
Abstract
Enantiomeric excesses of
1. Introduction
Amino acids are the fundamental building blocks of proteins, and only 19 kinds of them with the
For the amino acid component, the meteoritic amino acids possess α, β, γ, and δ amino structures that present the carbon moiety C2–C9. The most abundant amino acid in the Murchison meteorite is glycine (∼3 ppm), whereas other relatively abundant amino acids are alanine, β-alanine, and isovaline, all detected at a concentration of ∼1.7 ppm. The diverse composition of the meteoritic amino acids suggests a prebiotic synthesis of amino acids in the meteorites' parent body and/or the interstellar medium (ISM).
The Rosetta mission was able to detect, via mass spectrometry, the presence of volatile glycine and precursor molecules in the coma of comet 67P/Churyumov-Gerasimenko in 2014 (Altwegg et al., 2016; Grady et al., 2018). However, mass spectrometry cannot quantitatively distinguish between structural isomers in the same molecular weight. Therefore, the mechanisms of the chemical evolution of glycine and amino acids still remain unsolved.
The proposed formation pathways for the amino acids are summarized in Fig. 1. The Strecker synthesis in the parent body of meteorites is a typical mechanism (Elsila et al., 2016). The Strecker mechanism has long been postulated for the formation of α-amino acids by the hydrocyanation of imines (RCH = NH) and a subsequent hydrolyzation via the formations of amino nitriles (Fig. 1). Similar to the Strecker mechanism, the cyanohydrin reaction can explain the formation of α-hydroxy acids, also detected in the Murchison meteorite, and they are distributed similarly to the α-amino acids.
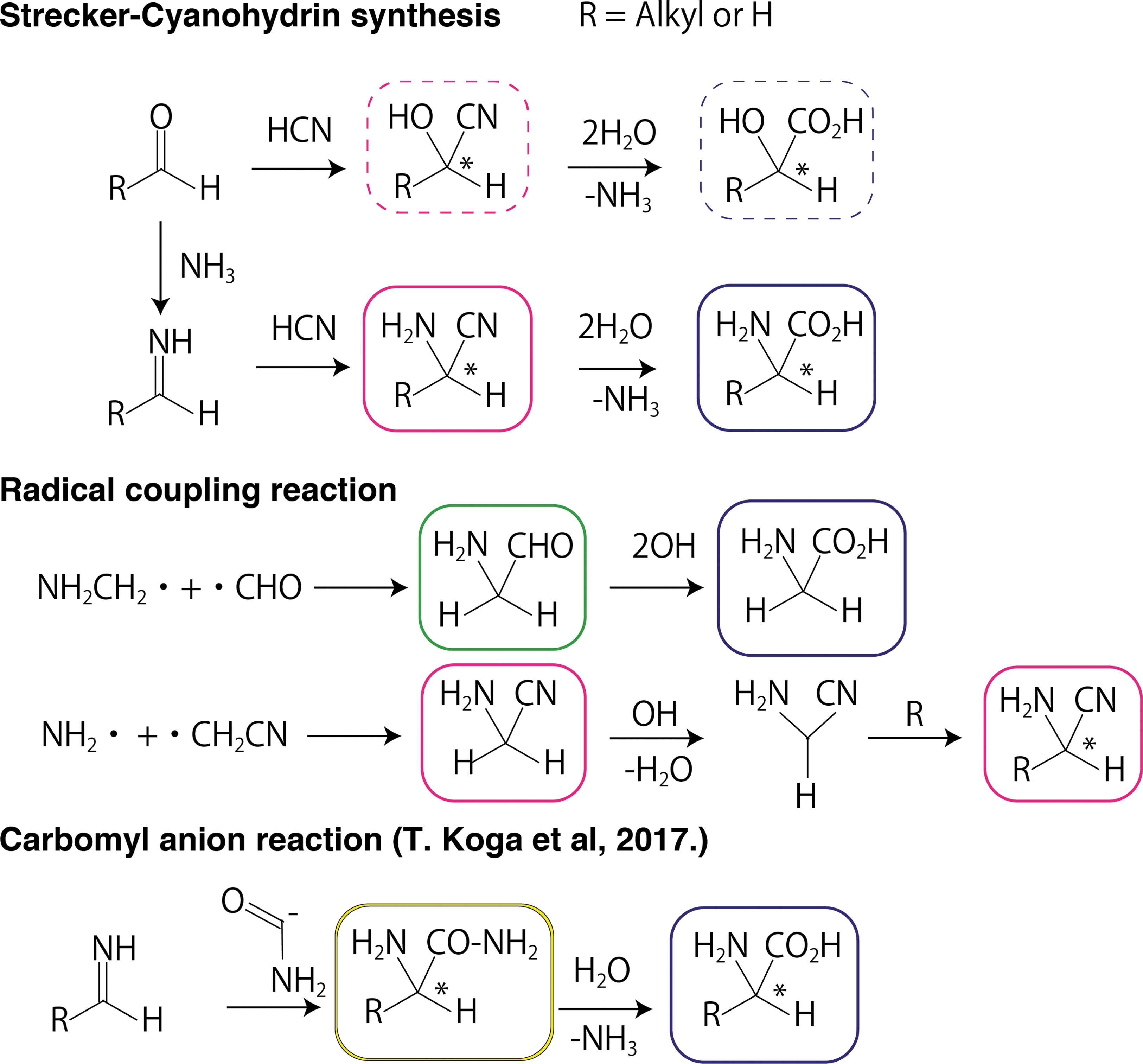
Proposed formation pathways for α-amino acids and α-hydroxy acids. Intermediate compounds of nitrile (pink), aldehyde (green), carbonylic acid (blue), and amide (yellow) are framed in different colors for the sake of clarity. Hydroxyl compounds are framed by the dashed lines in the corresponding colors. The chiral centers are indicated by an asterisk (*).
Another proposed mechanism worthy of attention is the reaction between the imine and carbomyl anion to form an amide intermediate (Koga and Naraoka, 2017). This scheme is consistent with the formations of a variety of meteoritic amino acids, soluble organic compounds, and alkylated pyridines present in the Murchison meteorite.
An alternative reaction pathway for the synthesis of amino acids is a radical coupling mechanism on grain surfaces in the ISM. Radical species are formed upon interstellar ultraviolet (UV) photolysis and cosmic rays irradiation, and their reaction barriers are low enough to proceed in the cold molecular cloud (T ∼ 10K). The suggested radical coupling mechanisms for the glycine formation can be realized according to three different paths (Garrod, 2013; Singh et al., 2013; Sato et al., 2018);
According to the chemical kinetic simulation in star-forming regions (Garrod, 2013), the dominant reaction processes are (1), (2), and (3) in the low (40K < T < 55K), middle (55K < T < 75K), and high (75K < T < 120K) temperature ranges, respectively. The simulation has demonstrated that the production of glycinal (NH2CH2CHO) is a key event for the glycine formation. For the formation of other amino acids, the radical coupling reactions of the type NH2CHCN + R → NH2CHRCN have been suggested. These reactions proceed through the formation of aminoacetonitrile (NH2CH2CN).
Based on these α-amino acid formation mechanisms, the origin of the molecular chirality can be traced back to their nitrile, aldehyde, and amino compounds with chiral centers, as highlighted in Fig. 1 by the color frames. Even though the generation of amino acids requires these multistep reactions, it is still unclear which formation pathways are dominant compared with other molecular formation reaction channels. If the radical coupling reactions are dominant in these chemical reactions, the molecular mechanism of homochirality is expected to be directly related to the formation processes.
The minimum energy principle (MEP) has been used to predict the presence of interstellar molecules in ISM (Lattelais et al., 2009, 2010). This principle is very efficient for many interstellar molecules with a small number of atoms (from two to six), although a few exceptional cases exist (Fourré et al., 2016, 2020). The MEP assumes that the most abundant molecule/isomer in ISM corresponds to the most stable one.
Although the number of isomers dramatically increase with the number of atoms, we enacted, in the present study, a computational effort in a comprehensive way to evaluate the stability of alanine and chiral alanine precursors within the MEP. The stability of related chiral molecules, namely, α-hydroxy compounds, as well as glycine, were also investigated to compare and test the present theoretical procedure. We implemented a new algorithm to generate isomers for a given chemical composition, and we refer to this approach as the random connection (RC) method.
The validity of this method was carefully assessed for glycine, whose isomers and isomerization channels were recently explored by Ohno et al. (2021) using their scaled hypersphere search-anharmonic downward distortion following the (SHS-ADDF) method. The group of Fourré et al. (2016, 2020) developed and benchmarked a software tool that was able to determine all possible isomers of a given composition.
Their approach uses a one-to-one correspondence between atoms with bonds and a weighted graph with vertexes within the graph theory framework. A thorough inspection of a specified vertex in a graph allows for extraction of all the possible isomers identified as unique graphs. Such a procedure is well suited for small-size molecules if the number of conformational degrees of freedoms are negligible. However, for larger molecules, the counting of all the possible graphs implies the risk of including unrealistic structures and connections, thus, requiring a more careful handling of the conformational degrees of freedom.
For this reason, our RC method represents a viable way to overcome these difficulties by introducing random samplings for both bond connections and initial atomic geometries and introducing a universal molecular force field that does not require adjustment according to the molecules that are targeted. The detailed computational procedure is described in Section 2. The advantages and disadvantages of our RC method compared with the previous approach by Fourré et al. (2016, 2020) are summarized in the Supporting Information (Supplementary Table S1).
In Section 3, the results of stable isomers for seven molecules, specifically glycine (
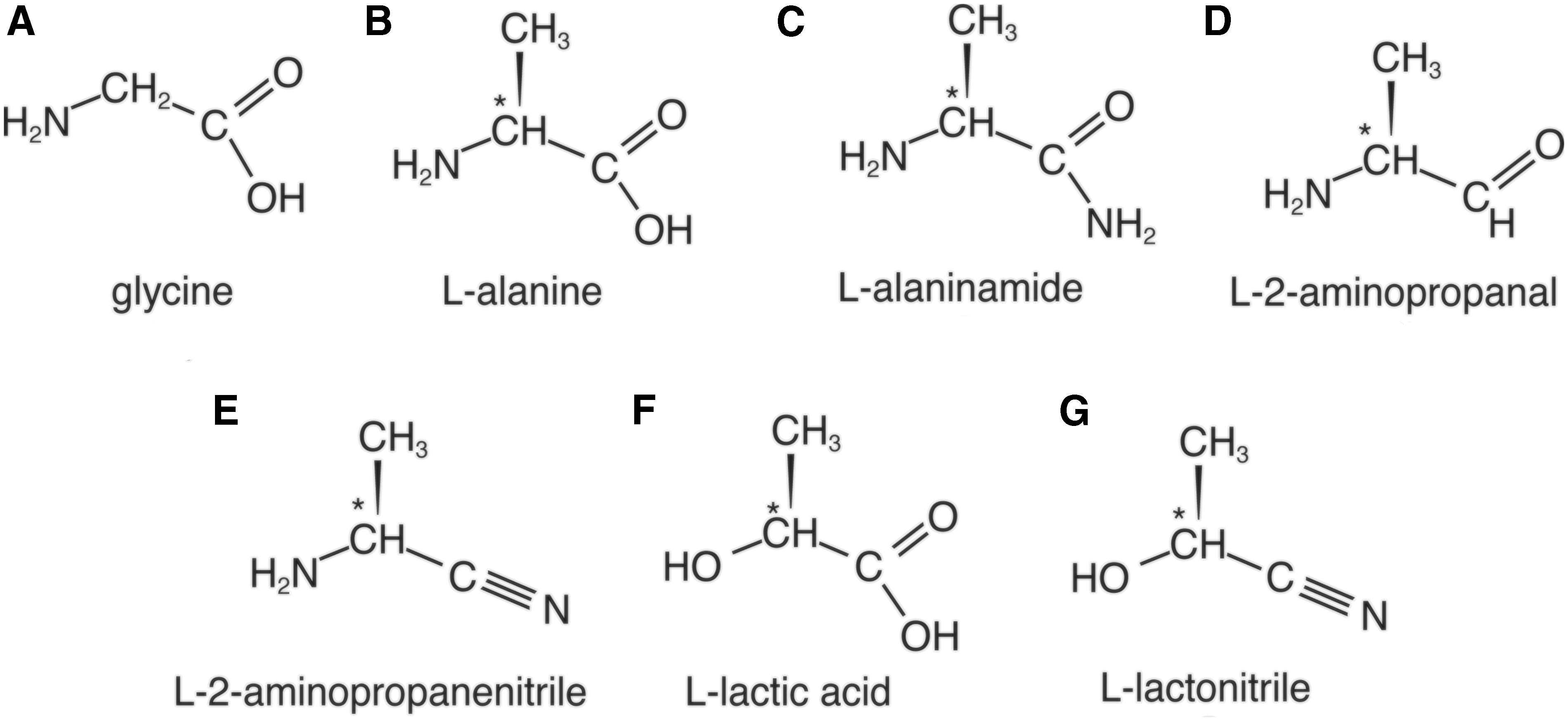
Molecular structures investigated in the present study (
2. Methods
The computational procedures of the RC method are detailed in this section. The overall flowchart is sketched in Fig. 3 and can be summarized as follows:
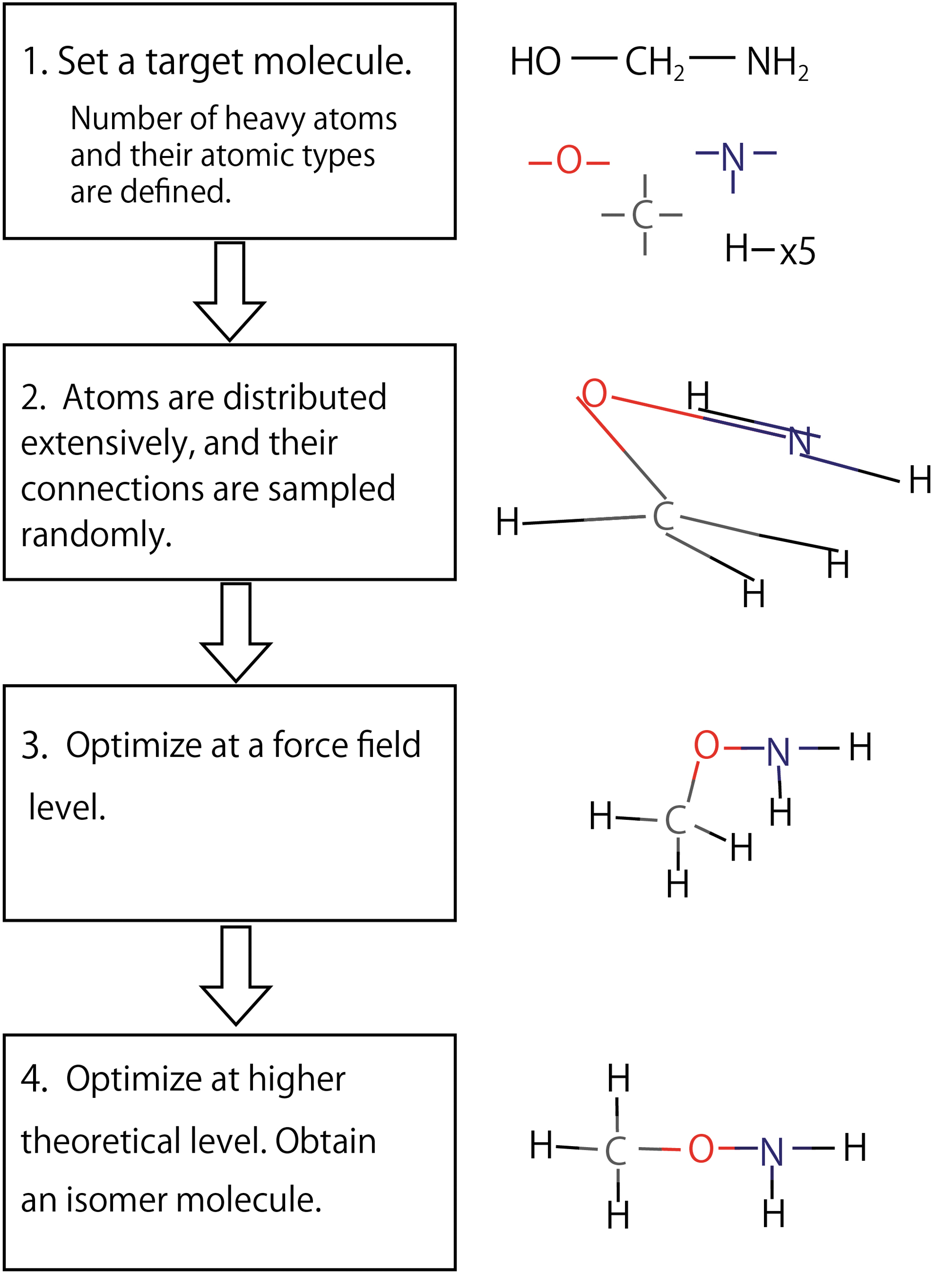
Schematic flowchart of the RC method proposed and implemented in the present work. By repeating the procedures 2–4, a number of isomers in different conformations are generated. RC = random connection.
A target molecule is initially identified. The number of atoms and their chemical types are then counted by virtually eliminating all their covalent bonds. For example, the molecule shown in Fig. 3 contains (1) one oxygen atom with two covalent bonds, (2) one carbon atom with four covalent bonds, (3) one nitrogen atom with three covalent bonds, and (4) five hydrogen atoms with one covalent bond. This virtual splitting allows for the identification of all the main building blocks of related isomers.
To construct new isomers, the isolated atoms are redistributed uniformly in a cubic box of 10 Å side length. Despite looking a bit arbitrary, this procedure is necessary to sample different conformers. The atoms are then newly bond-connected to other atoms with unconnected bonds. Initially, heavy atoms are connected. The bond-connections are randomly selected from all the available possibilities. Then, unconnected bonds are saturated with hydrogen atoms. By this procedure, the bonds are drawn to form a single molecule saturating the number of covalent bonds of each atom, avoiding spurious formation of separate molecules. If the atom types are not unique, all the possible atom-type-sets are searched separately. For instance, the O atom in a carboxylic acid has two possibilities, either the double-bonded carbonyl oxygen ( = O) or the single-bonded hydroxyl/ether oxygen (-O-).
After the molecule has the complete information of bond connection, the molecular geometry is optimized at a force field level. Our specific force field consists of a harmonic and a van der Waals component, accounting for the bonding (E R) and nonbonding (E vdW) interactions, respectively
The indices i, j run over all the bonded atom pairs, and the indices k, l run over the nonbonded atom pairs. This force field is a simplified form of the universal force field (UFF) (Rappe et al., 1992). The other terms of the original UFF, that is, bond angle bending (E q), dihedral angle torsion (E q), and electrostatic terms (E el), are not considered in the present study, because an exploratory rough optimization is needed in this stage. Further and more accurate optimizations within electronic structure calculations are carried out in the next optimization step, where the bond information is not valid under the calculation. All the parameters used in the present study (r ij, D ij) are reported in the supporting information (Supplementary Table S2).
The accurate structural optimization within higher-level theoretical approaches is performed at this step. We stress the fact that, in practical applications of this protocol, steps 2–4 should be carried out many times for sampling. It is not always granted that the former step succeeds in the generation of new bond-connections and different conformations of the isomer. It is recommended to carefully check the sets of the generated molecular structures before performing higher-level theoretical calculations, which is the most time-consuming. In our case, we adapted a procedure that makes use of two different levels of electronic structure calculation. In a first instance, we used a density functional theory (DFT) approach at the theoretical levels of B3LYP/6-31G and B3LYP/6-311++G(d,p). The DFT can properly reproduce the stable molecular structures and has been shown to be a viable way to be applicable to a high number of samplings. Zero-point vibrational energy (ZPE) corrections were evaluated at the B3LYP/6-311++G(d,p) theoretical level by performing frequency calculations. In the RC method, random samplings were performed for both connections and geometries. If the number of samplings is insufficient, a complete search for isomers is not pursued. Duplications that resulted in an identical molecule were used to check whether the sampling was sufficient. We found that 1000 samplings were sufficient to generate stable isomers for the present molecules (
All the electronic structure calculations were performed using the Gaussian16 program package (Frisch et al., 2016). Molecular structures shown in the figures were drawn using the visual molecular dynamics program (Humphrey et al., 1996).
3. Results and Analysis
3.1. Glycine isomers (C2H5NO2, A )
Glycine is the simplest amino acid and is a fundamental constituent of the main chain of proteins. Moreover, glycine can be synthesized in the laboratory and has been found in meteorites. Theoretical studies have been performed to inspect the formation and molecular properties of glycine, with special attention to conformers and isomers. This makes glycine the best reference molecule with which to check the validity of our RC method. Ohno et al. (2021) showed that there are two isomers that are lower in energy, and hence more stable, than glycine by using their SHS-ADDF method.
The most stable isomer was identified as CH3NHCOOCH (N-methylcarbamic acid,
The RC method allowed for reproduction of the reported results, and three stable conformers of glycine were obtained (Table 1 and Fig. 4). These results suggest that glycine is not the most stable isomer in C2H5NO2, and its energy is higher by 10.0 kcal mol−1 relative to the most stable isomer. The formation of the Cα carbon, -NH-Cα-CO-, requires higher energy compared with the formation of the direct carbonyl-amino groups linkage, that is, the peptide bond (-NH-CO-). All these stable isomers contain the carbonyl group (-C = O-), and the other unsaturated bonds of the ethylene group (C = C) and imine group (C = N) are not relatively stable. NH2CH2OCHO (aminomethyl formate,
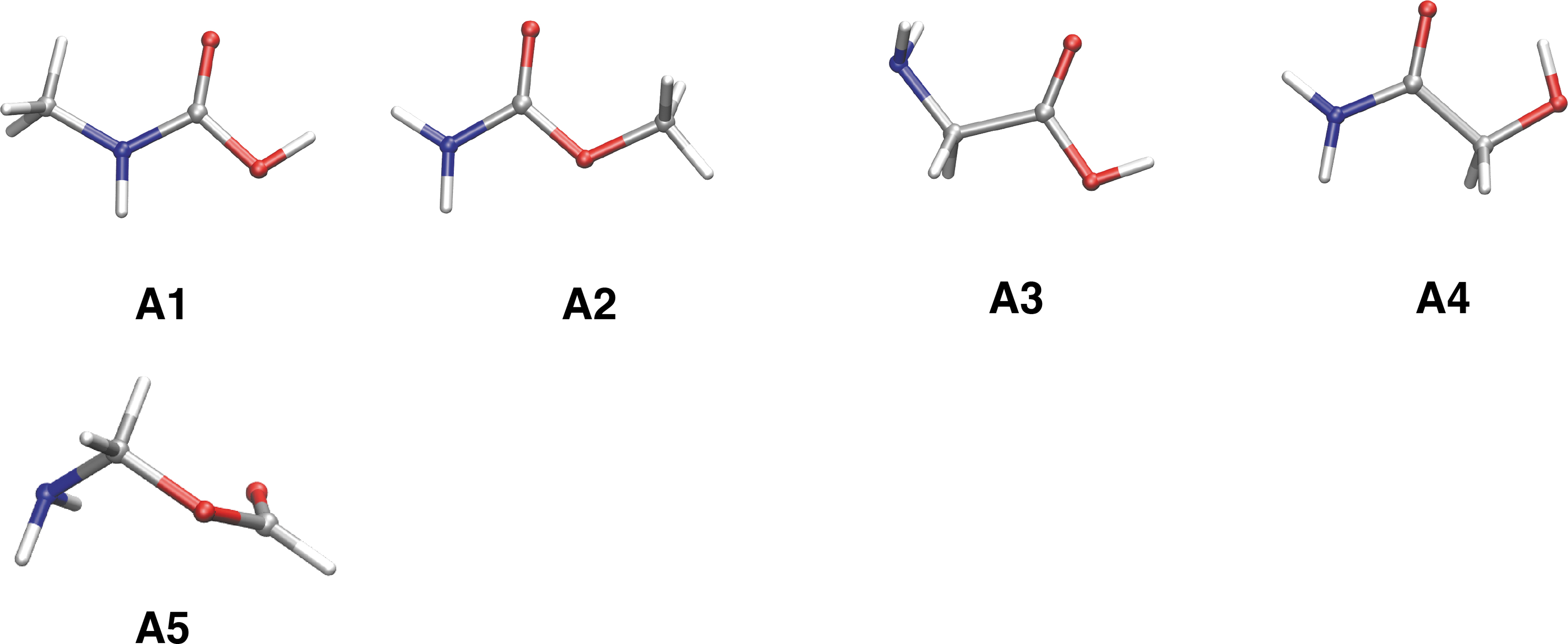
Stable isomers of the glycine composition (C2H5NO2). Most stable conformers for each isomer (
Relative Energies (ΔE) and Number of Stable Conformations (N conf.) in Isomers in Glycine Composition (C2H5NO2) as Obtained by the Random Connection Method
ZPE corrected relative energies (ΔE) with respect to the most stable isomer at the theoretical level B3LYP//6-311++G(d,p). Relative energies computed at the CCSD(T)/cc-pVTZ level are shown in parentheses.
Number of stable conformations with respect to the most stable conformation of glycine (
ZPE = zero-point vibrational energy.
3.2. Alanine isomers (C3H7NO2, B )
Given the reliability of the RC method in generating the stable isomers of glycine, we used this approach to search for alanine isomers (C3H7NO2). After 1000 samplings, we obtained 4 isomers that were more stable than alanine: N-ethylcarbamic acid (CH3CH2NHCOOH,
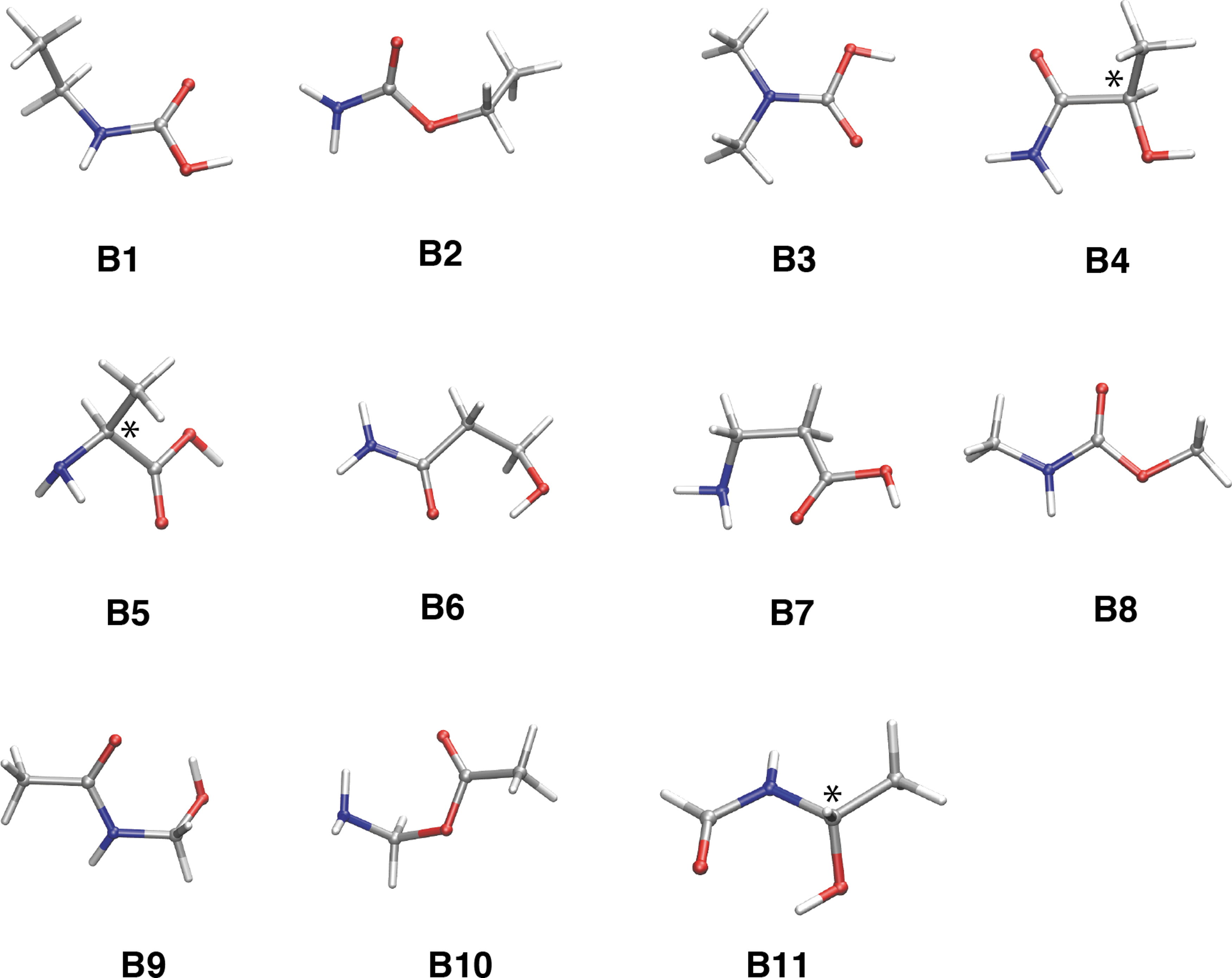
Stable isomers of the alanine composition (C3H7NO2). Most stable conformers for each isomer (
Relative Energies (ΔE) and Number of Stable Conformations (N conf.) in Isomers in Alanine Composition (C3H7NO2) as Obtained by the Random Connection Method
Isomers contain a chiral center are marked by asterisk.
ZPE corrected relative energies (ΔE) with respect to the most stable isomer at the theoretical level B3LYP//6-311++G(d,p). Relative energies computed at the CCSD(T)/cc-pVTZ level are shown in parentheses.
Number of stable conformations with respect to the most stable conformation of alanine (
Alanine (
In an analogous way, other stable isomers of alanine can be realized as follows:
It should be mentioned that there are no stable isomers that contain the ethylene group and imine in the stable isomers of alanine. It is also expected that all the monomer forms of the amino acids are characterized by an instability in comparison with the isomers, because the main chain part of the amino acid, NH2-CH-COOH, is energetically located at a higher value with respect to the methylcarbamic acid part (-CH2-NH-COOH).
3.3. Alaninamide isomers (C3H8N2O, C )
As alanine is not the most stable isomer in C3H8N2O, we turned our investigation to the alanine derivative, alaninamide. For this system, Fig. 6 and Table 3 show the molecular structures and summarize their relative energies. We found that only the N-ethylurea (
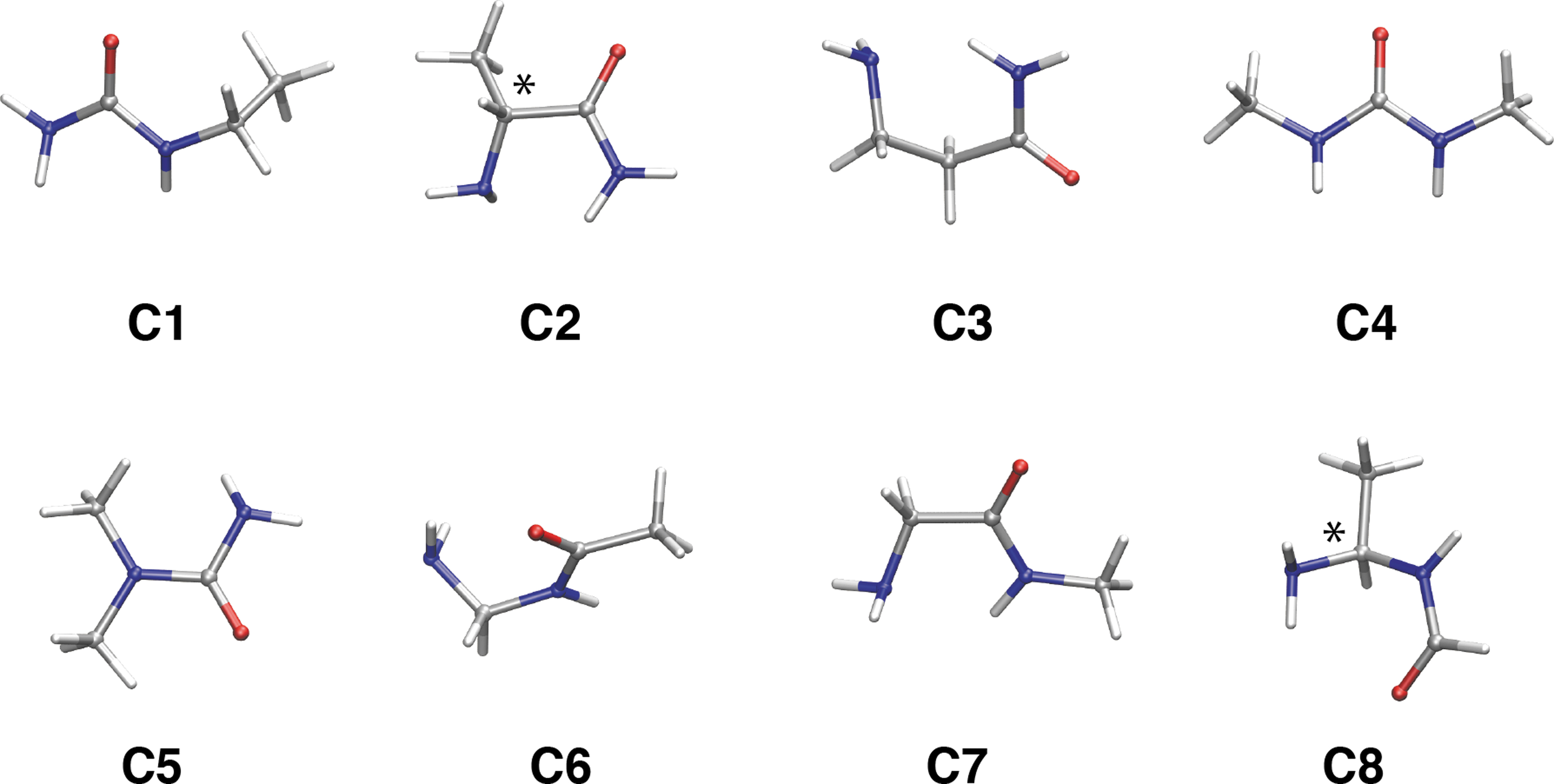
Stable isomers of the alaninamide composition (C3H8N2O). Most stable conformers for each isomer (
Relative Energies (ΔE) and Number of Stable Conformations (N conf.) for Isomers in Alaninamide Composition (C3H8N2O) as Obtained by the Random Connection Method
Isomers contain a chiral center are marked by asterisk.
ZPE corrected relative energies (ΔE) with respect to the most stable isomer at the theoretical level B3LYP//6-311++G(d,p). Relative energies computed at the CCSD(T)/cc-pVTZ level are shown in parentheses.
Number of stable isomers with respect to the most stable conformation of alanine amide (
The isomers whose stability is comparable to
3.4. 2-Aminopropanal isomers (C3H7NO, D )
In the ensemble of systems that have the C3H7NO composition, we found that there are eight isomers more stable than 2-aminopropanal (
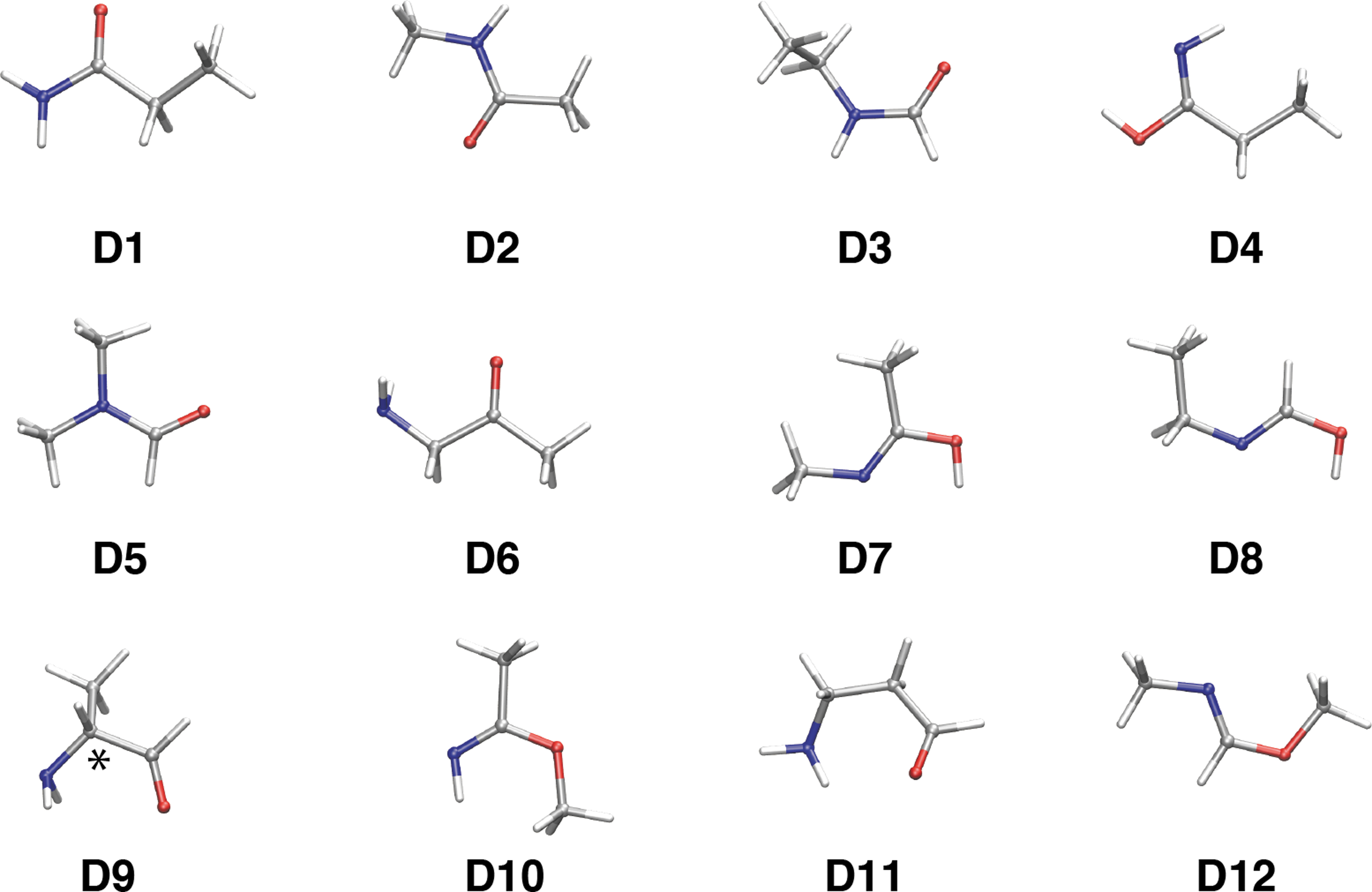
Stable isomers of the 2-aminopropanal composition (C3H7NO). Most stable conformers for each isomer (
Relative Energies (ΔE) and Number of Stable Conformations (N conf.) for Isomers in 2-Aminopropanal Composition (C3H7NO) as Obtained by the Random Connection Method
Isomers contain a chiral center are marked by asterisk.
ZPE corrected relative energies (ΔE) with respect to the most stable isomer at the theoretical level B3LYP//6-311++G(d,p). Relative energies computed at the CCSD(T)/cc-pVTZ level are shown in parentheses.
Number of stable isomers with respect to the most stable conformation of 2-aminopropanal (
The number of conformations of the stable isomers is 2, 3, 2, 4, 1, 4, 2, and 1 for all the systems from
Other stable isomers above
Another correspondence worthy of note is the tautomerization of amide (-NH-CO-) into imidic acid [-N = C(OH)-]. The analogies that arise in this case are
3.5. 2-Aminopropanenitrile isomers (C3H6N2, E )
As a further step in our investigation, we focused on the isomers of amid nitril, NH2CH(CH3)CN. We found that the most stable form is 2-aminopropanenitrile (
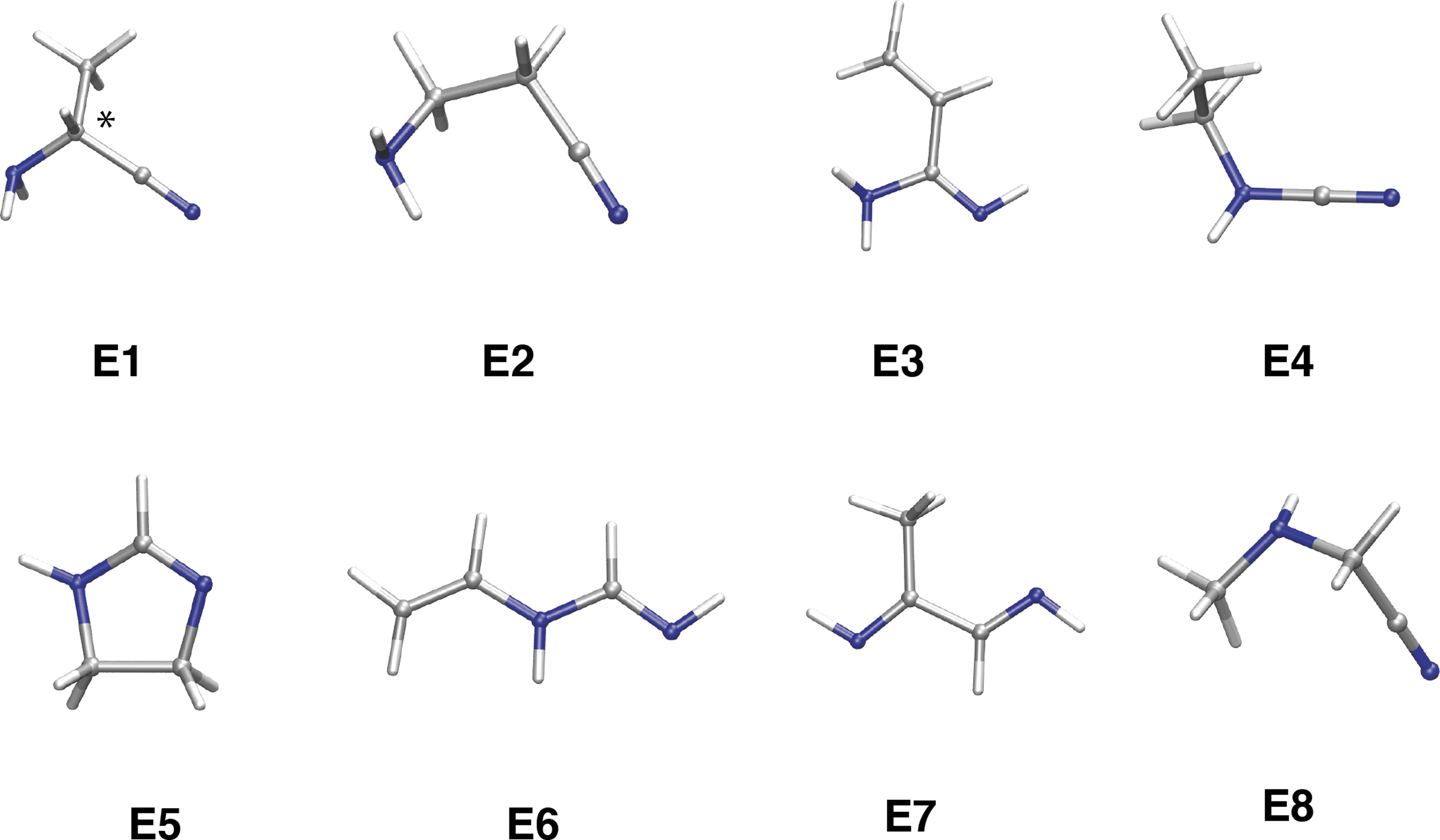
Stable isomers of the 2-aminopropanenitrile composition (C3H6N2). Most stable conformers for each isomer (
Relative Energies (ΔE) and Number of Stable Conformations (N conf.) for Isomers in 2-Aminopropanenitrile Composition (C3H7NO) as Obtained by the Random Connection Method
Isomers contain a chiral center are marked by asterisk.
ZPE corrected relative energies (ΔE) with respect to the most stable isomer at the theoretical level B3LYP//6-311++G(d,p). Relative energies computed at the CCSD(T)/cc-pVTZ level are shown in parentheses.
Number of stable conformations with respect to the most stable conformation of 2-aminopropanenitrile (
Imidazoline (
3.6. Lactic acid isomers (C3H6O3, F )
Isomers of lactic acid, 2-hydroxypropanoic acid [HOCH(CH3)COH], were our next target. We found that only etabonic acid (
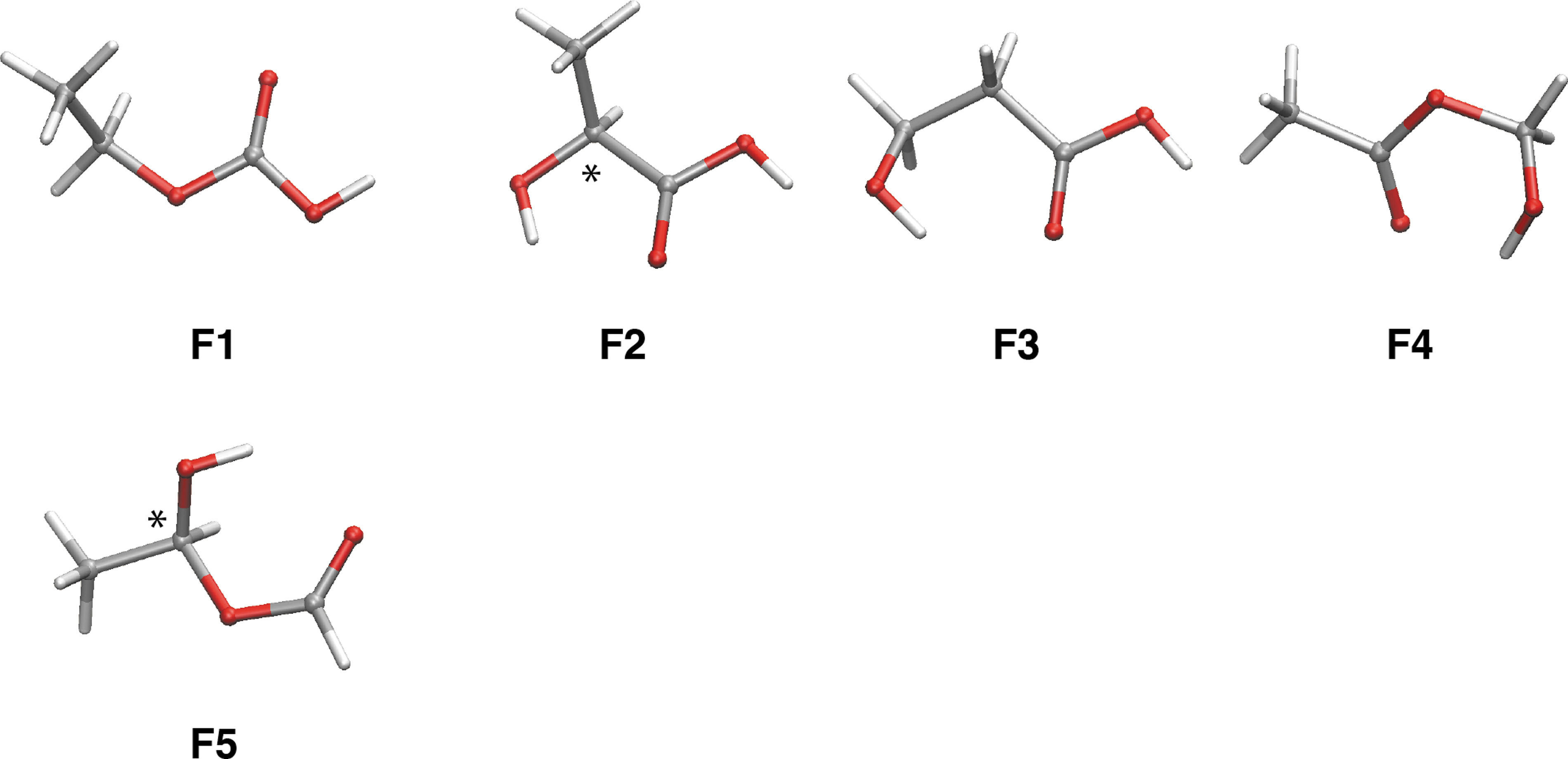
Stable isomers of the lactic acid composition (C3H6O3). Most stable conformers for each isomer (
Relative Energies (ΔE) and Number of Stable Conformations (N conf.) for Isomers in Lactic Acid Composition (C3H6O3) as Obtained by the Random Connection Method
Isomers contain a chiral center are marked by asterisk.
ZPE corrected relative energies (ΔE) with respect to the most stable isomer at the theoretical level B3LYP//6-311++G(d,p). Relative energies computed at the CCSD(T)/cc-pVTZ level are shown in parentheses.
Number of stable conformations with respect to the most stable conformation of lactic acid (
In this substitution,
3.7. Lactonitrile isomers (C3H5NO, G )
To complete our study, we sought the isomers of hydroxy nitril, HOCH(CH3)CN. In this case, we found that two isomers are more stable than 2-hydroxypropanenitrile (
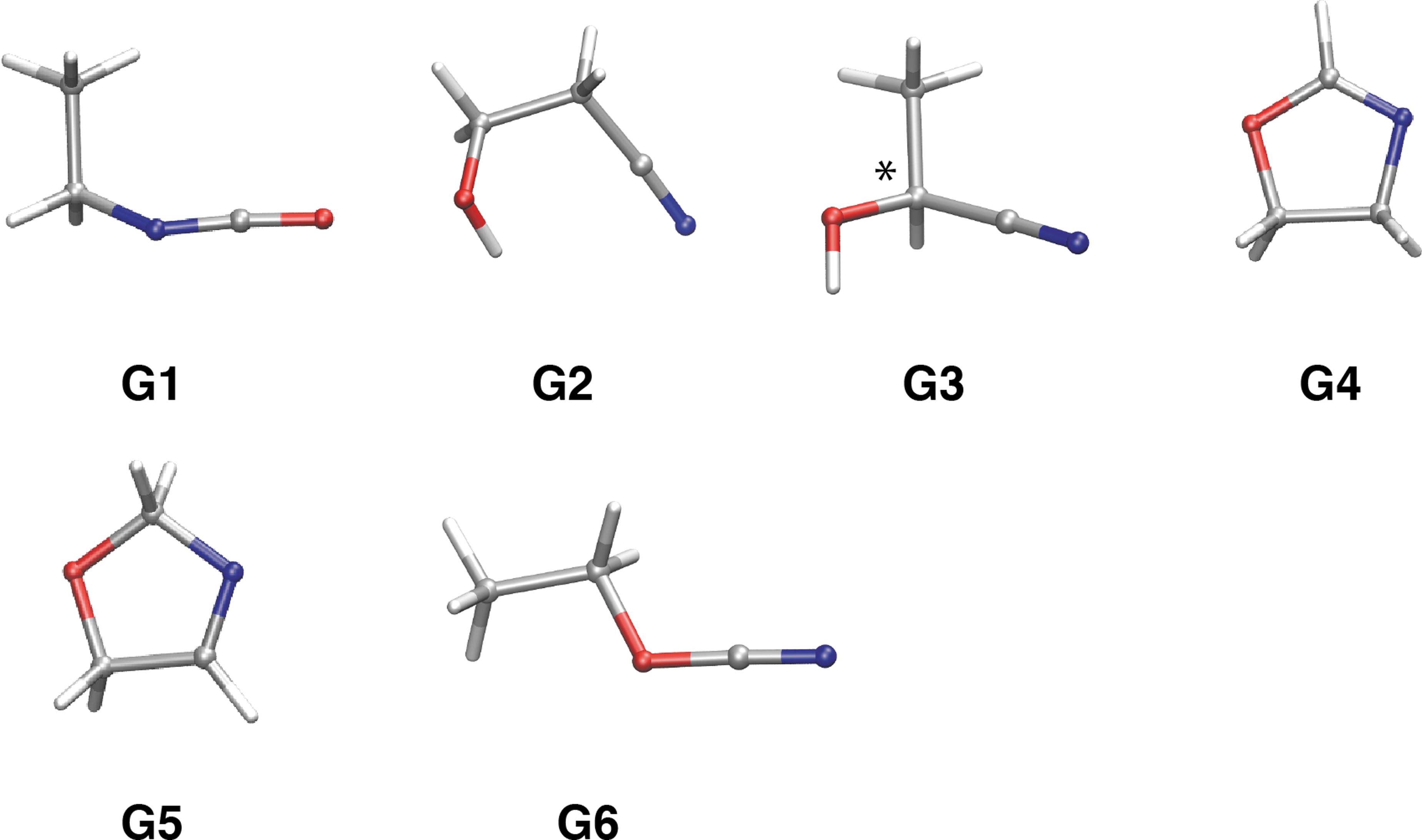
Stable isomers of lactonitrile composition (C3H5NO). Most stable conformers for each isomer (
Relative Energies (ΔE) and Number of Stable Conformations (N conf.) for Isomers in Lactonitrile Composition (C3H5NO) as Obtained by the Random Connection Method
Isomers contain a chiral center are marked by asterisk.
ZPE corrected relative energies (ΔE) with respect to the most stable isomer at the theoretical level B3LYP//6-311++G(d,p). Relative energies computed at the CCSD(T)/cc-pVTZ level are shown in parentheses.
Number of stable conformations with respect to the most stable conformation of lactonitrile (
Hydroxypropanenitriles (
3.8. Origin of homochirality
The present results indicate that the thermodynamical stability of amino nitrile is remarkably advantageous, and that amino nitrile is a key chiral molecule in comparison with other precursors and amino acids in the interstellar and circumstellar medium. Recently, the first chiral molecule, propylene oxide, was detected in Sagittarius (Sgr) B2(N) (McGuire et al., 2016). Theoretical investigations carried out by Hori and coworkers suggest that the photolysis of propylene oxide or a cation precursor can induce the homochirality (Hori et al., 2022).
If the enantiomeric excess of chiral isomer generates from photoreactions under circular polarized light (Fukushima et al., 2020), the most stable and longest-living species, aminonitriles, become the best candidates responsible for the enantiomeric excess of amino acids. Therefore, we expect that the 2-aminopropanenitrile (3-aminopropanenitrile) might be a crucial molecule that contributes to the origin of homochirality for all the chiral α-amino acids (β-amino acids). Although 2-aminopropanenitrile has not yet been detected in Sgr B2(N) (Møllendal et al., 2012), observations and analyses for aminonitriles are worth consideration as targets.
On the other hand, aminoacetonitrile, the precursor of glycine, has been detected in Sgr B2 (Belloche et al., 2008). Amino acids are much less stable than nitriles on UV irradiation and cosmic rays in interstellar regions (Bernstein et al., 2004). Previous theoretical studies have shown that the formation of aminoacetonitrile requires the overcoming of a relatively low energy barrier (ΔU = 8–9 kcal mol−1), though the hydrolysis of aminoacetonitrile to glycine is characterized by a higher electronic energy barrier (ΔU = 27-34 kcal mol−1), which cannot proceed at cryogenic temperatures (Woon, 2008; Rimola et al., 2010).
Assuming a typical temperature of T < 100K in the molecular cloud, the energy differences of ΔE = 3.2 and 5.7 kcal mol−1 correspond to the population differences of less than 1.0 × 10−7 and 3.5 × 10−13 according to the Boltzmann distribution. It should be noted that abundance in the ISM is not only determined exclusively by thermal stability but also by environmental effects, such as adsorption and reactivity with atomic hydrogen, which can contribute to abundance as well (Fourré et al., 2020). Nonetheless, it becomes clear that a small relative energy difference of even ΔE = 3.2 kcal mol−1 contributes to the relative probability in the ISM.
These reactions are deeply different from ordinary chemical reactions that occur in the laboratory at T = 300K, which can overcome energy barrier up to the values of about ΔE = 20 kcal mol−1 by thermal activation (Shoji et al., 2022).
3.9. Limitation of the present theoretical approach
In the present theoretical investigation, amino acids and their chiral precursors are assumed to be in the gas phase. In the ISM, the dominant molecular formation occurs on the surface of dust and ice, and the processes of adsorption and diffusion of molecules on the surfaces can play a role in the chemical evolution of interstellar molecules (Vidali, 2013; Wakelam et al., 2017; Kayanuma et al., 2017). Indeed, isomers of isocyanic acid (HNCO) have been shown to be characterized by different adsorption energies with an upper limit of 19.4 kcal mol−1 (Lattelais et al., 2015).
Nonetheless, even for the large perturbation on relative energies due to surfaces adsorption, HNCO and HOCN isomers remain the most and the second-most stable isomers, respectively. Thus, this energetic order is not jeopardized by the vicinity of an adsorbing surface. This is consistent with our results, and it is confirmation that the relative energies obtained in the gas phase (MEP) are reliable and represent a reliable predictive quantity for the identification of molecules. Further theoretical investigations on the relative stabilities on icy surfaces will be desirable and represent the target of forthcoming studies.
4. Conclusion
To investigate the stabilities of alanine and the related chiral molecules among their isomers, six chiral molecules formed during prebiotic processes and achiral glycine were targeted in the present theoretical study. The molecular mechanism of the formation of homochirality of amino acids is intimately related to the origin of life on Earth. The molecules of amino acids and their precursors are of a large size compared with molecules detected in ISM, and their conformational isomers as well as their structural isomers should be searched explicitly for their relative stabilities.
The approach originally proposed by Fourré et al. (2016, 2020) has been demonstrated to be efficient for the search for ISM molecules provided that the number of conformers is not too large. Nonetheless, it can fail in finding exhaustively all the relevant conformers and unexpected bonding, especially for larger and complex molecules. To overcome this difficulty, we developed a new isomer search algorithm, referred to as the RC method, by introducing random selections for the degrees of freedoms of bond connection and conformation.
The successful application of the RC method to glycine allowed us to move to the actual target of this study. More precisely, two isomers, N-methylcarbamic acid (
The RC method was applied to chiral molecules, including alanine and alanine precursors. Based on the relationship of the molecular frameworks in different compositions, we found that there are structural similarities between the stable isomers. Among the stable isomers of alanine and alanine precursors (
The higher energies of α-amino acid and the precursors can be explained by the group substitutions; the main chain part of the amino acid, NH2-CH-COOH, is higher in energy with respect to the methylcarbamic acid part (-CH2-NH-COOH).
Analogously, the hydroxy acids and the precursors (
Despite their structural similarities to
The intrinsic stability of 2-aminopropanenitrile is a convincing piece of evidence on which the origin of homochirality of amino acids can be rationalized. To generate the enantiomeric excesses of
In this respect, this work can stimulate forthcoming theoretical and experimental investigations to consolidate or disprove the outcome of our modeling.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by JST, PRESTO Grant Number JPMJPR19G6, Japan and JSPS KAKENHI grant numbers 19H00697, 20H05453, 20H05088, and 22H04916. This work used computational resources of (1) Cygnus system through Multidisciplinary Cooperative Research Program in CCS, University of Tsukuba, and (2) supercomputer Fugaku provided by the RIKEN Center for Computational Science (Project ID: hp210115). Mauro Boero thanks the HPC Center at the University of Strasbourg funded by the Equipex Equip@Meso project and the CPER Alsacalcul/Big Data, and the Grand Equipement National de Calcul Intensif (GENCI) under allocation DARI-A0100906092.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Abbreviations Used
Associate Editor: Michael C. Storrie-Lombardi
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.