms on data sets from different SSN runs under various conditions and instrumentation to ensure that accurate results are consistently obtained. Since the data set for each biomolecule class was generated by a single experimental run of that specific class, the experimental conditions (e.g., pore size, buffer chemistry) may influence event data independently of the influence of the specific biomolecule type. Generation of more data by analyzing different biomolecule types, structures, and sizes under various buffer conditions and with different nanopores will be instrumental in refining such an ML-based classification.
There are a few major caveats for applying this laboratory-based detection of terran biomolecules toward astrobiological life detection. First, the utility of ribosomes or ribosome-like translational machinery as a biosignature relies on the assumption that such “life as we don’t know it” synthesizes biomolecules for cellular functioning via translation or an analogous mechanism. Although it is not known how life may exist without ribosomes or ribosome-like particles, we acknowledge the possibility that alternate biochemistries, cellular structures, and life strategies could be associated with life-forms that would not be detectable by searching for translational machinery. In addition, this work assumes that such a translational apparatus will remain stable during cellular lysis, extraction, and analysis conditions. If translational machinery from an alien life-form were to be destroyed or otherwise disrupted by such processes, this would increase the chance of false-negative results. Finally, unknown abiotic conditions could produce confounding structures with similar signatures, resulting in false-positive readings. This possibility highlights the need for stringently using negative controls.
Furthermore, SSN instrumentation has particular considerations for spaceflight that would need to specifically be addressed. The technology currently requires human manipulation, so automation of sample acquisition, preparation, loading, and measurement would be necessary. The shelf life of fabricated nanopores would need to be constrained, and damaging effects of spaceflight radiation, temperatures, pressures, and other environmental conditions would need to be considered. SSNs typically need cleaning (often using Piranha solution, ultraviolet light, plasma, or other caustic methods) and conditioning, so it would be important to select effective cleaning methods that are compatible with spaceflight, long-term storage, and automated methods. This technique is highly sensitive and capable of detecting low abundances of biomolecules—appropriate use of negative controls and overall avoidance of contamination from Earth would be absolutely critical to ensure the fidelity of these measurements.
We argue that ribosomal detection could serve as one line of evidence for life, and such measurement could be conducted in conjunction with other life detection analysis. Such detection could be particularly useful as a prescreening tool for samples, subjecting it to this biologically agnostic particle measurement technique to determine size distribution and potential biogenicity of samples. This could aid in sample selection or prioritization before passing it forward to more targeted analyses (e.g., DNA sequencing, microscopy, mass spectroscopy, or other compositional analyses). Furthermore, we demonstrate how ML can be utilized to classify biomolecules based on various features of translocation events measured by an SSN device. Overall, SSN presents one strategy to interrogate various components of a sample and search for patterns or features that may indicate biogenicity. Negative results from an attempt to detect ribosomes would not necessarily preclude the possibility for life-forms to be present within the sample. Opportunities for in situ sample preparation may be limited by lack of knowledge of the biochemistry of any putative biomolecules, and so, it is possible that chemical incompatibilities could disrupt the structure of any molecules of interest. In addition, “life as we don’t know it” may sustain cellular processes through another mechanism or structure. However, detection of ribosomes or ribosome-like structures with a SSN instrument could support a hypothesis that samples are from a biologically active ecosystem, especially if both ribosomes and abundant molecules with RNA-like properties are identified.
Perhaps the most compelling astrobiological application of SSN instrumentation is the search for signs of life in potentially habitable saline environments on Mars, Europa, or Enceladus. Salinity exerts a major effect on the ability of an organism or a biomolecule to persist in a system: microbial activity can be detected throughout wide ranges of salinity levels and salt compositions, and biological structures such as intact cells and biomolecules can be preserved even beyond the saline limits for active life (Klempay et al., 2021; Oren, 2008). Thus, many have postulated that saline environments should be a major target for astrobiological exploration, given their biological relevance and presence throughout the solar system (Davila and Schulze-Makuch, 2016; Neveu et al., 2020; Pappalardo et al., 2013; Phillips et al., 2023). Although saline environments are biologically relevant, they can also pose challenges for conducting science in such environments. Indeed, many analytical techniques are impeded by salt and require desalination treatments (Lawrence et al., 2023). Given SSN’s intrinsic requirement that samples be suspended in a conductive solution, it may be possible to use this technique to analyze environmental biomolecule samples within their original saline solutions or diluted with a high salt buffer of choice, without complex processing such as salt removal. Furthermore, Ocean Worlds are expected to be low in available free energy; thus any existing biosphere would likely be low in biomass (Jones et al., 2018). SSN’s low limit of detection and high sensitivity make it a promising technology for achieving the single-molecule resolution that would be necessary to effectively search for intact biomolecules in such environments.
SSNs are of interest for future astrobiological space exploration due to their ability to detect biomolecules with high sensitivity and low false-positive rates, as well as their potential utility in searching for agnostic biosignatures. Beyond its applications for astrobiological life detection, SSN has other qualities that overall make it well-suited for space payloads in general, including high portability, minimal resource requirements, low-mass and low-power requirements, potential for analysis on minimally prepared samples, and capability for rapid analysis. Interplanetary space missions with the explicit goal of searching for extraterrestrial life are expected in the coming decades (National Academies of Sciences, Engineering, and Medicine, 2022). With further research and development, an SSN instrument may be a compelling addition to a future mission that seeks to detect signs of extant life in the solar system.
Footnotes
Acknowledgments
The authors thank Chen Chen for valuable contributions and comments on the ML components of this article. The authors also thank the anonymous reviewers for invaluable feedback throughout the peer review process.
Authors’ Contributions
J.M.M. and C.E.C. developed the concept. J.M.M. and C.E.C. performed the experiments. J.M.M. and C.E.C. analyzed the NanoCounter data. J.M.M., M.K., and C.E.C. performed the ML analysis. J.M.M. and C.E.C. wrote the article. All authors edited and approved the article. Biomolecule graphics (appearing in Figs. 1, 3, ![]() ) were created with BioRender.com
) were created with BioRender.com
Data Availability
Data can be downloaded at https://osf.io/56qkv/ and the code for analysis can be downloaded at ![]()
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
This work was supported by NASA award 80NSSC22K0188 to C.E.C. and NASA FINESST award 80NSSC22K1320 to J.M.M. and C.E.C. This work was also supported by NASA award 80NSSC19K1028 to C.E.C.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Associate Editor: Sherry L. Cady
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.