
Abstract
Samples returned from Mars would be placed under quarantine at a Sample Receiving Facility (SRF) until they are considered safe to release to other laboratories for further study. The process of determining whether samples are safe for release, which may involve detailed analysis and/or sterilization, is expected to take several months. However, the process of breaking the sample tube seal and extracting the headspace gas will perturb local equilibrium conditions between gas and rock and set in motion irreversible processes that proceed as a function of time. Unless these time-sensitive processes are understood, planned for, and/or monitored during the quarantine period, scientific information expected from further analysis may be lost forever.
At least four processes underpin the time-sensitivity of Mars returned sample science: (1) degradation of organic material of potential biological origin, (2) modification of sample headspace gas composition, (3) mineral-volatile exchange, and (4) oxidation/reduction of redox-sensitive materials. Available constraints on the timescales associated with these processes supports the conclusion that an SRF must have the capability to characterize attributes such as sample tube headspace gas composition, organic material of potential biological origin, as well as volatiles and their solid-phase hosts.
Because most time-sensitive investigations are also sensitive to sterilization, these must be completed inside the SRF and on timescales of several months or less. To that end, we detail recommendations for how sample preparation and analysis could complete these investigations as efficiently as possible within an SRF. Finally, because constraints on characteristic timescales that define time-sensitivity for some processes are uncertain, future work should focus on: (1) quantifying the timescales of volatile exchange for core material physically and mineralogically similar to samples expected to be returned from Mars, and (2) identifying and developing stabilization or temporary storage strategies that mitigate volatile exchange until analysis can be completed.
Executive Summary
Any samples returned from Mars would be placed under quarantine at a Sample Receiving Facility (SRF) until it can be determined that they are safe to release to other laboratories for further study. The process of determining whether samples are safe for release, which may involve detailed analysis and/or sterilization, is expected to take several months. However, the process of breaking the sample tube seal and extracting the headspace gas would perturb local equilibrium conditions between gas and rock and set in motion irreversible processes that proceed as a function of time.
Specialist members of the Mars Sample Return Planning Group Phase 2 (MSPG-2), referred to here as the Time-Sensitive Focus Group, have identified four processes that underpin the time-sensitivity of Mars returned sample science: (1) degradation of organic material of potential biological origin, (2) modification of sample headspace gas composition, (3) mineral-volatile exchange, and (4) oxidation/reduction of redox-sensitive materials (Figure 2). Consideration of the timescales and the degree to which these processes jeopardize scientific investigations of returned samples supports the conclusion that an SRF must have the capability to characterize: (1) sample tube headspace gas composition, (2) organic material of potential biological origin, (3) volatiles bound to or within minerals, and (4) minerals or other solids that host volatiles (Table 4).
List of Findings
Table of Contents
Abstract
Executive Summary
List of Findings
Introduction
Scope of the report
Time-Sensitivity of MSR Investigations: Background
Time-Sensitivity of MSR Investigations: Rationale
Time-Sensitive Processes and Their Characteristic Timescales
Degradation of organic material (including volatile compounds)
Modification of sample headspace gas composition
Mineral-volatile exchange
Hydrous sulfate minerals
Poorly crystalline and X-ray amorphous materials
Phyllosilicate minerals
Hydrous carbonate minerals
Oxidation/reduction of redox-sensitive minerals
Estimating the Loss of Scientific Information as a Function of Time: Implications for Sample Handling and Curation Workflow within an SRF
Recommendations for SRF Sample Processing and Analytical Capabilities
Goal 1: Characterize sample tube headspace gas composition
Goal 2: Characterize organic material of potential biological origin (including volatile hydrocarbons)
Goal 3: Characterize mineral-bound volatiles
Goal 4: Characterize solid-phase volatile hosts
Recommendations for Future Work
Acknowledgments
Disclosure Statement
Funding Information
References
Acronyms Used
T
Once a set of return-worthy samples is collected and cached in the vicinity of Jezero Crater by the Perseverance Rover, MSR is currently planned to continue with two subsequent missions currently scheduled to launch as early as 2026. These missions would collect the cached samples and store them in a capsule on what is referred to as the Mars Ascent Vehicle (MAV). This vehicle would then launch from the martian surface and release a capsule referred to as the Orbiting Sample container (OS) for subsequent capture by a spacecraft that would return the samples to Earth. As early as 2031, that spacecraft could then release the OS (bound within the Earth Entry System (EES)) into Earth's atmosphere. After landing, the Earth Entry System would be collected and transported to a biocontained Sample Receiving Facility (SRF). Once contained within an SRF, the EES and OS would be opened, and the samples would eventually be extracted from their tubes and characterized. Once initial sample characterization and the sample safety assessment protocol are complete, the samples could be distributed to the international scientific community for objective-driven investigation.
The OS container should prevent its contents from exceeding 30°C. Nevertheless, from the moment that samples are collected and sealed within the Perseverance rover to the time that they are opened on Earth, the samples would have experienced a complex temperature history spanning several years. Although this temperature history would be monitored by a variety of on-board and remotely acquired temperature measurements, it is likely to involve transfer of volatile compounds to and from minerals contained within the geological samples anticipated at Jezero Crater and the sample headspace gas. This, in turn, may induce irreversible changes to sample mineralogy and chemical and isotopic composition, as well as changes to the headspace gas. However, if local conditions are known at the time of sampling, along with the temperature history of the tubes, mineralogy of the samples, and gas composition of the headspace soon after they are opened, the complex processes of volatile transfer between rock and gas can be unraveled with thermodynamic and kinetic constraints to determine the state of the samples at the time they were collected at the martian surface.
Equally important, however, is the expectation that, assuming the sample tube seals are still intact, breaking that seal and extracting the headspace gas will perturb local equilibrium conditions between gas and rock and set in motion volatile exchange processes that proceed as a function of time. Therefore, unless these processes are understood and/or accounted for or monitored once the sample tubes are opened, irreversible changes may occur to both geological samples and headspace gas that could jeopardize scientific information gained from further analysis. For this reason, an MSR SRF should have the capabilities to measure these properties inside biocontainment. Understanding these “time-sensitive” processes and their capacity to jeopardize Mars returned sample science and identifying strategies to maximize the retrieval of scientific information within biocontainment are the focus of this report.
Scope of the report
This report is the outcome of a series of meetings held between specialist members of the second MSR Planning Group (MSPG-2). The group (the Time-Sensitive Focus Group; TS-FG) was charged with (1) identifying which MSR science measurements should be considered time-sensitive, (2) determining the
Time-Sensitivity of MSR Investigations: Background
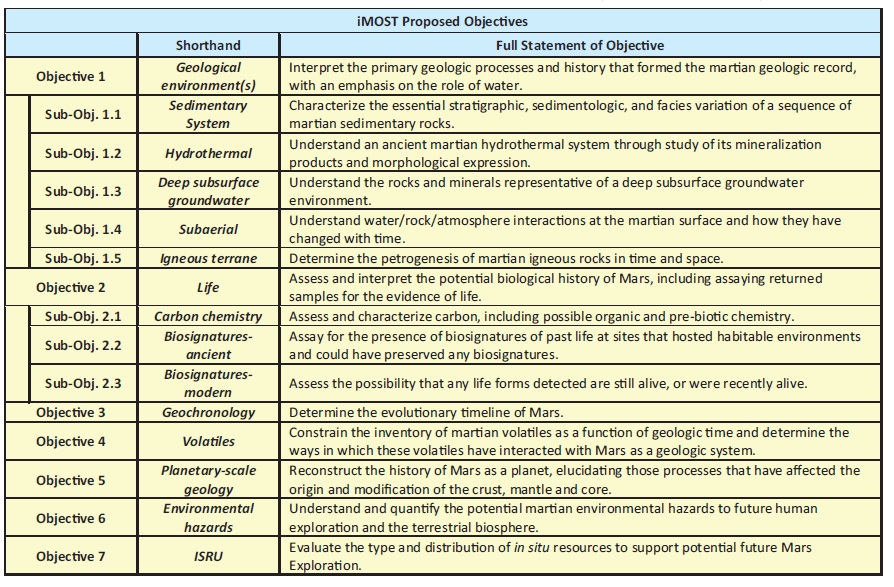
The scientific objectives of MSR have been re-evaluated in many iterations over the last few decades by the National Research Council (e.g., National Research Council, 2011) and the Mars Exploration Program Analysis Group (e.g., MEPAG 2015). Most recently, in 2017, the International Mars Exploration Working Group chartered an international group of scientists to re-evaluate and define the scientific and engineering objectives of MSR. That group (the International MSR Objectives and Samples Team; iMOST), along with defining the scientific objectives of MSR, identified the types of samples and the specific measurements required to best address each objective in a report released in 2018 (Beaty et al., 2019). Those objectives are listed in Table 1. Importantly, iMOST concluded at the time that the final four Mars 2020 landing sites all have the potential to address all objectives outlined in their report, including Jezero Crater, the final landing site of the Mars 2020 Perseverance Rover.
Scientific Objectives of Mars Sample Return (Beaty et al., 2019)
Scientific Objectives of Mars Sample Return (Beaty et al., 2019)
In 2019, the Mars Sample Return Planning Group (MSPG, 2019) established planning assumptions and potential requirements of MSR science performed in the sample receiving facility (SRF) under biological quarantine (accepting iMOST scientific objectives as their terms of reference). One important aspect of the MSPGs work on science in containment was to identify time-sensitive measurements, or those that are sensitive to physical and chemical changes that occur upon opening the sample tubes as samples eventually re-equilibrate with new conditions. The identification of a time-sensitive aspect to measurements in support of MSR implies that some measurements must be made on samples as quickly as possible or, in other words, before the Sample Safety Assessment Protocol (SSAP) could be completed (therefore executing them in biocontainment would be necessary; Figure 1).
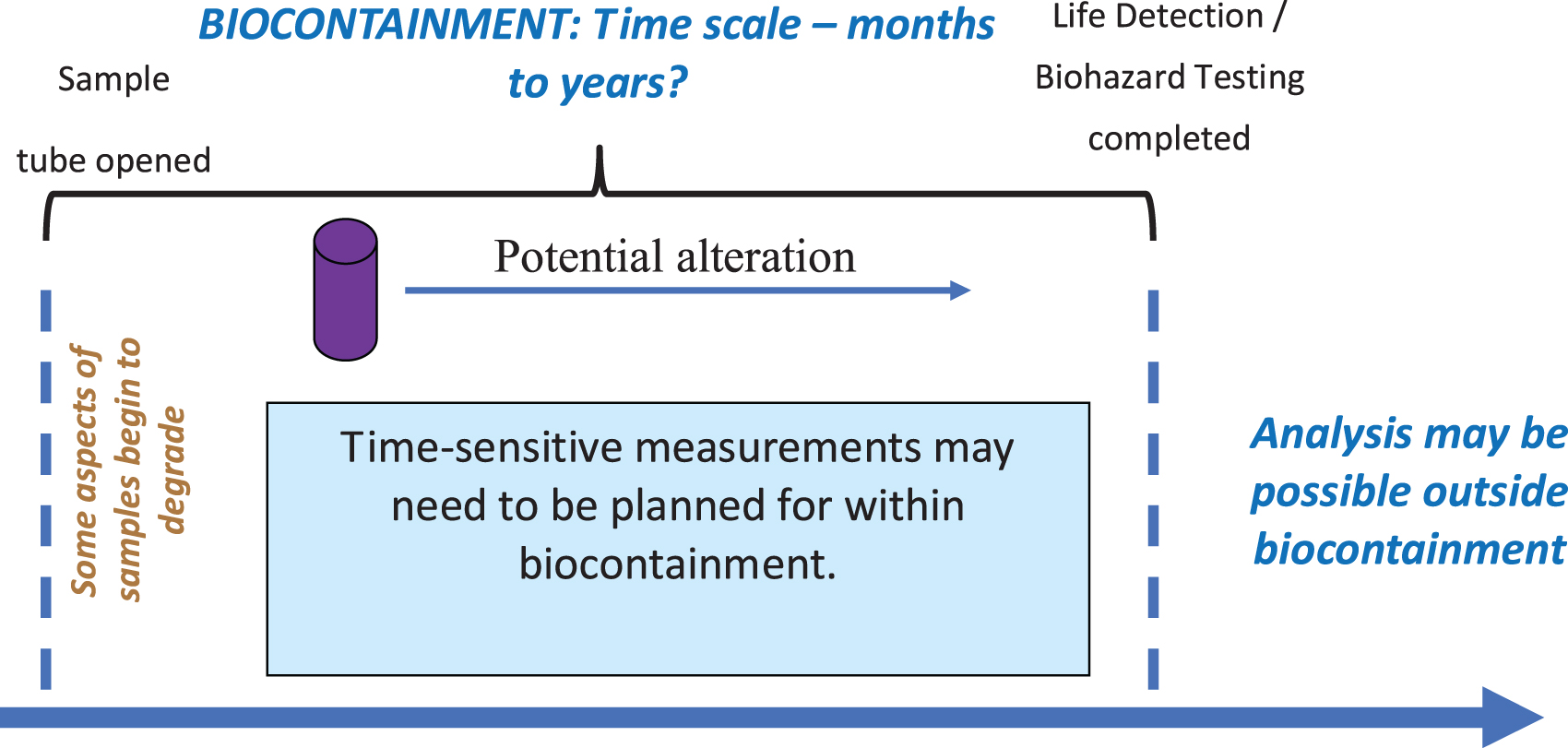
Illustration of relationship of time-sensitive measurements to biohazard testing and biocontainment.
Of the measurements identified by iMOST in 2018 that support the principal science objectives of Mars Sample Return, the MSPG identified 26 as having some degree of time-sensitivity (Supplement-Table 1). However, when those measurements should be made, in what order, and the degree to which scientific information may be lost as a function of time elapsed, was not considered in the report, though it was highlighted as a critical next step in MSR planning.
The MSPG further subdivided the 26 time-sensitive measurements into five categories (MSPG, 2019) as follows: (1) Headspace gas measurements include chemical and isotopic measurements made on headspace gas extracted from sample tubes. The time-sensitivity of these analyses arises because, due to the small molar quantities anticipated, they are deemed especially vulnerable to leakage and contamination during quantitative gas collection, sample transfers, contamination, etc. (2) Hydrated minerals that reflect chemical and isotopic equilibria from Mars include mineralogical, chemical, and isotopic measurements of minerals that incorporate a volatile component derived from, for example, the martian atmosphere or hydrosphere. This includes minerals such as phyllosilicates and hydrated sulfates that are known to exchange volatiles with their surroundings on relatively short timescales. (3) Measurements sensitive to gas-exchange chemistry include those measurements that target the chemical and isotopic composition of redox-sensitive gases (i.e., O2, CO2, H2, H2S, SO2, NOx, ClOx), which are known to exchange or be released from their host phases (i.e., mineral surfaces or lattices) over relatively short timescales. (4) Surface chemistry and reactivity of regolith or dust samples includes measurements that target reaction between regolith materials and H2O or various reagents. The time-sensitivity underpinning these measurements arises because of volatile-exchange processes among minerals and mineral surfaces in materials where finely particulate material (and therefore high total surface area) may be present. (5) Sample preparation processes include various solvent extraction procedures that may generate short-lived reactive intermediate compounds, which may degrade or otherwise react with sample materials.
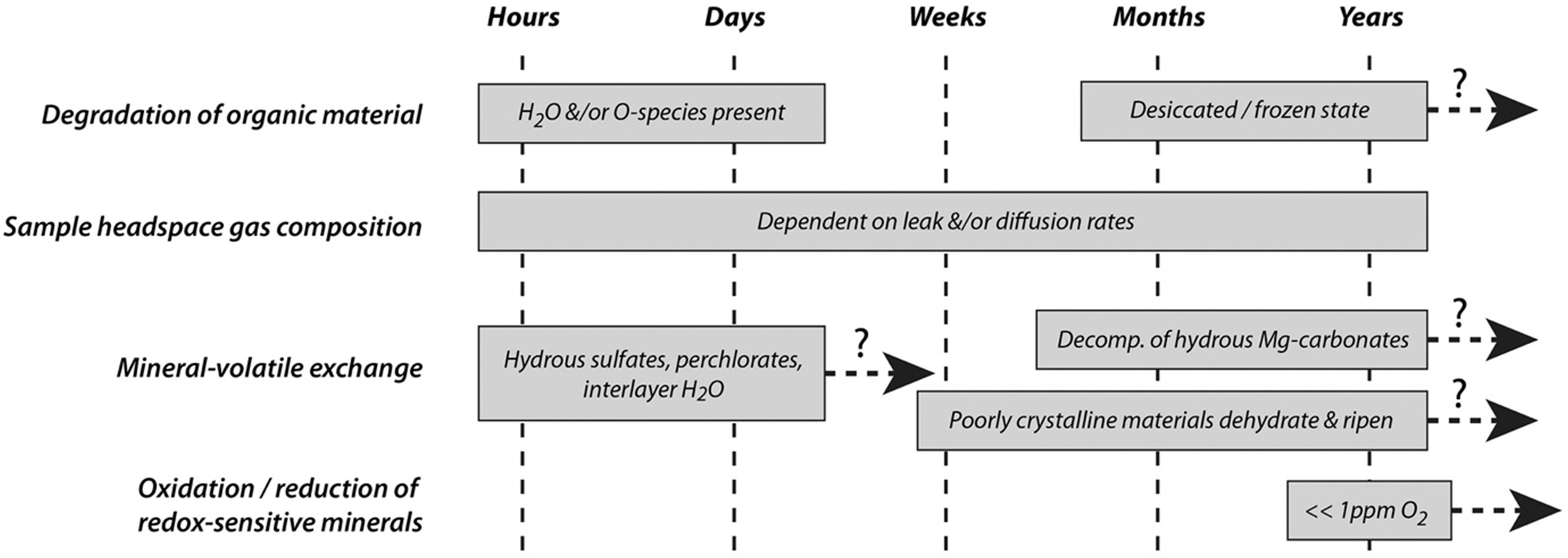
Characteristic timescales of processes that underpin the time-sensitivity of MSR measurements. Some processes (such as the degradation of organic material and mineral-volatile exchange) are associated with different timescales depending on other factors such as environmental conditions and mineralogy.
The initial list of 26 time-sensitive iMOST measurements includes a wide range of specific measurement types to be conducted on a variety of sample types. To identify and focus on the physical and chemical processes that underpin time-sensitivity and estimate their characteristic timescales, the Time-Sensitive Focus Group (TS-FG) has identified four key processes that underpin the time-sensitivity of all 26 time-sensitive iMOST investigations as follows: (1) degradation of organic material (including volatile compounds), (2) modification of sample headspace gas composition, (3) mineral-volatile exchange, (4) oxidation/reduction of redox-sensitive minerals. In what follows, this report summarizes current understanding of each of these four processes in the context of characteristic timescales (Figure 2), and the degree to which scientific information may be lost as a function of time if specific iMOST measurements cannot be completed on very short timescales (Table 4). The report traces the 26 time-sensitive measurements in the context of these four processes to ensure that no measurements were disregarded during the study.
Time-Sensitive Processes and Their Characteristic Timescales
Degradation of organic material (including volatile compounds)
Cells are constructed from many different molecules, but their architecture is dominated by four major classes of macromolecules: lipids, proteins, nucleic acids, and carbohydrates. As all these molecules are chains whose length (i.e., the number of monomers linked together to make the completed molecule) is generally much greater than can be achieved abiotically. When they are not actively repaired by a cellular apparatus, they tend to fragment. This rate of fragmentation/destruction varies and is greatly influenced by the chemical environment in which they are located. In this section, the term “macromolecule” is generally used to refer to the major classes of molecules in extant organisms, but the principles of degradation discussed also apply to some complex organic molecular structures, potentially including biosignature molecules such as kerogen and its components.
An example of the environmental dependence of biological macromolecule stability is the nucleic acid, DNA. In natural environments on Earth, once an organism dies, the DNA is degraded by microorganisms. However, even in the absence of biological activity, DNA undergoes spontaneous hydrolysis (depurination), which results in the loss of purine residues (adenine and guanine) and results in DNA strand cleavage. This leads to an inexorable decay of DNA (Figure 3). These depurination reactions, in the absence of specific catalysts, have the shortest half-lives of DNA degradation reactions (hundreds to thousands of years). However, other pathways include the hydrolysis of the phosphodiester bonds along the DNA backbone and the conversion of cytosine residues to uracil residues (deamination reactions). Specific conditions vary the rates of the reactions. For example, deamination reactions have half-lives of centuries in single stranded DNA on account of the more accessible nature of the bases compared to double stranded DNA. The presence of transition metals, which may be relevant for martian samples, can greatly accelerate the rate of phosphodiester bond breakage (e.g., Gates, 2009).
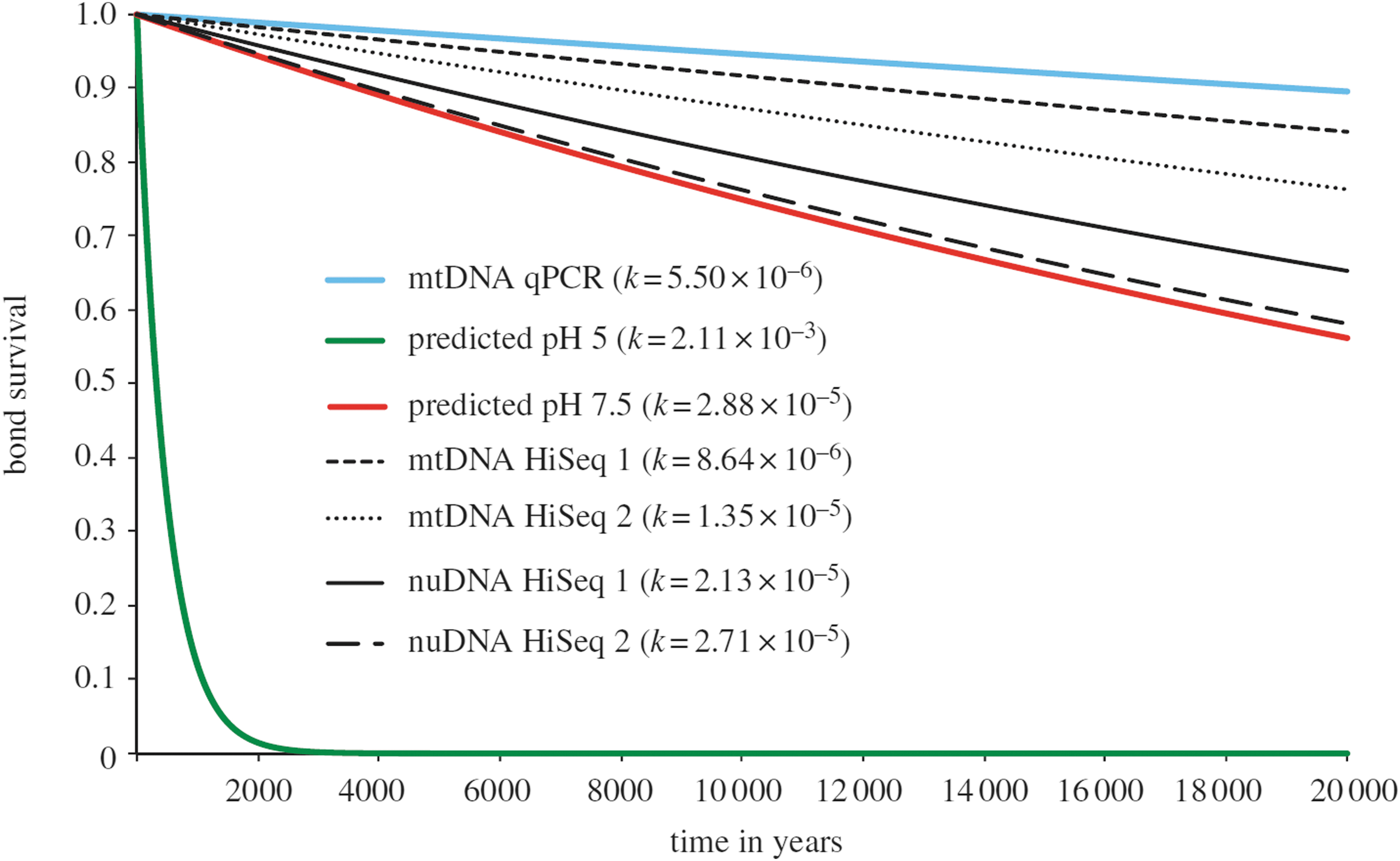
DNA half-life exceeds hundreds of years in bone when storage conditions are appropriate (from Allentoft et al., 2012). The green line shows the predicted rate of DNA degradation at pH 5.0 based on depurination calculations illustrating the change in rate of degradation depending on chemical environment.
The rate of degradation of DNA, as with all molecules, is strongly dependent on the chemical environment. Quantifying these factors and predicting the extent to which they change the half-life of the molecule is complex (Allentoft et al., 2012). Factors that influence the rate of degradation include pH, ionic environment, and the presence of chemical species such as oxidants. Temperature is a strong determinant of stability, as expressed by the Arrhenius relationship, which describes the reduction in chemical reaction rates at lower temperatures. This is thought to account for the detection of DNA in ∼500 kyr ice cores (Willerselev et al., 2007).
There are two specific factors of relevance to the case of martian samples, which are important in accelerating the potential rate of decay of all macromolecules. First, the presence of water in a sample can accelerate decay since it provides a liquid medium for the movement of radicals and other chemical species deleterious to macromolecules. In the absence of water, molecules largely remain in a chemically inactive state, although they will still be subject to radiation damage, for instance, in the natural environment. In a martian sample, the presence or release of liquid water into the sample creating an aqueous environment around macromolecules can, in principle, allow chemical reactions to occur more rapidly. Regardless of the status of water in any given sample, this point would also apply to sample material into which water was added for analytical procedures.
Second, the rates of destruction of macromolecules can be greatly enhanced by the presence of reactive oxygen species (Imlay et al., 1988). For example, the presence of hydrogen peroxide can result in destruction of DNA within minutes. The mechanisms of degradation are not fully elucidated (Linley et al., 2012); however, single-strand breakage caused by the formation of reactive oxygen species, hydroxyl radicals and associated species are likely pathways. Significantly, with respect to martian samples, the presence of iron can facilitate Fenton-mediated reactions that increase degradation rates (Linley et al., 2012).
Similar to DNA, proteins can degrade over periods with half-lives corresponding to hundreds of years under certain conditions, which is far greater than the time periods associated with Mars sample analysis (Radzicka and Wolfenden, 1996; Smith and Hansen, 1998) (Figure 4).
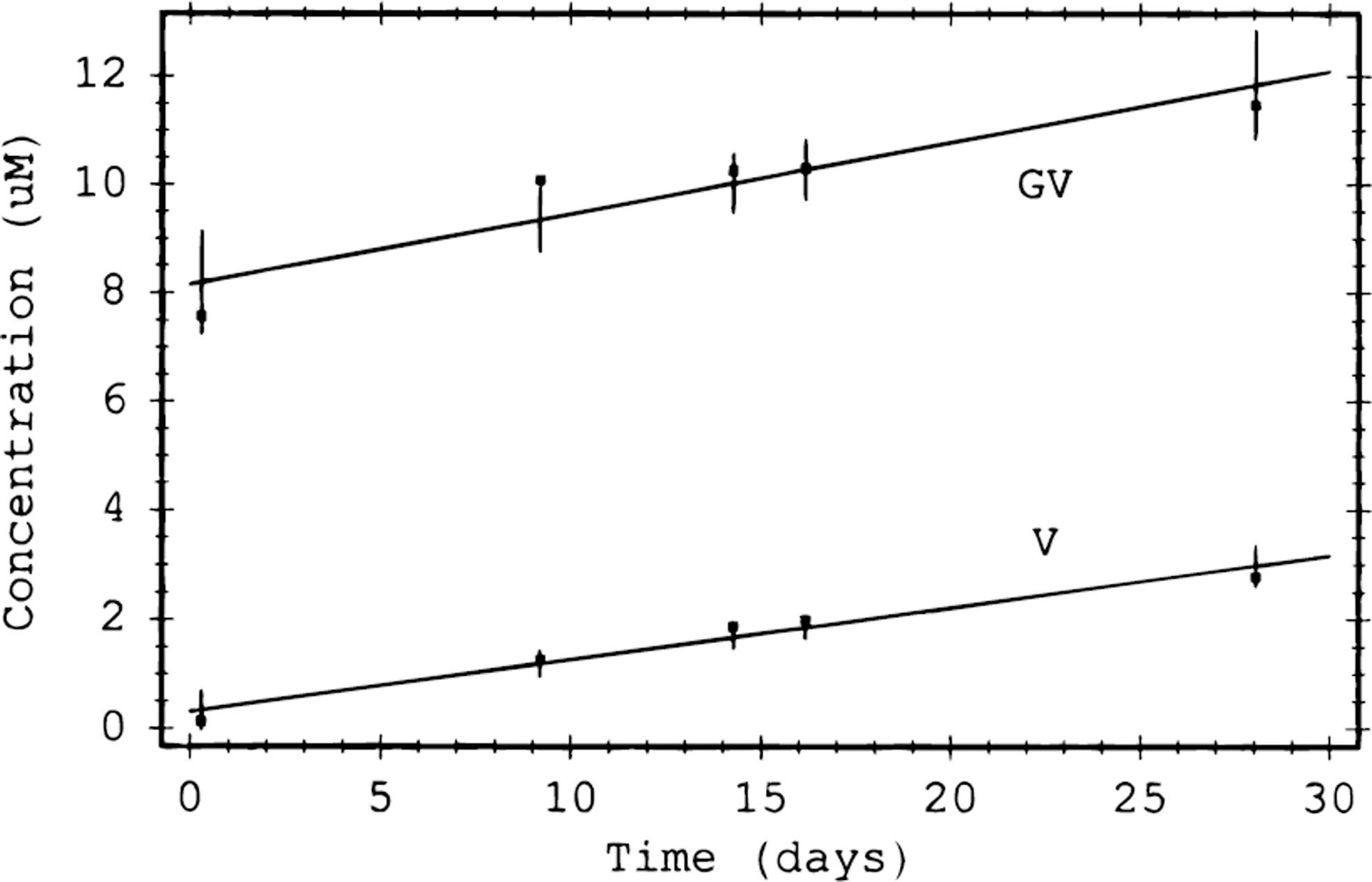
Production of glycyl-valine (GV) and valine (V) from their N-(phenylacetyl)glycyl-D-valine precursor (splitting of peptide bond) at a pH of 7 in water (Smith and Hansen, 1998). These data lead to half-lives at neutral pH conditions of ∼250 years.
However, as with DNA, the presence of reactive oxygen species in aqueous conditions increases the rate of protein destruction (Dean et al., 1996) by direct chemical interaction with amino acids and their connecting peptide bonds. These reactions are influenced, potentially enhanced, by the presence of transition metals, with relevance to martian samples. The order of magnitude increase of the reaction rate cannot be easily predicted for any natural system since, as with DNA, it is determined by a variety of parameters (pH, ionic environment, temperature, concentration of transition metals, radiation environment, etc.), but destruction could occur, in principle, at room temperature and in the presence of oxidants such as hydrogen peroxide on the order of minutes/hours (e.g., Kim et al., 2000).
Qualitatively, from the point of view of time-sensitive science and martian samples, the points raised above illustrate an important principle that motivates our recommendations: macromolecular stability is generally on the order of many years under optimal or near-optimal storage/preservation, but in the presence of chemically reactive species that are known to be relevant to the martian surface, half-times of many key molecules can be reduced to the order of minutes. Although it is currently not possible to quantify these rates exactly, as they depend on the diversity of chemical and physical parameters associated with any given samples, we can state that the minimization of the exposure of samples to conditions (e.g., reactive oxygen species, temperatures greater than ambient room temperature) known to cause macromolecular destruction should be our goal.
Of course, if samples do contain oxidants, then the destruction may have already occurred on the martian surface prior to collection or within the enclosed micro-environment of the sample tube. However, once we have the samples in an SRF, it is prudent to investigate and store the samples in such a way as to minimize their exposure to conditions known to accelerate destruction of biomolecules.
We do not know what organic macromolecules could exist in a martian sample because of the unknown status of any putative life in them. Given the potential for martian life to have evolved with different molecular architectures, we cannot surmise a priori how similar its component molecules would be to those of life on Earth. Hence we propose that the analytical capability should be sufficiently capable to allow for the analysis of a range of different classes of known bioorganic molecules, as well as enable agnostic detection of a range of organic molecular structures.
The visual detection and localization of cellular material and its associated organic signature can be initially carried out by confocal microscopy equipped with Raman/FTIR/UV fluorescence to investigate for the presence of discrete cell-size concentrations of organic matter or cell-like objects and once pliable extracellular relicts. DNA extraction and analysis are specifically focused on the key information molecule of life on Earth, and the capacity to carry out this analysis should be present in case any other evidence of life was detected. We suggest a range of mass spectrometry methods capable of analyzing proteins, lipids, and small molecular weight metabolites associated with life (Section 6.2). These latter methods provide a high degree of agnostic analytical capability since they can detect generic organic molecules.
In situ measurements of the martian atmosphere and evolved gases released from near-surface sedimentary rocks and aeolian materials in Gale crater by Curiosity (e.g., Conrad et al., 2016) and analyses of SNC meteorites (e.g., Ott, 1988; Bogard et al., 2001) indicate that volatile compounds could be present in the samples and headspace gas collected by Perseverance in Jezero Crater. Variable levels of methane that exhibit a seasonal variation have been measured in the atmosphere in Gale crater by the Sample Analysis at Mars (SAM) tunable laser spectrometer (TLS) with an average value of 0.4 ppbv (Webster et al., 2018) and temporary elevated spikes up to 7 ppbv (Webster et al., 2015). These levels of methane are consistent with small sources of methane released from the martian surface or subsurface reservoirs. Higher abundances of methane evolved from drilled rock powders during pyrolysis heating (ppm levels) were also detected by the SAM TLS instrument (Webster et al., 2018), and martian methane that is believed to reside in fluid inclusions or along crystal boundaries has also been released from several martian meteorites by crushing at room temperature (Blamey et al., 2015). A variety of other volatile compounds that include chloromethanes, chlorobenzenes, simple alkanes, dimethylsulfide, methanethiol, and thiophenes have been identified by SAM above background levels during evolved gas analyses and GCMS analyses of sedimentary rocks in Gale crater (Ming et al., 2014; Freissinet et al., 2015; Eigenbrode et al., 2018; Szopa et al., 2020). Although some of these volatiles detected by SAM may be derived from the breakdown of less volatile macromolecular organic matter or from chemical reactions during pyrolysis heating, it is possible that near-surface martian materials in Jezero Crater will contain volatile compounds that could be lost from the samples during sample processing after the tube seals are opened or their concentrations reduced over time due to exposure to elevated temperatures (i.e., >20-25°C) in curation.
The analysis of volatile compounds returned from the Moon during Apollo missions and from comet Wild 2 by the Stardust probe have also showed evidence of loss of sample volatiles under positive pressure in an N2 glove box at ∼20°C. For example, evolved gas measurements of Apollo 16 soil 61221 soon after the lunar sample was returned to Earth showed that the sample was volatile-rich, with HCN comprising ∼5-10% of the total evolved gas released (Gibson and Moore, 1973). Gibson and Moore suggested that the HCN in the lunar soil was derived from a nearby cometary impact while other investigators have suggested that HCN could be derived from solar wind implanted precursors (Holland et al., 1972). HCN and other volatiles, including NH3 and formaldehyde, are also likely chemical precursors for amino acids that have been identified in lunar samples (Fox et al., 1976; Brinton et al., 1996; Elsila et al., 2016). A later investigation of the same Apollo 61221 soil analyzed by Gibson and Moore did not detect HCN in the sample (Epstein and Taylor, 1993), which could be a result of sample heterogeneity or loss of HCN from the sample in curation. During the Apollo era, lunar curators and scientists recognized the fact that HCN and other volatiles could be lost from the samples during long term storage in a nitrogen-purged cabinet at room temperature, so they placed some of the lunar samples in special curation conditions. These include sample cores sealed under vacuum on the lunar surface in containers with indium knife-edge seals that could still hold vacuum today. Some of the Apollo 17 samples were also transferred to a freezer at -20°C within a month after their return to Earth. The vacuum-sealed, standard room temperature, and cold curated Apollo samples are all currently being processed for analyses as part of the Apollo Next Generation Sample Analysis (ANGSA) Program. For example, a sample portion taken from the bottom of the Apollo 73002 upper drive tube core (Figure 5) within 2 days after tube opening was shipped to NASA Goddard for volatile organics analyses that includes HCN, aldehydes/ketones, amines, carboxylic aids, and amino acids (personal communication, J. Elsila on 11/7/19). Gas extraction and headspace measurements of volatiles in the vacuum-sealed Apollo drive tube will also be performed during ANGSA. The results of these measurements could help inform the curation and analysis strategy for Mars Sample Return.

Photo of the Apollo 17 upper drive tube core 73002 that is being processed for analysis under the ANGSA Program. Image credit: NASA/James Blair.
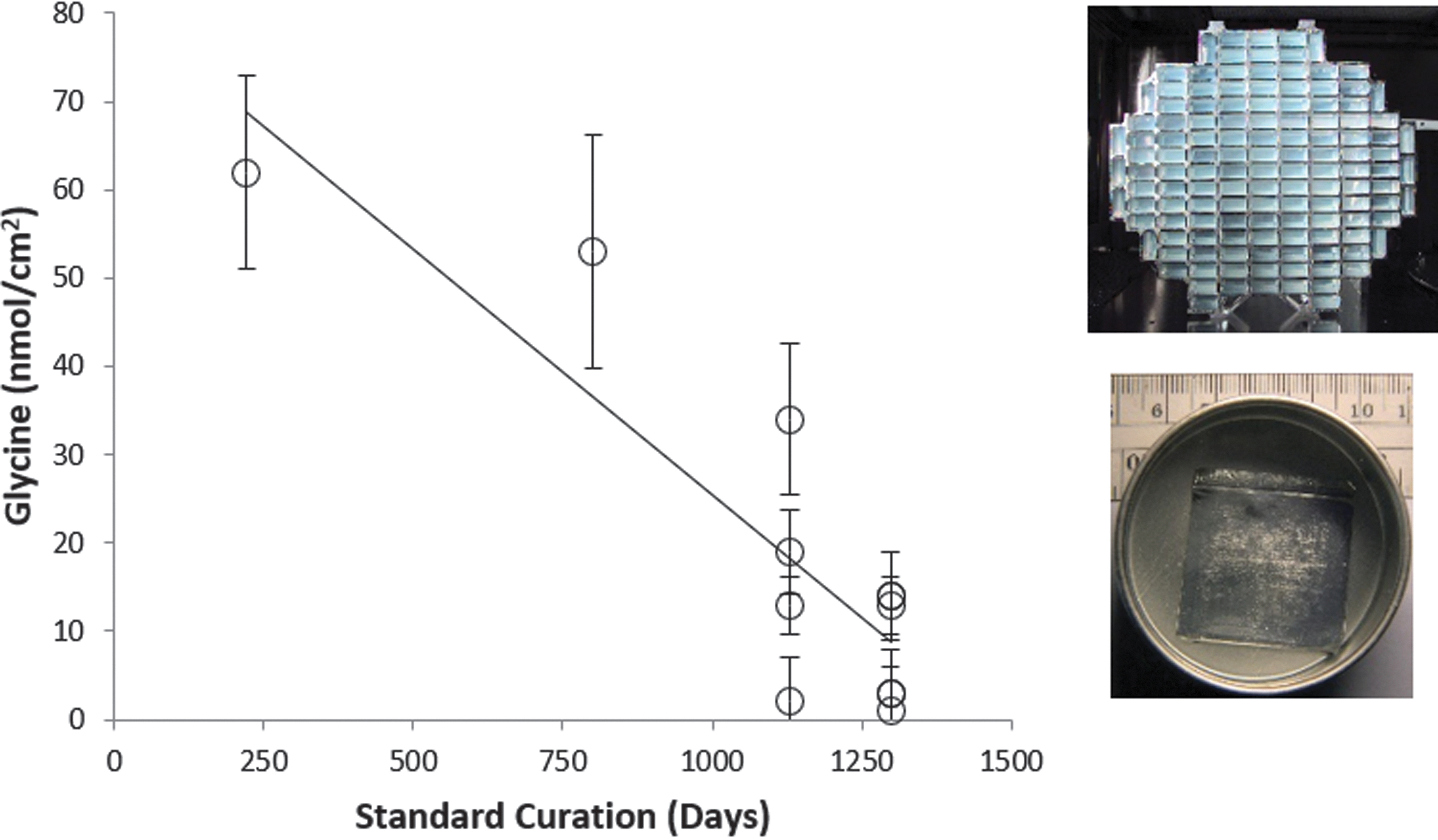
Volatile loss has also been inferred through the analyses of amino acids extracted from comet-exposed samples returned from comet Wild-2 by the Stardust mission and stored unsealed at room temperature (Elsila et al., 2009; Glavin et al., 2008). The work involved analyses of 13 different samples of aluminum foil from the Stardust collector over the course of 37 months (July 2006 to August 2009). A strong negative correlation was observed between the concentration of glycine detected from acid-hydrolyzed water extracts of the foils and the number of days in curation (Figure 6). Given these results and the fact that glycine itself is not volatile under these conditions, it seems plausible that the decrease in observed abundances of glycine in the water extracts is due to the loss of volatile precursors on the foil, including HCN, formaldehyde, and possibly other molecules. The apparent glycine abundance from Stardust foils stored under standard curation conditions, which decreased over time (60 pmol/cm2/day) equates to a reduction of ∼0.1% of the volatile glycine precursor each
Oxidants and other reactive species have been detected or inferred to be present on Mars in both the atmosphere and regolith by previous missions. The formation and presence of pathways for oxidants in the atmosphere and martian near surface have been an area of significant scientific interest given that oxidizing substances can rapidly destroy or degrade organic compounds, which has implications for strategies to search for evidence of life on Mars (see Figure 7, Lasne et al., 2016). The main oxidants that have been detected or suggested to be present on Mars include iron and magnesium perchlorate salts, which were identified during the Phoenix lander mission (Hecht et al., 2009; Kounaves et al., 2014) and present in samples analyzed by Curiosity in Gale Crater (Stern et al., 2017). Iron-bearing species such as Fe2O3 or FeO4 2- are much less stable (Christenson et al., 2004; Tsapin et al., 2000), and reactive oxygenated species that include peroxides, superoxide, O2 - superoxide radical ions (Oyama et al., 1977; Yen et al., 2000), and H2O2 (Clancy et al., 2004; Encrenaz et al., 2004) have been detected in the atmosphere and estimated to be present at part-per-million levels in the regolith (Zent and McKay, 1994; Yen et al., 2000). Although perchlorate salts are thermally stable under martian ambient conditions and will not decompose or react with organic compounds until they are heated to 200C or higher (Glavin et al., 2013; Sutter et al., 2017), peroxides and other reactive oxygenated species in a martian sample are much less stable and can rapidly oxidize and degrade organic compounds, especially at temperatures above 30°C and in the presence of liquids. For example, the oxidation of aliphatic hydrocarbons by t-butylperoxy radicals at 60°C is ∼16x faster than at 30°C (Korcek et al., 1972). In general, chemical reaction rates double for every 10°C increase in temperature.
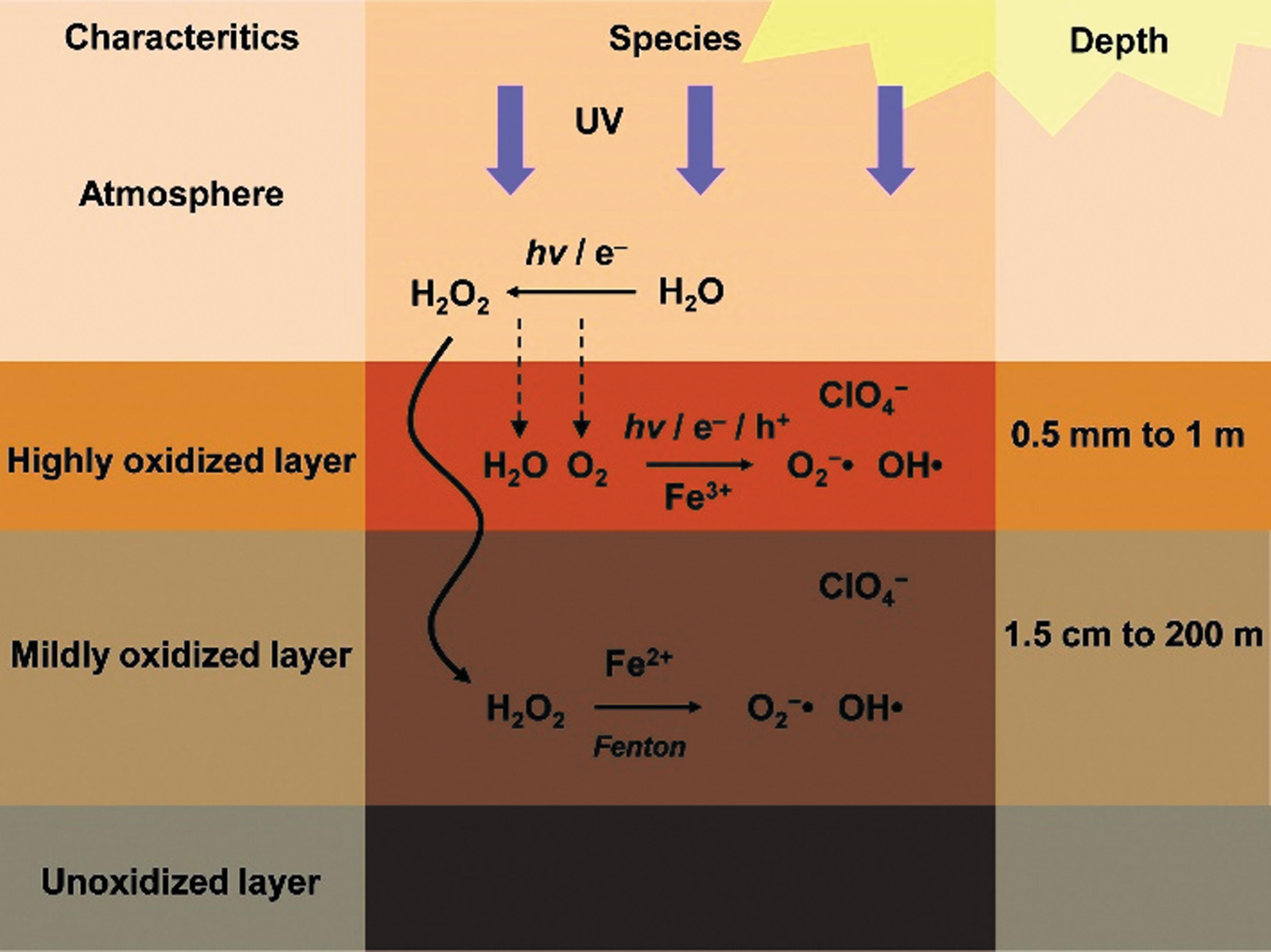
Simplified schematic of the oxidizing layers and formation of reactive O-species on Mars (from Lasne et al., 2016)
The most direct evidence for the chemical reactivity of near-surface martian samples and the impact of elevated temperatures on the activity of the samples comes from the Viking Labeled Release (LR) experiments. In the LR biology experiments, scooped soil from the upper few centimeters at the Viking 1 and 2 lander sites was transferred into a sealed and pressurized cell, and a nutrient solution containing 14C labeled glycine, alanine, formate, and glycolate was added to the soil to determine whether there was any evidence for metabolic activity that would produce volatile byproducts such as CO2. The radioactivity from any 14C labeled byproducts in the headspace was then measured as a function of time after soil exposure to the nutrient mix. As shown in Figure 8, radioactivity of the headspace gas that was measured by the Viking 1 LR instrument above background levels indicated some activity in the soil. However, no radioactivity was recorded after preheating the soil to 160°C for 3h, which presumably inactivated the soil prior to injection of nutrients into the cell. The soil was mostly inactivated after 50°C heating for 3h, though it continues to be further inactivated after storage in the dark at temperatures from 10-26°C over several months (Figure 8).Levin and Straat (1976; 1977; 1979, 2016) argue that these LR results are consistent with biological activity in the soil, while others argue that the LR measurements are best explained by the reaction of organics in the nutrient solution with non-biological substances, such as hypochlorite (ClO-) or other oxidants in the martian soil that could have been generated by the degradation of perchlorates by ionizing radiation (Quinn et al., 2013). Regardless of the source of the soil activity (biologic or non-biologic), it is clear from the LR experiments that both elevated temperatures and time will lead to a reduction in soil activity.
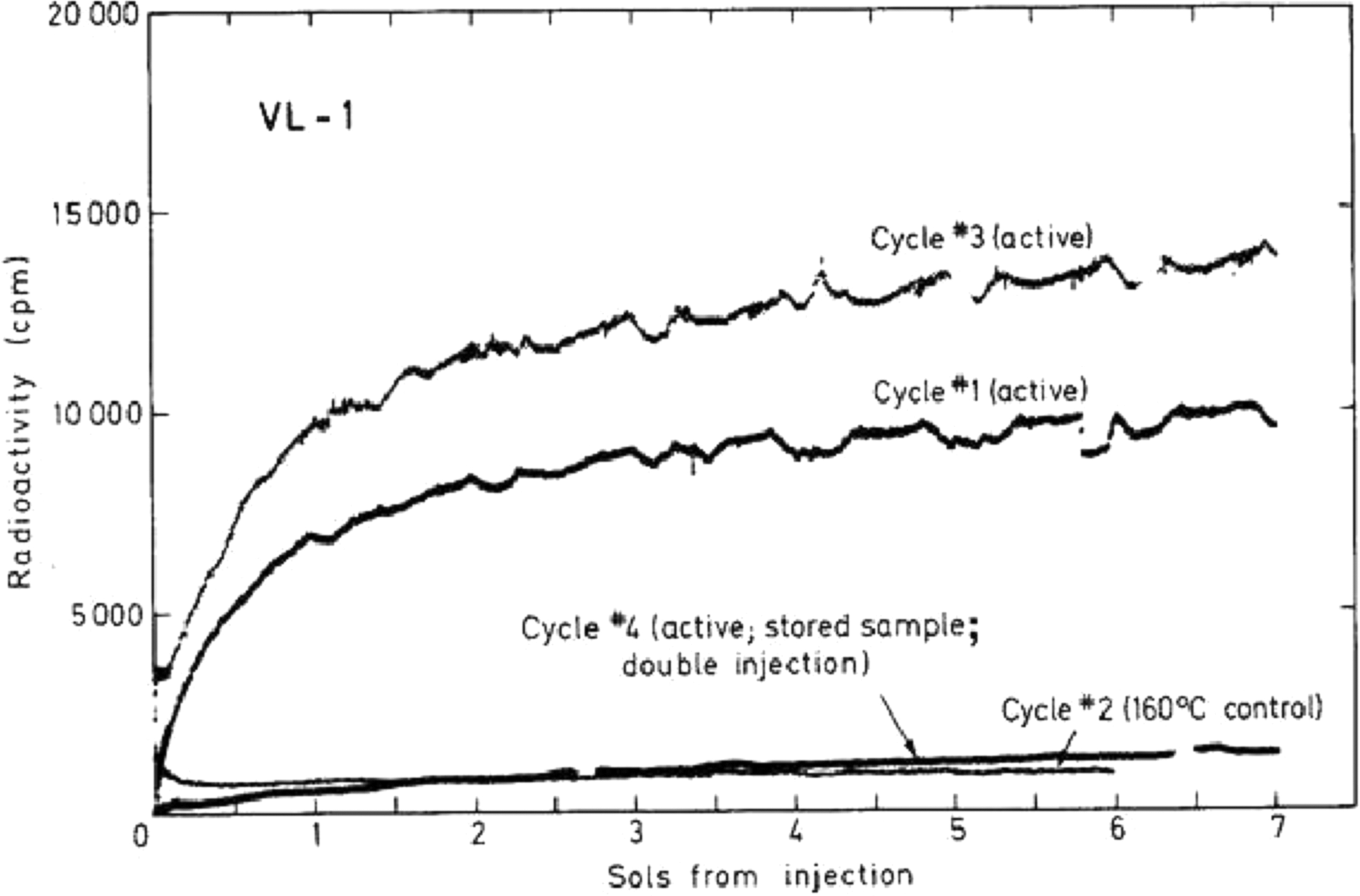
Viking Lander 1 Labeled Release (LR) data indicates the active agent(s) in the soil are not stable at elevated temperatures (Levin and Straat, 1979)
Detailed biological and chemical analyses of unconsolidated regolith and other rock samples returned from Jezero Crater will be crucial in answering the question as to the nature of the active agent(s) in the soil samples analyzed by the Viking Lander. Given that regolith samples in Jezero Crater may have already been heated to a maximum temperature of 10°C prior to collection and the cached sample tubes could see periodic temperature excursions up to ∼30°C on the surface and during return to Earth, it is unclear whether reactive species in the samples will survive. Nevertheless, reactive oxygenated species have been identified on the martian surface and in the atmosphere and could be present in the returned rock or soil samples or tube headspace. These species will rapidly degrade organic molecules, are unstable at elevated temperatures and humidity, and should be measured as soon as possible in the headspace gas and in the samples themselves after the sample tube seals are opened at the SRF.
If elevated abundances of volatile compounds such as methane, ethane, chloromethanes, along with others, and/or reactive O-containing species such as hydrogen peroxide are detected above background levels (background relative to levels measured in the headspace gas of the flight witness tubes) in the sample tube headspace gas after extraction, then a portion of the remaining sample core should be hermetically sealed and stored in a freezer (at -20°C or lower) to prevent additional volatile loss and chemical reactions in the sample. Time-sensitive analyses targeting trace volatile species that could be present in sample tube headspace gases, such as water, methane, ethane, formaldehyde, chloromethanes, peroxides, hydrogen chloride, hydrogen sulfide, sulfur dioxide, hydrogen cyanide, nitrogen oxide, and ammonia, should be done initially by using non-destructive techniques such as cavity ring-down spectroscopy. In addition, quantification of the nature and abundance of any adsorbed reactive oxygenated species in the solid sample when using non-destructive methods such as electron paramagnetic resonance spectroscopy should be made prior to exposure of the sample to solvents or elevated temperature and humidity.
The quantity of gas contained within the sample tube headspace will be small, that is, ∼13 cc, 7 mbar, equiv. to ∼4 micromoles max for an empty tube, an order of magnitude less for tubes filled with solid samples. Although this amount would be sufficient to make noble gas and some stable isotope measurements (e.g., C in CO2), it would not be sufficient to make all high-quality isotope measurements (e.g., N, triple O isotopes, C and N isotopologues, noble gas elemental and isotopic ratios; Swindle et al., 2022). Such a small amount of gas would make it sensitive to post flight contamination.
While no detectable leaks of the seals on the sample tube test units have been observed after environmental testing under a variety of conditions in the laboratory (He leak rate <1x10−10 atm-cc/sec), the leak rate is not constrained to be low enough to prevent loss of a substantial amount of an atmospheric sample (Cockell et al., 2022). Seals made under laboratory conditions show minimal loss, though it is not known how dusty the conditions on Mars will be, or how other conditions may affect the leak rate of individual seals. The precise leak rates for each seal will not be known until they have been analyzed on Earth. Thus, the risk that much, or all, of the headspace gas in some of the sample tubes could be lost to leakage or contamination, rendering it useless, is considered substantial.
After disassembly of the Earth Entry Vehicle (EEV) inside the SRF to remove the OS and its sample tubes, the ambient pressure in the facility will be two orders of magnitude higher than that of the interior of the sample tubes. The difference in the partial pressure of nitrogen between the sample tube exterior and interior is expected to be even higher because nitrogen,a trace constituent in the martian atmosphere, is the major constituent in Earth's atmosphere and the facility where the tubes would be curated. Contamination, even associated with a very small leak, could render some of the most important measurements (i.e., of chemical composition) impossible (Swindle et al., 2022).
As discussed above, and examined in more detail below, volatile exchange between minerals and ambient surroundings (whether in the sample tube or the curation environment once sample headspace gas has been extracted) may also modify sample headspace gas composition. Depending on the minerals present within the sample tube and the material's volatile content and exchange history, solid samples may continue to de-gas within biocontainment even after headspace gas has been extracted and analyzed (see Section 4.3 on loss of mineral-bound volatiles over time). This has the potential to jeopardize scientific investigations that target mineral-bound volatile components and/or the solid phases with which they are associated. The outcome of the Time-Sensitive Focus Group's investigations of mineral-volatile exchange (discussed at length below in Section 4.3) lead to the following findings and recommendations for the treatment and analysis of sample headspace gas:
Mineral-volatile exchange
Volatile exchange between solid phases and their immediate surroundings encompasses a range of processes that can occur on various timescales. The approach taken here is to examine the processes whereby volatiles are exchanged among minerals that have been identified or inferred to exist at the martian surface or within the shallow crust, particularly within Jezero Crater and its surroundings. This section focuses on mineral groups that are specifically referenced in the iMOST report and are known to exchange volatiles over timescales that may be relevant to the sample handling and curation workflow.
Hydrous sulfate minerals
The stability, particularly the hydration states, of many hydrated sulfate minerals are dependent upon relative humidity (RH) and temperature (T) (e.g., Chou et al., 2013). At the martian surface, the RH changes from 5% to 100% over the course of a diurnal cycle (Figure 9), and the stability fields for various hydration states of sulfate minerals change considerably over this RH range at martian surface temperatures and pressures (Chou et al., 2013; Wang et al., 2009). Given that sulfate minerals have been detected in the Jezero Watershed (Salvatore et al., 2018), they are among the possible mineralogical constituents that will be collected and cached in sample tubes by the Perseverance rover that would be brought back to Earth during the Mars Sample Return Campaign.
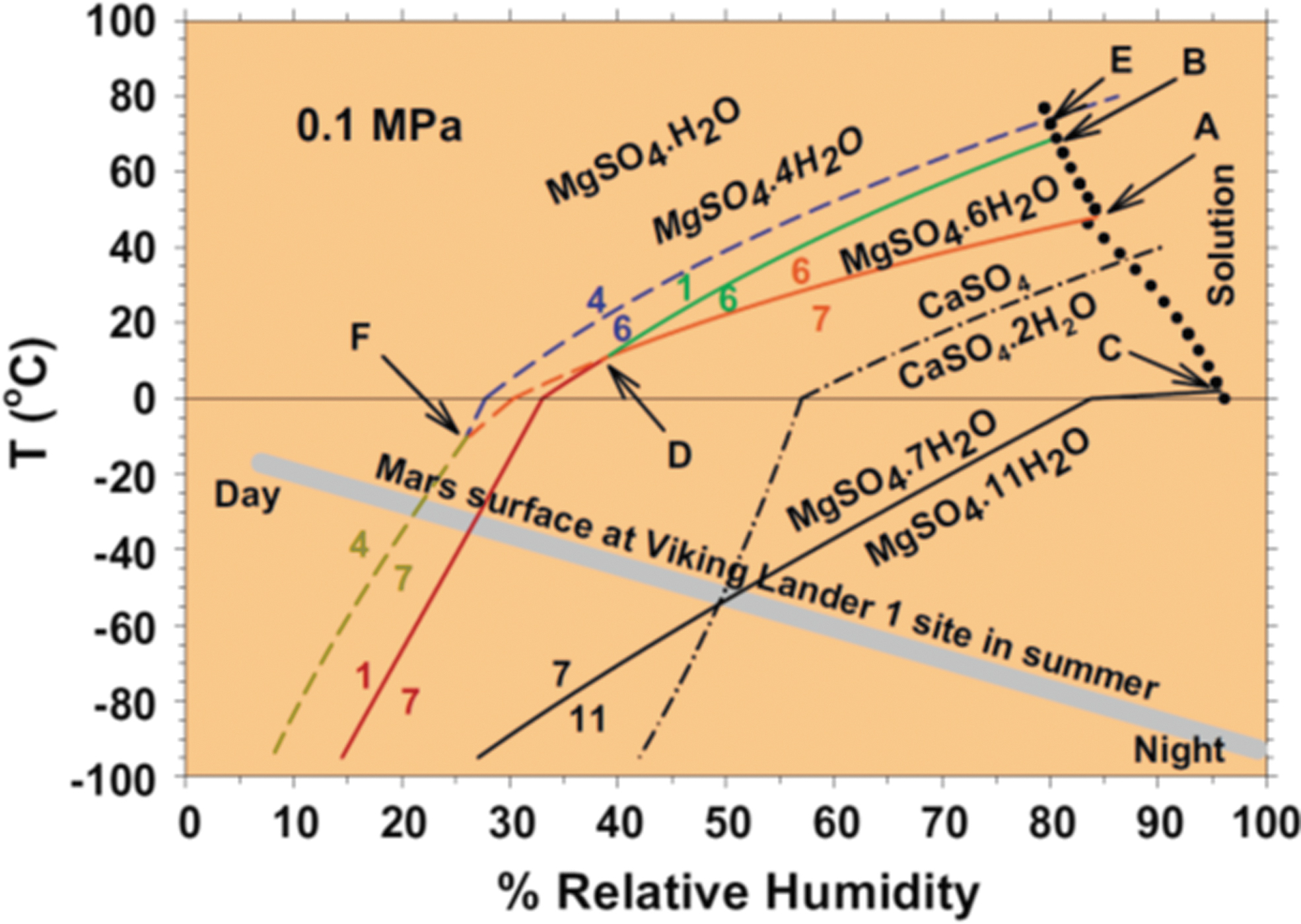
Phase relations in the system MgSO4–H2O at 0.1 MPa. Stable boundaries are shown by heavy solid curves, and metastable boundaries are shown by dashed curves. T-RH conditions of a martian day at the Viking Landing site in martian summer is shown with a thick solid gray line. Adopted from Chou et al. (2013).
In contrast to the pressure (P), T, and RH conditions at the martian surface, the P, T and RH conditions of typical pristine curation gloveboxes like those that service the Apollo sample collection at NASA Johnston Space Center (JSC) are much less variable and largely outside of the range of the martian surface (Allen et al., 2011; McCubbin et al., 2019). In fact, the Apollo gloveboxes are under a constant purge of gaseous N2 that has ≤10 ppm H2O (McCubbin et al., 2019), which would establish a highly desiccating environment for any hydrated sulfate minerals.
The differences between martian surface conditions and those in the pristine sample environment in curation labs on Earth, coupled with the stability fields of hydrated sulfates as a function of P, T, and RH, indicate that there could be problems with preserving hydrated sulfates long term in the curation environment, although the degree to which hydrated sulfates represent a time-sensitive analysis in a sample return facility (i.e., time-scale of 2-3 years) requires additional knowledge about the kinetics of hydration and dehydration reactions. Knowing that sulfate phases will likely be out of equilibrium with the SRF isolator conditions (Tait et al., 2022) underscores the time-sensitive nature of analysis of these phases.
To date, experimental studies that have investigated the rates of dehydration of hydrated sulfate minerals have shown that it is dependent on T, RH, sulfate phase (composition), and grain size (Okhrimenko et al., 2020; Okhrimenko et al., 2017; Ritterbach and Becker, 2020; Wang et al., 2009; Wang et al., 2012). Although T and RH can be controlled by SRF facility requirements, the identity of the phases and their respective grainsizes will be intrinsic properties of the samples and unlikely to be known prior to opening of the tubes.
With respect to the effect of sulfate mineralogy on the rates of dehydration, magnesium sulfate hydrates/dehydrates on the order of hours or days at room temperature and low RH conditions (Okhrimenko et al., 2020; Okhrimenko et al., 2017; Wang et al., 2009; Table 2), similar to that of the pristine Apollo sample gloveboxes (McCubbin et al., 2019). Iron sulfates dehydrate/rehydrate on similar time scales to Mg sulfates (Wang et al., 2012). In contrast to Mg- and Fe-sulfates, Ca-sulfates are less reactive and hydrate/dehydrate on the order of months or longer (Ritterbach and Becker, 2020).
Reaction Rate Ratios of Five Dehydration and Rehydration Processes of Mg-Sulfates Relevant to Mars Sulfate Mineralogy. Adopted from Wang et al. (2009).
With respect to the effects of grainsize on the kinetics of hydration/dehydration of sulfate minerals, the rate-limiting step is related to transfer at the surface of the grain (transfer coefficient) rather than diffusion within the crystal structure (Okhrimenko et al., 2020). Consequently, the rate of dehydration is inversely correlated with grainsize and is highly dependent on the total surface area of the sulfate minerals present (Okhrimenko et al., 2020).
Although the sulfate phases are likely to evolve within the sample tubes after collection, being able to back out that history to determine the state of the sulfates at the time of collection is predicated on knowing the state of the sulfates once the samples are first opened. If the sulfates dehydrate in the SRF, that history will be difficult, if not impossible, to discern. In addition, the characterization of mineral-bound volatiles, including those bound in hydrous sulfate minerals, is central to achieving several objectives central to returned sample science that relate to geological environments and the evolution of climate (Beaty et al., 2019). Consequently, phase transitions among hydrous sulfate minerals that may take place in biocontainment not only jeopardize investigations that target mineralogical properties but also those that target the chemical and isotopic composition of volatiles.
Currently, we do not know of a viable mitigation strategy for preserving the state of the sulfates in the samples at the time of sample tube opening other than storing the samples at temperatures below the lowest sulfate closure temperature. Mitigation strategies at room temperature could be developed by buffering the RH of the surrounding atmosphere. However, the RH value that would be needed is dependent on the specific sulfate phase that is to be preserved, and knowledge of the specific sulfate phase(s) present in the tubes will not be known at the time of tube opening. If the identity of sulfate phases can be determined quickly, RH could be used to preserve those sulfates after their identification.
The CheMin instrument (an X-ray diffractometer) on board the Mars Science Laboratory has shown that every single sample analyzed thus far contains a measurable amount of X-ray amorphous material (Smith et al., 2018). This result is consistent with previous spectroscopic observations (Singer 1985) of the martian surface as well as laboratory analysis of martian meteorites (McSween Jr., 1994). In situ analyses by CheMin indicate that the X-ray amorphous content ranges from 15-70 wt% in these samples (Figure 10; Smith et al., 2018). Several processes, including volcanism, aqueous alteration, and impact metamorphism, can induce X-ray amorphization of crystalline materials (Rampe et al., 2014). Volcanic and impact processes also drive the formation of silicate melts that solidify as X-ray amorphous glasses (Friedman and Long 1984). Aqueous alteration, which can amorphize minerals in situ and facilitate dissolution/reprecipitation reactions, and aqueous precipitation (e.g., from hydrothermal fluids) can produce X-ray amorphous and poorly crystalline nanominerals (minerals that only exist at the nanoscale), colloids, and mineral nanoparticles. Candidate X-ray amorphous phases include opals, Fe, Al, Ti, Ca, and Mg oxides, and hydrated salts such as perchlorates (Hochella et al., 2008; Banfield and Zhang 2001; Cornell and Schwertmann 2003; Navrotsky 2007; Carrier and Kounaves 2015). Such X-ray amorphous phases will react at different rates when the sample tubes are opened. Perchlorate salts and X-ray amorphous colloids (with high surface area) are expected to be the most reactive phases, followed by nanomaterials and poorly crystalline oxides. Characterization of these phases is a time sensitive measurement. Volcanic and impact glasses and hydrated opals react more slowly, hence their characterization is not considered time sensitive.
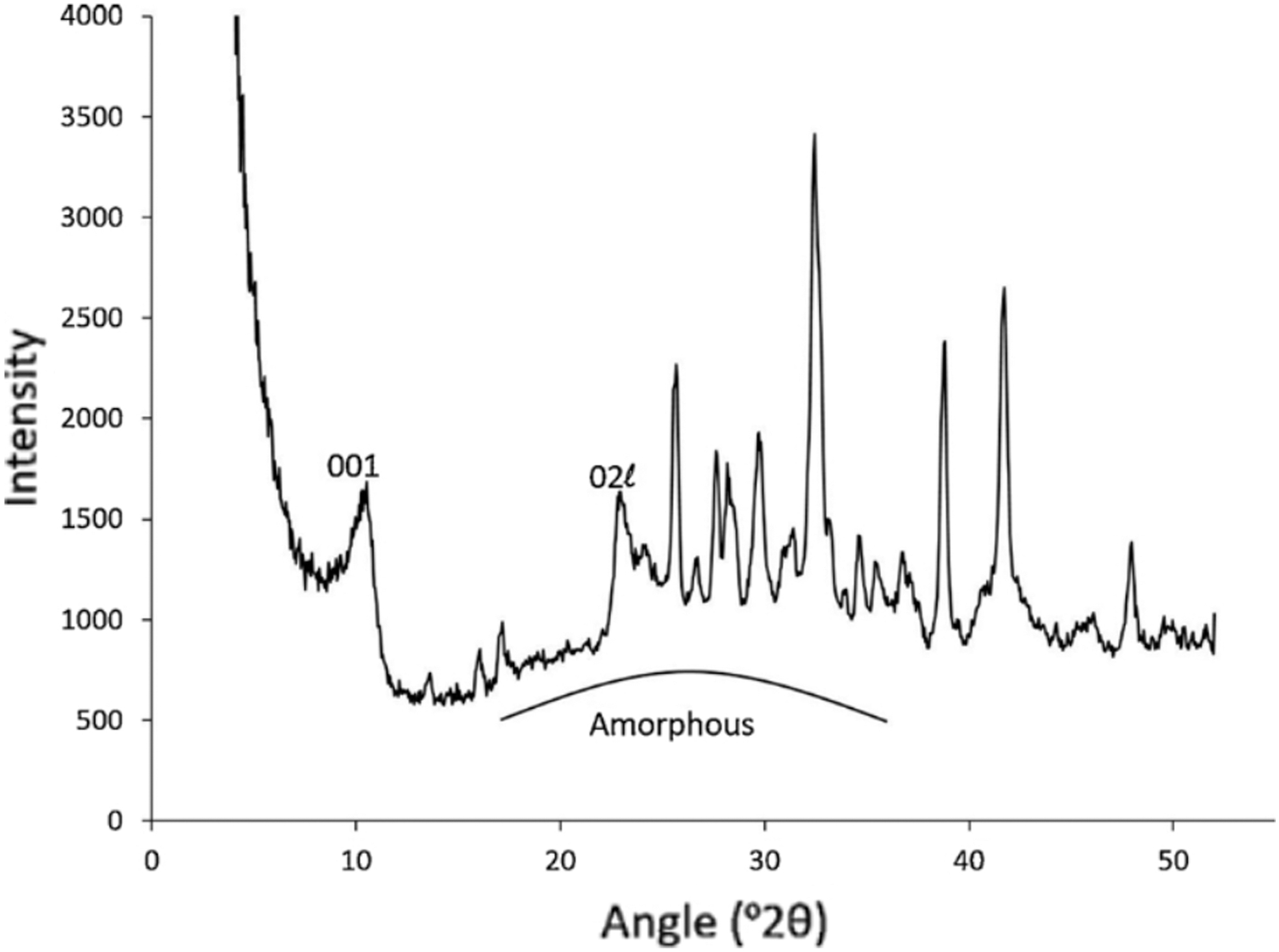
A CheMin X-Ray diffraction pattern of the Marimba sample from the Murray formation, Gale Crater. The broad increase in the baseline between 18˚ and 35˚ 2θ is attributed to the presence of X-ray amorphous phases. 001 and 02l refer to diffraction peaks attributed to clay minerals. Figure adapted from Rampe et al. (2020).
Perchlorate salts are probably the fastest reacting X-ray amorphous phases under typical laboratory conditions (i.e., low RH and T between ∼15-25°C). These salts are capable of hydrating and dehydrating in a matter of hours under martian surface conditions (Gough et al., 2011). Their initial hydration state will depend on the martian T and RH when the sample tube is sealed (Figure 11). After the tube is sealed, it will become a closed system, and the perchlorates will re-equilibrate to these new conditions. As the T inside the tubes fluctuates during the caching period on Mars' surface and during the return flight, so too will the perchlorate hydration state. Although the initial hydration state may not be preserved, it would be important to characterize the perchlorates at their equilibrium state inside the sealed tube because, like other minerals that host volatile components, they offer important constraints on volatile cycling on Mars, which underpins several iMOST objectives (Beaty et al., 2019). This equilibrium state may be altered within a matter of hours (Gough et al., 2011) when the sample tubes are opened under curation conditions (likely <10 ppm H2O). However, although data are currently limited, the timescales over which hydrous perchlorate phases may re-equilibrate and/or exchange volatiles with their surroundings is likely a function of gain size, lithification, or cementation (all of which influence grain boundary diffusion of H2O) and the relative humidity of the ambient environment as well (see Section 4.3.1 on hydrous sulfate minerals; Robertson and Bish, 2011). Thus, it may be possible that hydrous perchlorate minerals could be stabilized against phase transitions with relative humidity buffers, but this also requires a priori knowledge of the hydrous perchlorate phase in question.
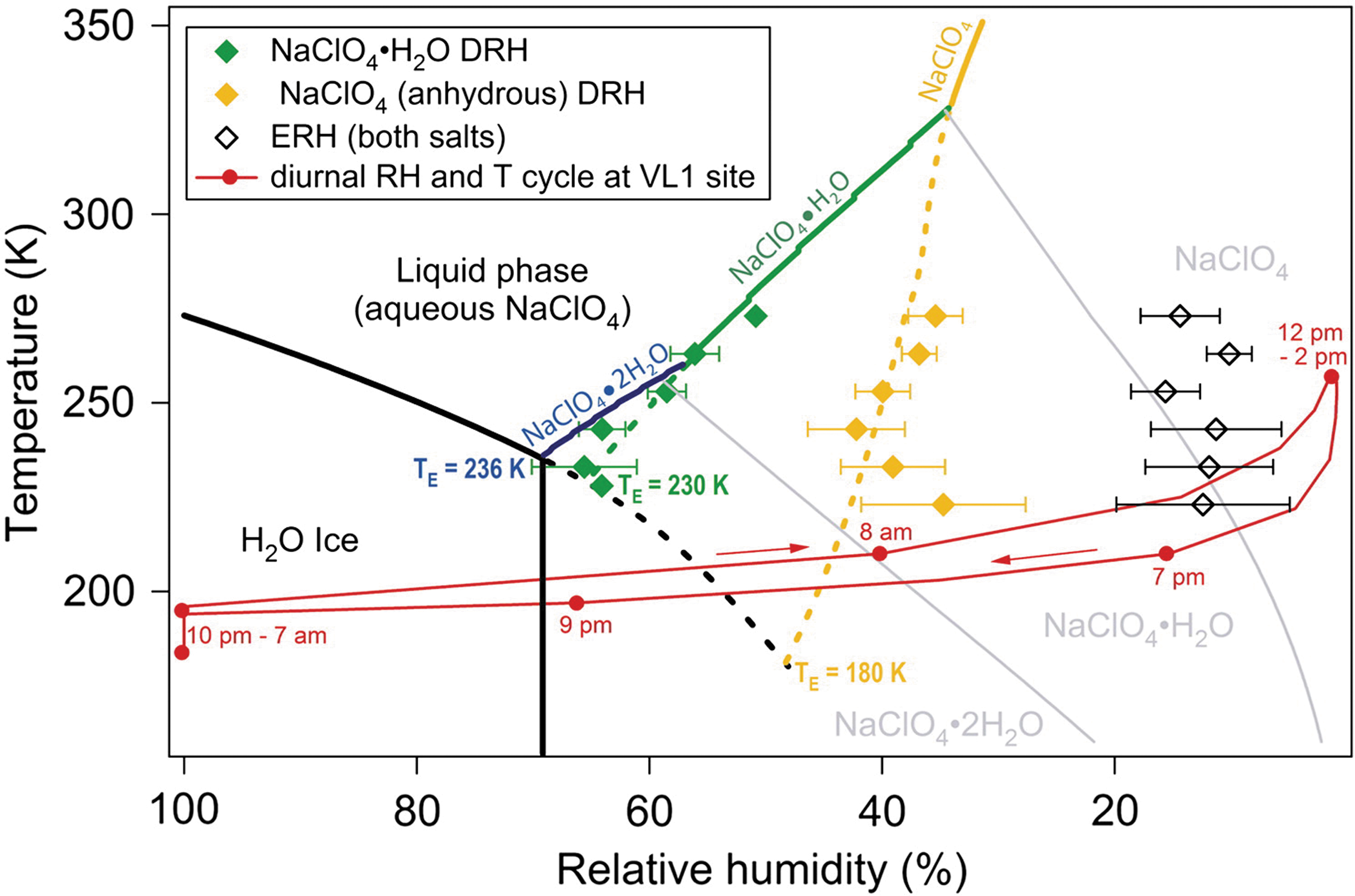
A temperature vs. relative humidity phase diagram for water and Na-perchlorate at the Viking 1 Landing Site (VL1) showing how changes in temperature and relative humidity can cause perchlorate deliquescence relative humidity (DRH) and efflorescence relative humidity (ERH) to rapidly fluctuate. Figure adapted from Gough et al. (2011).
Nanominerals and mineral nanoparticles are characterized by a very high surface area to volume ratio (Hochella et al., 2008). In general, nanoparticles behave differently than bulk minerals with a lower surface area to volume ratio (Banfield and Zhang, 2001). For example, nanocrystalline iron oxides are characterized by higher enthalpies of water adsorption and higher Gibbs free energies of adsorption than bulk iron oxides. These differences stem from a higher relative abundance of surface sites and crystal lattice defects in nanomaterials than for bulk materials. As a consequence, nanocrystalline iron oxides such as ferrihydrite and hematite adsorb more volatiles per unit mass than bulk minerals (Wang et al., 2013; Hiemstra et al., 2019). However, although nanocrystalline iron oxides are capable of adsorbing more volatiles than bulk minerals, those volatiles are less tightly bound (Navrotsky et al., 2008). Under curation conditions, which may result in the loss of surface adsorbed H2O, nanocrystalline iron oxides are likely to aggregate and recrystallize to eventually form more coarsely crystalline bulk minerals. This recrystallization will, in turn, result in a reduction in mineral surface area and a corresponding loss of adsorbed volatiles. These reactions have been described in the context of Ostwald ripening (Hiemstra et al., 2019), and their characteristic rates have been interpreted by using kinetic theory (Heaney et al., 2020)(Johnson and Mehl, 1939); these processes are likely to occur over a matter of hours to weeks, depending on the crystallite size and identity of iron oxides present in the samples (Schwertmann and Cornell, 2000).
Phyllosilicate nanoparticles are also associated with higher surface areas than their bulk counterparts, but laboratory experiments suggest that smectite nanoparticles bind H2O more strongly than bulk smectite. As this bound H2O desorbs, the nanoparticles aggregate and coarsen, in turn, reducing the total surface area (Elprince et al., 2015). The kinetics of this reaction are not well characterized, but on analogy to other nanoparticle systems, dehydration may occur over the course of weeks to months under typical laboratory conditions.
Nanocrystalline Ti, Al, and some Fe oxides will undergo irreversible phase transitions as they dehydrate and coarsen. Ti oxides may convert from anatase to brookite (Navrotsky, 2007), and Al oxides may convert from the ɣ-Al2O3 phase to the α-Al2O3 (corrundum) phase (McHale et al., 1997). Metastable iron oxide phases such as ferrihydrite may transform into hematite or goethite depending on ambient conditions (Cornell and Schwertmann, 2003; Heaney et al., 2020). All of these reactions are expected to occur on the order of days to months after opening the sample tubes under typical laboratory conditions. Therefore, the properties of nanocrystalline materials should be characterized in detail within ∼1-3 months of opening the sample tubes. Most importantly, this should include structural analysis (e.g., by total X-ray scattering and pair distribution function analysis) and replicate Brunauer-Emmett-Teller (BET) surface area measurements as a method to monitor the evolution of these phases as they are stored under curation conditions.
Volcanic and impact glasses are subject to devitrification, but this is expected to proceed slowly under typical laboratory conditions (Marshall, 1961; Yanagishima et al., 2017). Replicate analyses of Apollo samples after storage for 40 years confirms the stability of glass phases (Taylor et al., 2018). However, devitrification rates are temperature dependent, and small increases in temperature of 10-20°C could increase reaction rates (Rébiscoul et al., 2015). Similarly, the reordering of opal-CT and its subsequent transition to quartz will proceed slowly under curation conditions (Duffy, 1993). Consequently, we do not consider characterization of these materials to be time sensitive.
Orbital and in situ data indicate that phyllosilicate minerals are relatively common and, in many cases, abundant within the martian crust (e.g., Ehlmann and Edwards, 2014), in particular at Jezero Crater and the surrounding Nili Fossae region. Phyllosilicate minerals may exchange volatiles in several ways, but mineralogical and geochemical studies have shown that H2O, the principal (but not the only) volatile associated with phyllosilicate minerals, may be accommodated through physisorption on crystallite surfaces and pores and within interlayers (Sposito and Prost, 1982; Schoonheydt and Johnston, 2006). Volatile signatures (i.e., isotopic compositions) may be recorded by clay minerals through structural incorporation of O, which is derived from the H2O the phyllosilicate initially crystallized from; structural O is generally stable and lost or exchanged at temperatures higher than a few hundred degrees Celsius and will not be considered further here.
A number of phyllosilicate minerals can also accommodate H2O within interlayer spaces, which arise through the electrostatic repulsion of negatively charged tetrahedral-octahedral-tetrahedral (or so-called “2:1”) layers present in smectites, some vermiculites, and mixed layered varieties (Sposito and Prost, 1982; Schoonheydt and Johnston, 2006). The negative charges on adjacent layers require charge compensation by cations, which are accommodated within interlayers, and because they are weakly bound, they are generally exchangeable (Sposito and Prost, 1982; Schoonheydt and Johnston, 2006). The H2O sorption behavior in smectite minerals has been extensively studied and is generally a strong function of the interlayer cation (i.e., Li, K, Na, Ca, Mg) and the relative humidity. In general, as relative humidity increases, interlayer cations may be hydrated by zero, one, or two layers of H2O molecules (Figure 12; Ferrage et al., 2005). In addition, experimental studies performed on powders have shown that the incorporation and loss of H2O from phyllosilicate interlayers and on surfaces involves significant hysteresis, largely because different mechanisms regulate the adsorption and desorption of H2O from phyllosilicate minerals (Ferrage et al., 2005; Schoonheydt and Johnston, 2006).
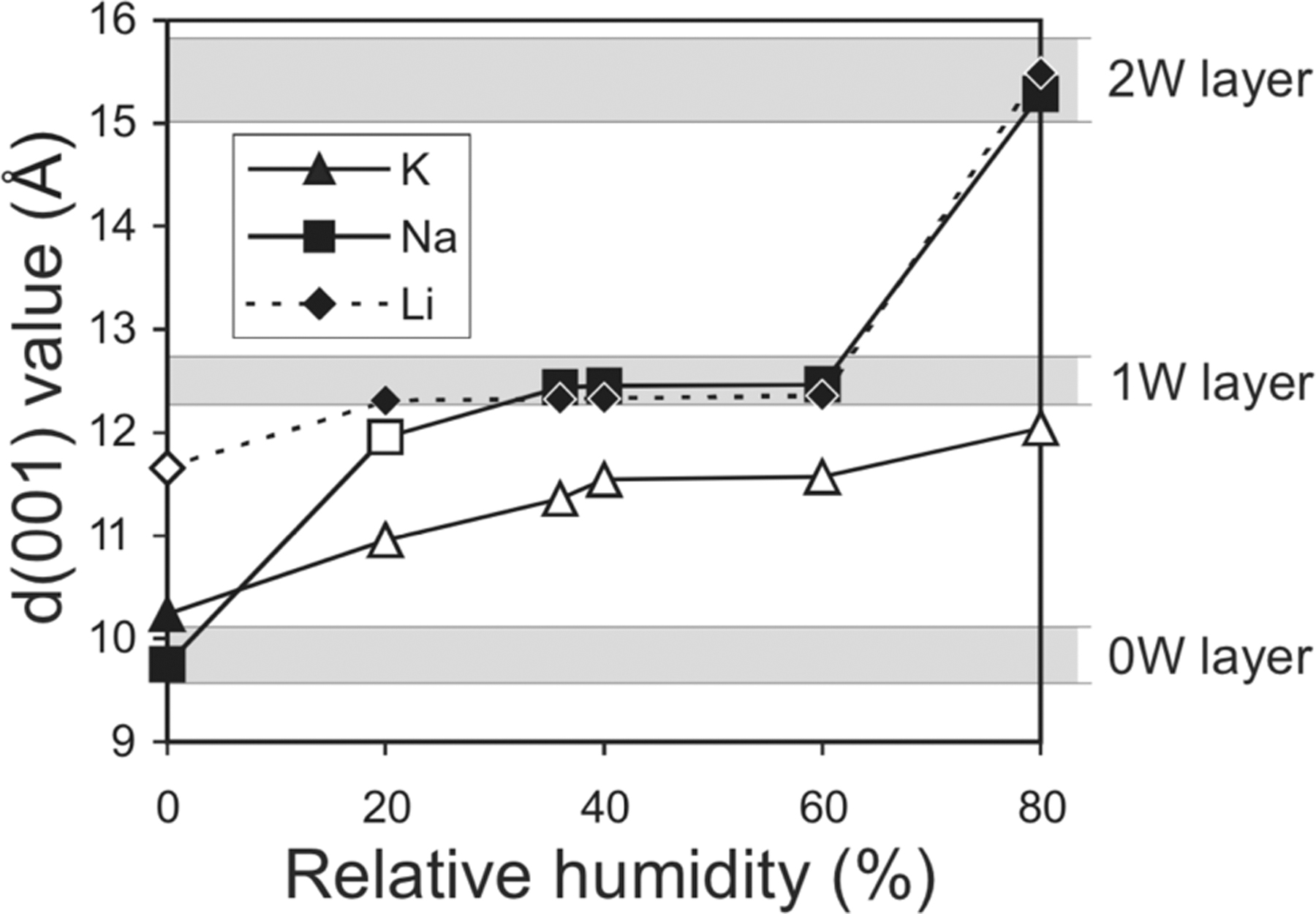
Basal spacing of montmorillonite (d(001)) as a function of relative humidity and interlayer cation (Ferrage et al., 2005). 0W, 1W and 2W refer to the number of H2O “layers” hydrating interlayer cations.
The accommodation of interlayer H2O in smectite minerals presently at the martian surface has been investigated from a thermodynamic point of view, which provides constraints on the amount and reactivity of H2O that may be accommodated in clay-bearing samples sealed and returned from Mars (e.g., Bish et al., 2003). For instance, although some experimental results published by Zent et al. (2001) suggest that Na-smectite would not be hydrated at martian surface conditions, thermodynamic estimates calibrated against laboratory measurements of H2O sorption isotherms (Bish et al., 2003) indicate a strong temperature dependence of H2O sorption for Na-smectite and Ca-smectite (Figure 13), largely arising from enthalpy of hydration. Thus, Bish et al. (2003) concluded that at 215K, smectite minerals would be partially, though not entirely, dehydrated during the day, whereas smectites are likely to be significantly hydrated at night as the relative humidity reaches maximum values. These results imply that, for smectite-bearing materials, the time of sampling will strongly dictate the hydration state of interlayers once the samples are sealed. However, even though the accommodation of H2O within smectite interlayers varies strongly across a diurnal timescale, this volatile component has the potential to provide valuable constraints on volatile cycling and long-term H2O budgets that address several objectives identified in iMOST (Beaty et al., 2019). Thus, regardless of the conditions of the time of sampling, interlayer-bound H2O itself carries high scientific significance, and its detailed characterization should be performed in line with relevant timescales over which it may be lost when sample tube seals are breached, and the headspace gas is removed.
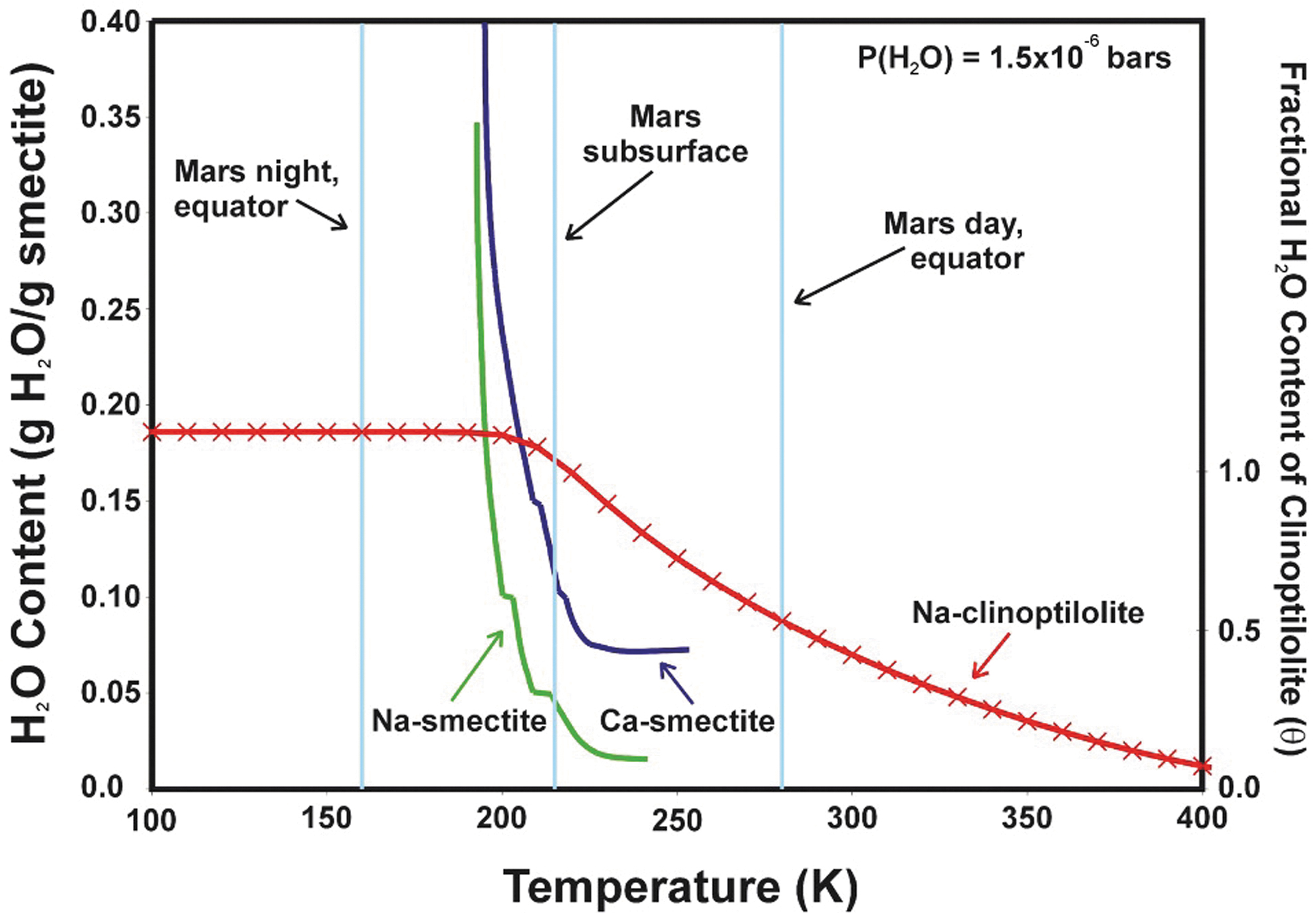
Estimated hydration states of smectite and clinoptilolite under current martian surface conditions (Bish et al., 2003).
The timescales of interlayer H2O exchange among phyllosilicate minerals has been studied extensively, though these have focused on powdered high-surface areas and often single-phase materials. In general, these studies show that, in response to changes in either temperature and/or relative humidity, smectite minerals re-equilibrate at timescales of minutes to several hours (Emerson, 1962, Likos and Lu, 2002) (Figures 14 and 15). Importantly, however, even though surface and interlayer-bound H2O are exchanged in response to temperature and relative humidity, most structural aspects of clay minerals (with exception of the interlayer spacing) remain intact.
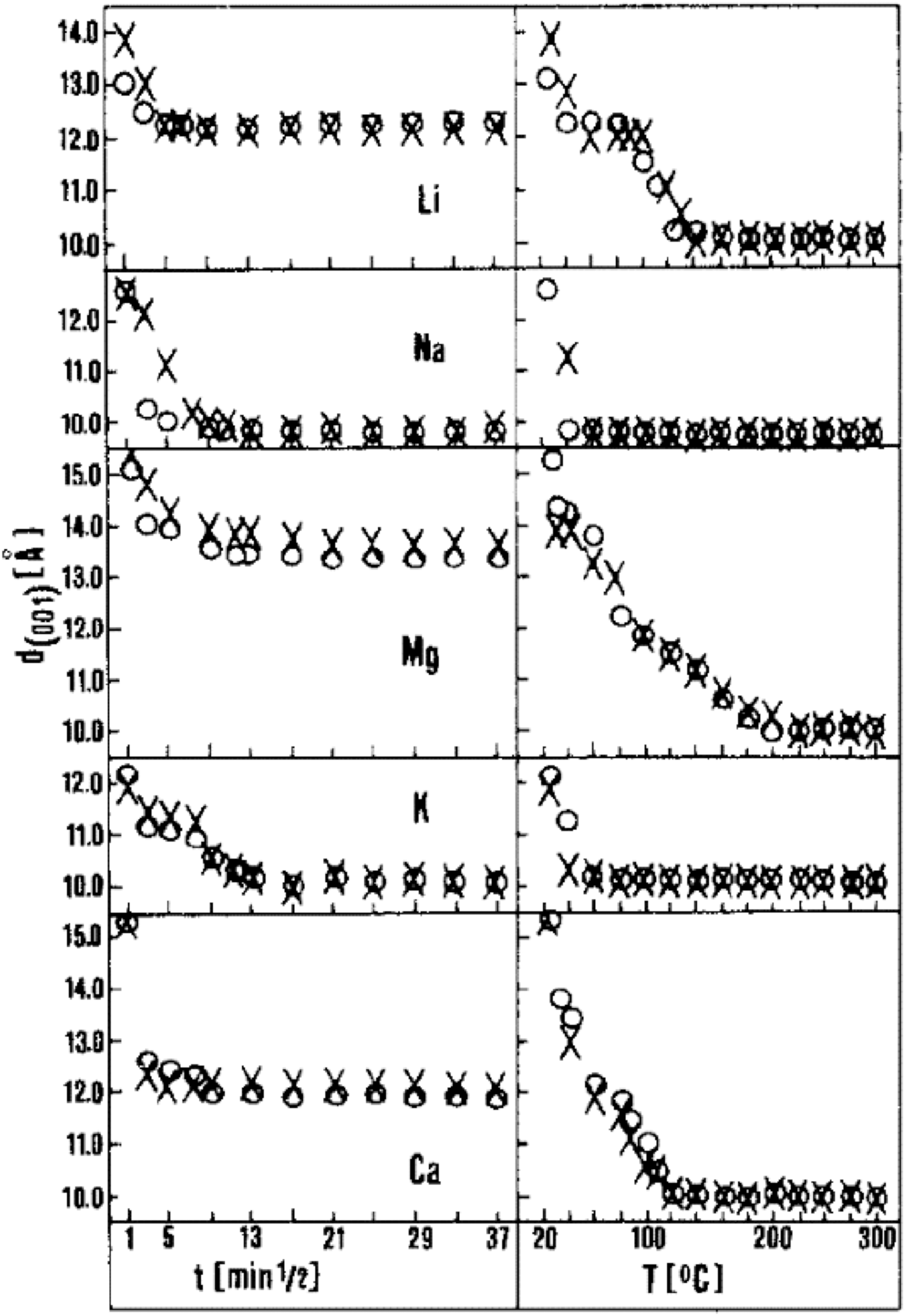
Basal spacing of montmorillonite as a function of relative humidity, interlayer cation, and temperature. In response to changes in relative humidity or temperature, interlayer H2O content adjusts (along with expansion or contraction of the interlayer spacing, d(001)) at timescales of minutes to hours in monomineralic samples (Emerson, 1962).
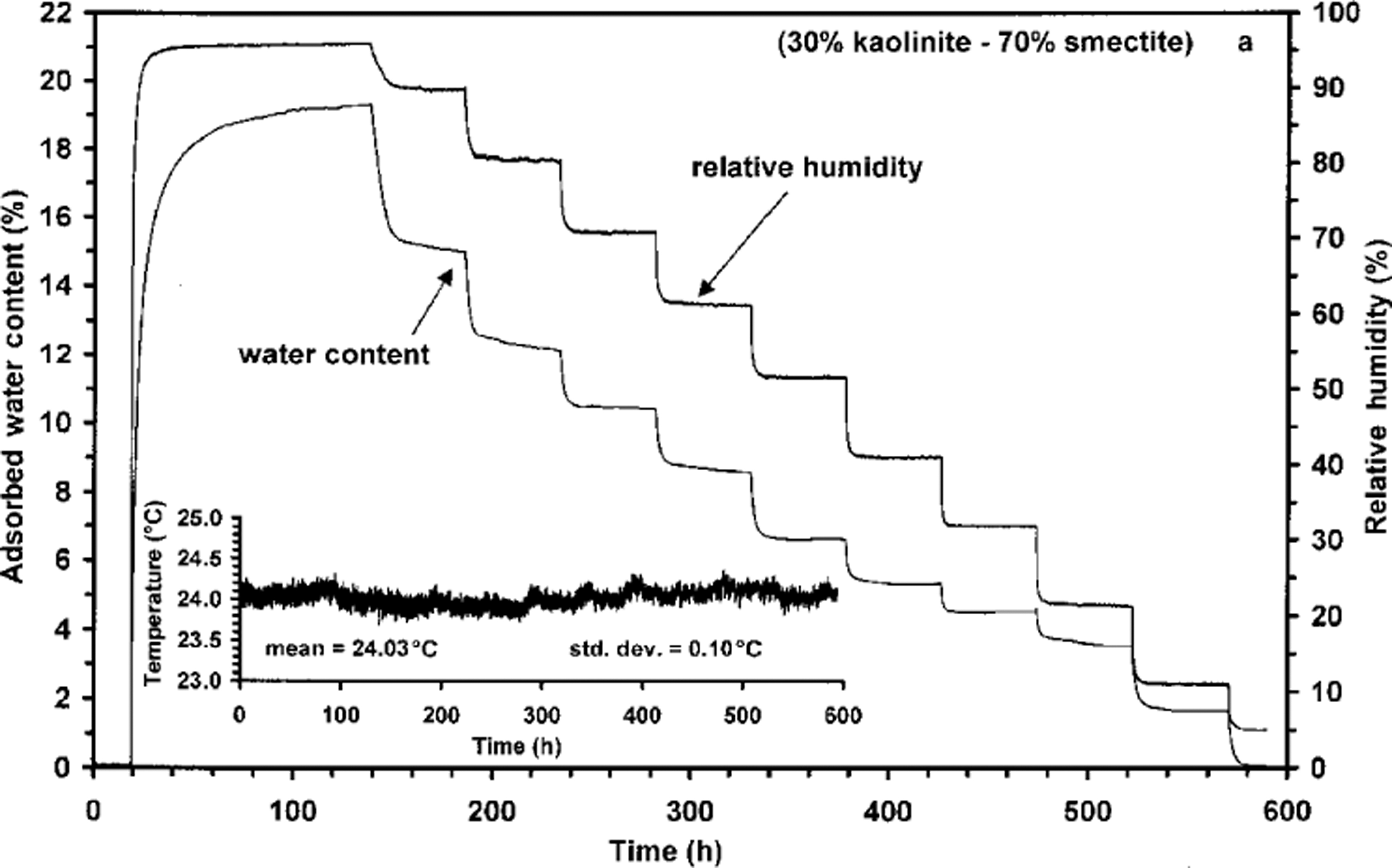
Changes in adsorbed H2O content of a 70% smectite / 30% kaolinite mixture at 24°C in response to stepped changes in relative humidity (Likos and Lu, 2002). The mixture re-equilibrates over timescales of hours to days.
Exploration and sample return from Jezero Crater and the surrounding Nili Fossae region, where significant accumulations of carbonate minerals have been identified, warrant consideration of hydrous carbonate minerals and their potential for exchange with their surroundings.
From a thermodynamic point of view, calcite, magnesite, and dolomite are the stable carbonate phases in their respective chemical systems. Thus, all other hydrous carbonate phases (Table 3) are metastable with respect to these three phases, which immediately establishes a thermodynamic driving force for transformation in the presence of H2O (i.e., as a solvent or vapor phase). The timescales over which these transformations occur, however, are poorly understood in “dry” systems as a function of RH. Volatile exchange and phase transitions among hydrous Mg-carbonates have received more attention than for hydrous Ca-carbonates. The hydrous Mg-carbonates lansfordite and nesquehonite are known to lose H2O and decompose to hydromagnesite in a dry state over a matter of months (Davies and Bubela, 1973; Ming and Franklin, 1985; Morgan et al., 2015), which is consistent with thermodynamic considerations in the Mg-carbonate system (Figure 16). However, the T-P dependence of these transformations is poorly understood. In addition, hydromagnesite has been observed to convert to stable magnesite in timescales of ∼10-100 years (Davies and Bubela, 1973; Ming and Franklin, 1985; Morgan et al., 2015). Although comprehensive studies are limited, highly hydrated Mg-carbonate phases are generally more stable at low RH, whereas higher RH and T values promote conversion to hydromagnesite (Wilson et al., 2013; Morgan et al., 2015). In addition to these studies, geochemical and mineralogical investigations have documented the formation of hydrous Mg-carbonate phases through interaction between thin films of water and Mg-silicate minerals over a period of several years at low temperatures characteristic of the Antarctic (Jull et al., 1988; Velbel et al., 1991; El-Shenawy et al., 2020). In general, this work supports the conclusion that hydrated Mg-carbonates are susceptible to volatile loss and recrystallization and transformation over timespans of months and perhaps longer.
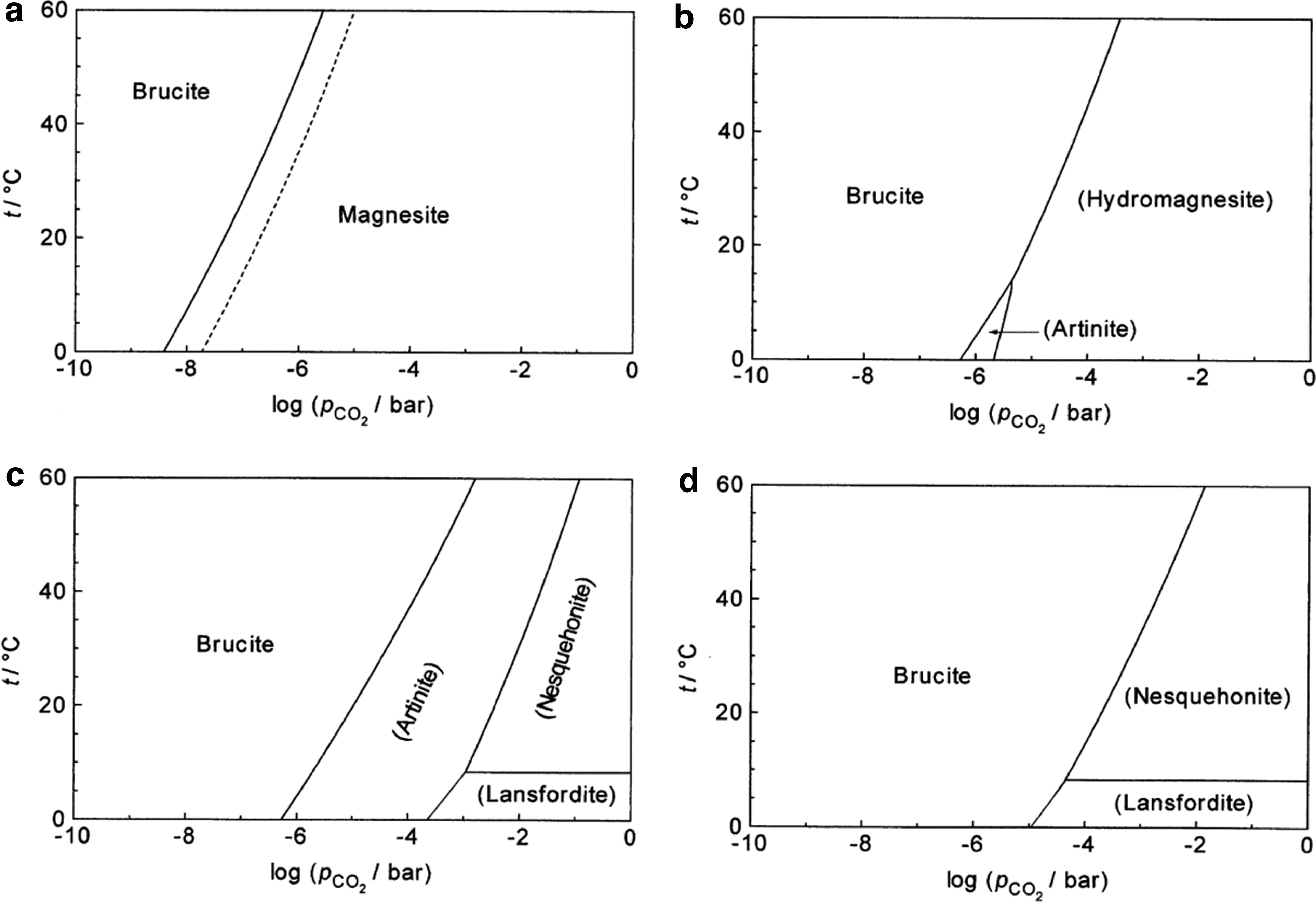
Phase diagrams for the system MgO-CO2-H2O, illustrating thermodynamic drive for recrystallization of hydrous phases (from Koenigsberger et al., 1999). (a) Stable equilibria of brucite with natural (solid line) and synthetic (dashed line) magnesite. (b) Magnesite was suppressed in the calculations. (c) Magnesite and hydromagnesite were suppressed in the calculations. (d) Magnesite, hydromagnesite and artinite were suppressed in the calculations.
Anhydrous and Hydrous Ca-, Mg- and Ca/Mg-Carbonate Minerals.
An important caveat for time-sensitive aspects of mineral-volatile exchange (including all mineral groups that are known to exchange volatiles) lies in assessing the limits to which timescales can be successfully extrapolated from the literature to samples of the type expected to be returned from Mars. For instance, most of the literature sources identified that constrain the timescales of mineral-volatile exchange are based on experimental measurements that utilize single minerals or small numbers of minerals in simple mixtures and focus on powdered material at high surface area. As noted above, because volatile exchange in lithified geological (or cemented regolith) samples is likely to be governed by diffusion along grain boundaries and/or thin films of water, the time constant associated with volatile exchange is almost undoubtedly longer than implied by experiments focused on powders (which forms the literal basis for constructing Table 4 below). To that end, we strongly recommend further studies on physically and mineralogically analogous materials to quantify the associated timescales of volatile exchange more precisely among lithified materials.
Estimation of the Loss of Scientific Information as a Function of Time for Various Targets (i.e., Properties, Materials and/or Substances) of Time-Sensitive MSR Investigations. iMOST Measurement Numbers Refer to the Order in which these Appear in Supplement-Table 1).
The principal process that jeopardizes investigations targeting redox-sensitive minerals involves the oxidation of mineral-bound reduced compounds as a function of time. The rates and mechanisms of the Fe-oxidation are well understood in this context. In general, in the presence of thin films of H2O (largely modulated by RH), oxidation of mineral-bound Fe(II) occurs by atmospheric O2(g) (e.g., Stumm and Morgan, 1996), though recent investigations have quantified the oxidation rates of Fe(II) by other chemical oxidants such as oxychlorine species (Mitra and Catalano, 2019). It is important to note that, if reactive O-bearing species are identified in a sample contained within the SRF, their liberation in aqueous solution is likely to result in some rapid oxidation of redox sensitive minerals over a timescale of hours.
Fe-oxidation by O2(aq) is understood to display a first order kinetic dependence on the concentration or partial pressure of O2(g), meaning that for every one-order of magnitude increase in pO2, the oxidation rate increases by one order of magnitude (Singer and Stumm, 1970). This process also involves a second order dependence on pH such that one pH unit increase translates to a two order of magnitude increase in oxidation rate. However, the oxidation rate of redox sensitive mineral grains bound within a sedimentary or igneous matrix will be controlled by diffusion of O2(aq) through the oxidation product. The timescales for this process can be evaluated by utilizing a “shrinking core model” whereby the diameter of the unreacted core of a redox-sensitive particle decreases with the extent of oxidation, which in turn results in a decrease in the interfacial area between the core and the coating (which then reduces the rate of delivery of the reactant) (Lasaga, 1998; Rimstidt, 2014). Assuming a spherical 100-micron redox-sensitive particle (of either pyrite or siderite) and a diffusion coefficient of 2.49 x 10−10 m2/sec for O2, solutions can be obtained that estimate the volumetric fraction of a redox sensitive grain that may oxidize as a function of the O2 content of the atmosphere (and assuming sufficiently high RH to support aqueous phase reactions). These simple relationships indicate that under ambient atmospheric conditions (i.e., pO2 = 0.21 bar), 100-micron particles of siderite and pyrite would completely oxidize in ∼35 days and 5 days, respectively (Figure 17). These simple calculations are broadly consistent with observations. For instance, complete oxidation of structural Fe(II) bound in clay minerals has been achieved in a matter of several hours to days in air-saturated solutions (Chemtob et al., 2017; Neumann et al., 2011).
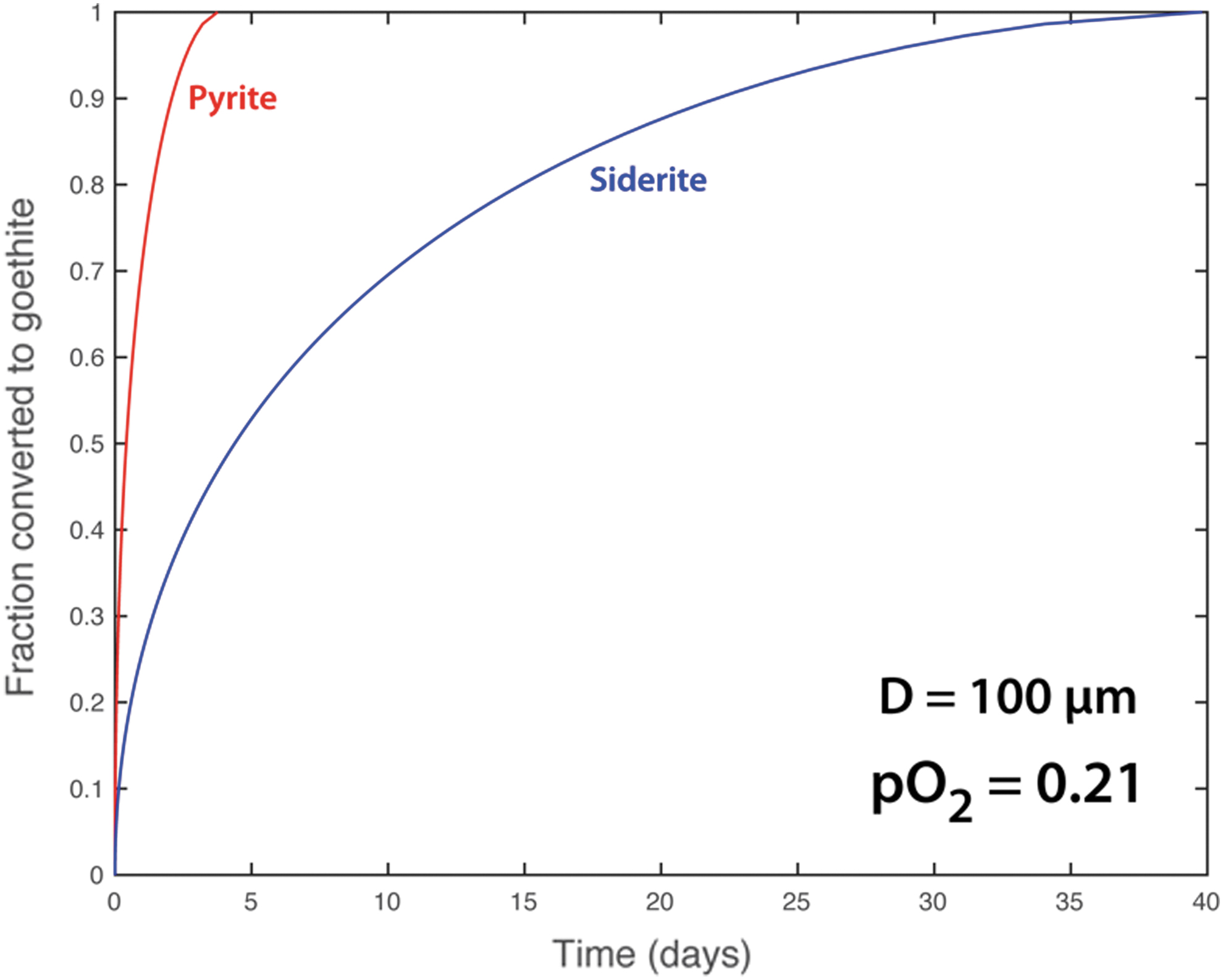
Oxidative transformation (in volume fraction) of a 100-micron spherical particle of a redox-sensitive grain to goethite at ambient atmospheric oxygen concentrations.
However, if redox-sensitive materials are stored in an anaerobic chamber where the O2 content of the atmosphere is very low, this timescale can be extended significantly. For instance, assuming commercially available O2-free nitrogen gas with an O2(g) content of 1ppm, 2 vol. % of a 100-micron particle of pyrite would oxidize in ∼3 months, which would oxidize ∼0.6 vol. % of a similar-sized particle of siderite (Figure 18). Using high-purity gas that ensures O2 content at 0.1 or even 0.01 ppm extends similar extents of oxidation out to the several year timescale. This in turn implies that as long as curation conditions involve ultra-pure low O2 (well below 1ppm O2 and preferably below 0.1 ppm) inert gas such as nitrogen, then sufficient oxidation should not occur during curation.
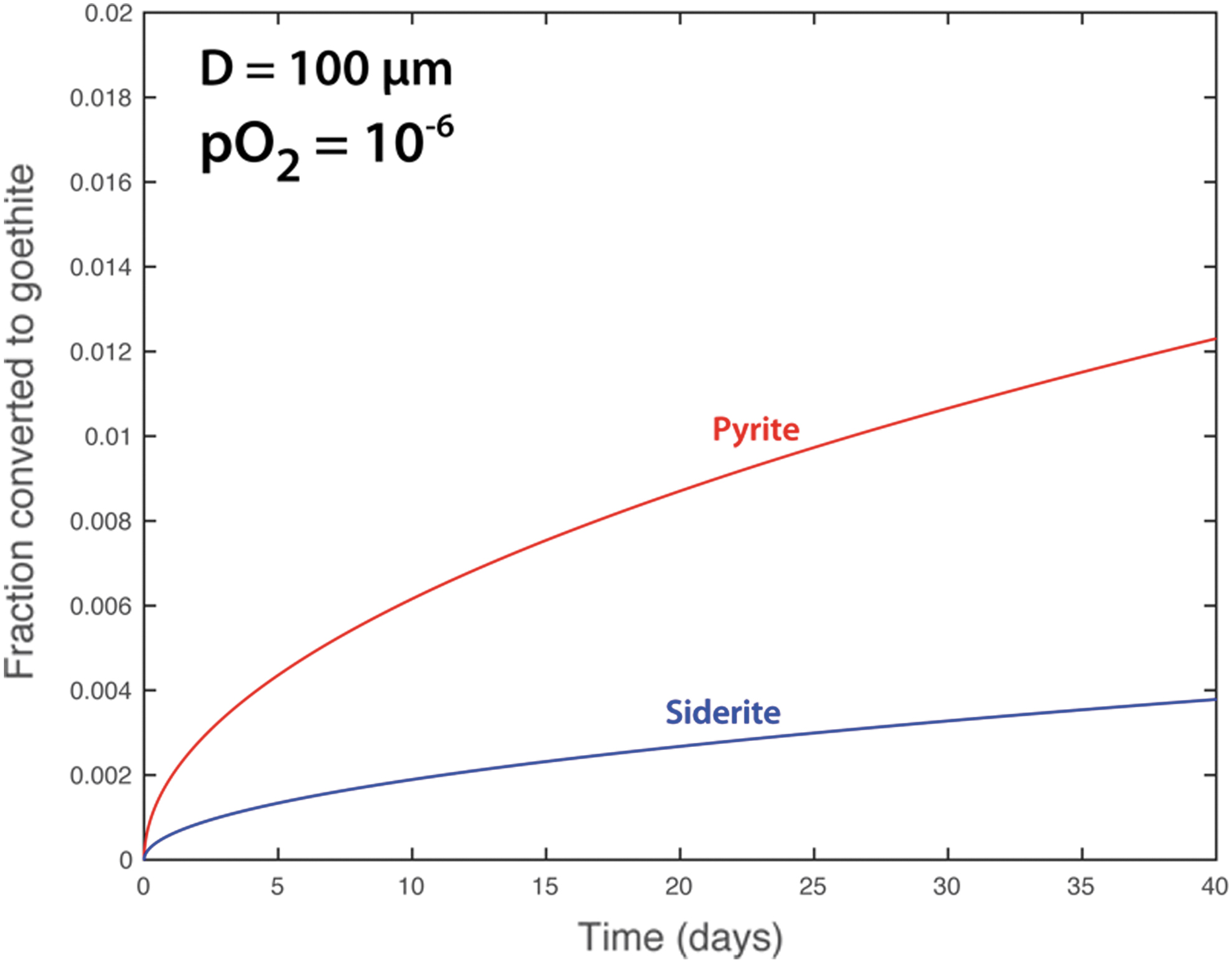
Oxidative transformation (in volume fraction) of a 100-micron spherical particle of a redox-sensitive grain to goethite at an ambient oxygen concentration of 1ppm O2(g).
Available constraints on the characteristic timescales that underpin time-sensitivity range from relatively coarse to uncertain. Nevertheless, in all cases, sufficient information is available to begin to estimate how scientific information may be lost from attempting time-sensitive MSR investigations as a function of time spent in biocontainment. This, in turn, highlights important implications for sample processing within the SRF.
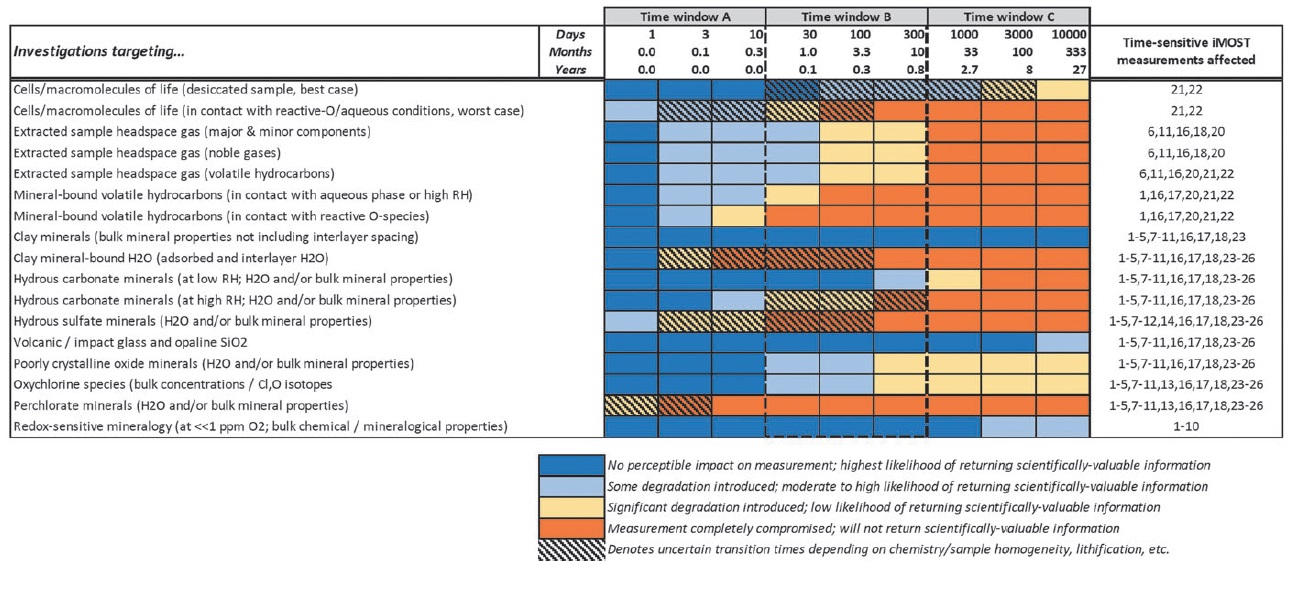
The loss of scientific information as a function of time can be estimated by first considering specific sample properties, minerals, solid phases, or compounds that the 26 time-sensitive iMOST investigation measurements target (Table 4). The available constraints that underpin time-sensitive processes during which a specific investigation target may degrade can then be used to evaluate the window of time available for scientific investigation. Here, time is divided into 0.5 order-of-magnitude increments from 1 to 10000 days. This partitioning establishes three windows of (A) 1-10 days, (B) 30-300 days (or ∼1-10 months), and (C) 1000-10000 days (∼3-30 years). Investigations that target a material or property that degrades to the point where they are rendered scientifically uninformative within time window A (1-10 days) may be difficult, if not impossible, to plan and execute within that timeframe. Investigations that target a material or property that completely degrades within time window C are here deemed likely to preserve scientifically useful information even if they are delayed until after the SSAP is complete and investigations can be pursued outside biocontainment (i.e., in the laboratories of individual principal investigators). Investigations that target a material or property that completely degrades within time window B may be accomplished within the sample handling / curation workflow inside an SRF if specific adjustments are made to accommodate them.
This analysis has identified three MSR targets that are likely to completely degrade within time window A. These are: clay mineral-bound H2O, including adsorbed and interlayer H2O, and hydrous sulfate and hydrous perchlorate minerals (for both hydrous minerals, the targets include the H2O bound within their structures and on their surfaces as well as their bulk mineralogical properties). A fourth investigation target, cellular materials (cells/macromolecules of life on Earth), may also fall within this category. However, if these materials are in contact with reactive O-containing species and aqueous conditions, the exact timescale associated with degradation is dependent on the nature of information sought from molecular analysis, T, and the organic or cellular material under investigation.
Time window B features at least three MSR investigation targets that may completely degrade within 1-10 months, including mineral-bound volatile compounds, whether in contact with an aqueous phase or reactive O-species; hydrous carbonate minerals, including the H2O bound on surfaces or within their structures and bulk mineralogical properties themselves; and possibly organic or cellular material in contact with reactive O-containing species and aqueous conditions, which again is dependent on the nature of information sought from molecular analysis, temperature, and the material in question. All of the other MSR investigation targets considered here are completely degraded within time window C, which can likely await analysis outside biocontainment with minimal to moderate impact on the investigation, if the samples are deemed safe to release in an unsterilized state.
Finally, this analysis provides objective information on which to base prioritization decisions within an SRF. Because each investigation target supports multiple MSR investigations and iMOST objectives, prioritization decisions are most appropriately made on the basis of which targets degrade the most quickly within an SRF under curation conditions. To that end, analyses characterizing organic molecules, the volatile components and bulk mineralogical properties of hydrated sulfate and carbonate minerals, as well as interlayer or surface adsorbed H2O in clay minerals, should be prioritized within an SRF and/or extended or mitigated where feasible.
Recommendations for SRF Sample Processing and Analytical Capabilities
The analysis of time-sensitive aspects of Mars returned sample science presented here highlights a number of sample processing and analytical considerations for maximizing the amount of scientific information retrieved from samples while in biocontainment. To that end, the Time-Sensitive Focus Group has considered, in detail, sample processing and analytical capabilities required to complete time-sensitive scientific investigations in biocontainment (i.e., those highlighted in Table 4). This analysis recognizes that new capabilities are likely to emerge in the coming years and so provides examples of procedures and/or analytical equipment that could conceivably meet the goals of time-sensitive science at the present time. The recommendations that follow are based on the following principles: (1) the sample receiving facility should be constructed as a “minimalist” facility; duplication of analytical functionality should be avoided where possible; (2) if more than one analytical capability is available to fully meet an analysis need, recommendations should favor simpler analytical procedures and equipment.
The investigation targets highlighted in Table 4 lead to four overall goals of time-sensitive science (Table 5), which must be successfully completed in biocontainment (though in no specific order): (1) characterization of the sample tube headspace gas composition, (2) characterization of organic material of potential biological origin (including volatile hydrocarbons), (3) characterization of mineral-bound volatiles, (4) characterization of solid-phase volatile hosts. These goals encompass the investigation targets identified in Table 4 that significantly degrade within window B (i.e., 1-10 months) or that may degrade within window B on the basis of estimated uncertainties due to sample lithification, cementation, etc. The goals also include characterization of the sample headspace gas which may not be inherently time-sensitive across window B but has been deemed so because results of these analyses inform the curation workflow (see also MSPG Science in Containment Report and Tait et al., 2022).
Traceability Matrix Outlining Goals of Time-Sensitive Science to be Completed in Biocontainment and Instrumentation Required to Meet Each Goal.
Traceability Matrix Outlining Goals of Time-Sensitive Science to be Completed in Biocontainment and Instrumentation Required to Meet Each Goal.
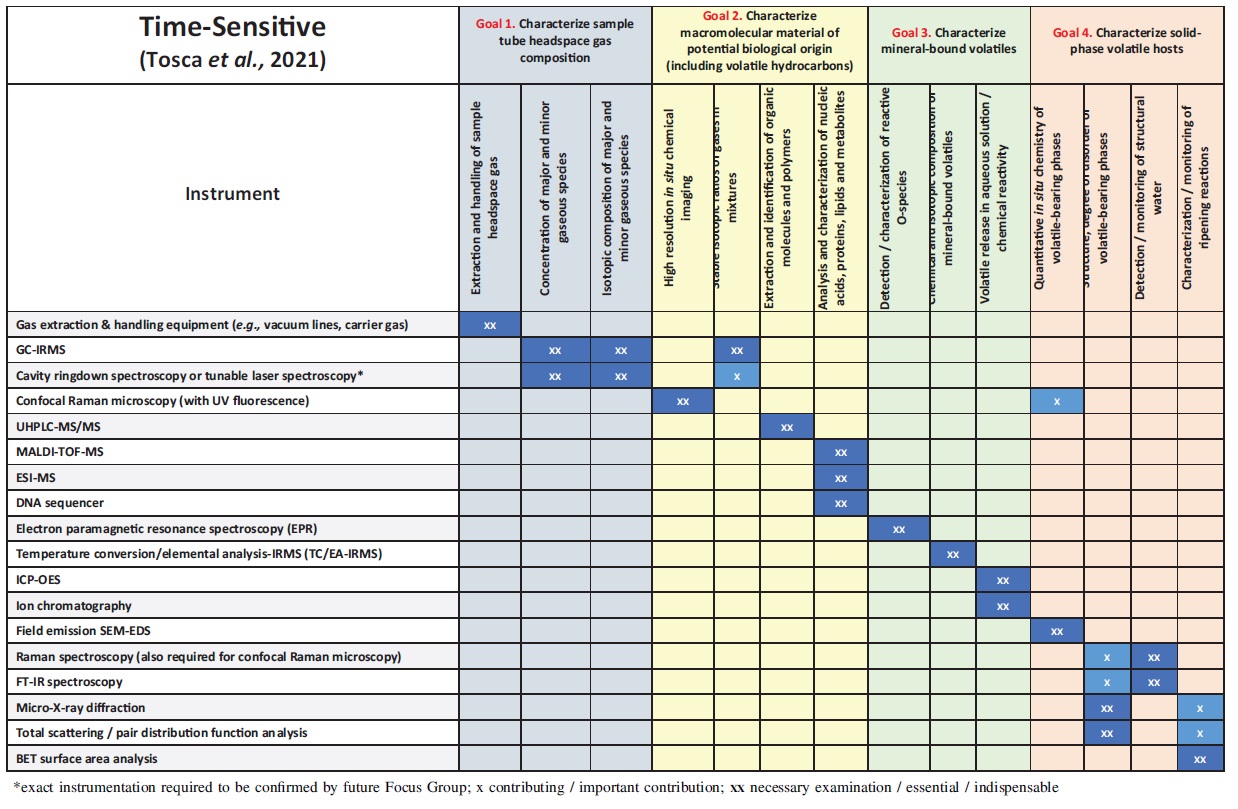
Each of the four goals of time-sensitive science can be further subdivided into a series of analytical requirements (Table 5). These requirements, in turn, inform sample processing and analytical equipment that the SRF should contain. Characterization of various sample attributes (i.e., organic material, mineral-bound volatiles, solid-phase volatile hosts, etc.) can be accomplished with a variety of analytical instrumentation and to varying degrees of specificity. For example, characterizing the specific solid phase hosting volatile compounds is an underlying requirement of Goal 4 in Table 5. However, while FT-IR spectroscopy provides information on hydration state and local bonding environment, it cannot not give unambiguous structural information. Conversely, while micro-X-ray diffraction provides structural information, in many instances it cannot unambiguously quantify the degree of hydration of some materials. Thus, both techniques provide important information that meet the stated analytical requirement. Accordingly, the Time Sensitive Focus Group has extensively discussed and agreed upon the minimum amount of data to credibly document sample characteristics and achieve each of the analytical requirements listed in Table 5. Because some instruments alone may be both necessary and sufficient to meet an analytical requirement, and others may contribute important information yet may not be sufficient to meet that requirement, this distinction has been made in Table 5.
Below, recommendations for sample processing and analytical capabilities are presented for each goal in the context of the processes discussed above that underpin time-sensitivity.
To meet this goal, the time-sensitive focus group recommends the following: Independent sampling of martian atmospheric gas should be prioritized to quantify volatile exchange history within the sample tubes, provide a reference composition to compare with headspace gases, aid in the detection of biosignatures, and permit enough material to support iMOST investigations. All sample tubes should be placed in a secondary container as soon as possible. The headspace of this secondary container should be subject to periodic compositional or isotopic monitoring of gaseous species that may have escaped the sample tube if a seal has been breached. This recommendation can be achieved, for example, by placement within a Sample Tube Isolation Chamber (STIC), as discussed by Tait et al. (2022). The headspace gas present in a sample tube selected for analysis should be retrieved and characterized compositionally and isotopically as soon as possible. The main objectives of these analyses will be to assess whether sample headspace gas has been contaminated by terrestrial atmosphere and characterize rapidly the sample headspace gas before further mineral-volatile exchange occurs. Time-sensitive analyses targeting sample headspace gases should aim to characterize, in a non-destructive way, the elemental composition of the gas and stable isotopic composition of key major elements (e.g., carbon, nitrogen, hydrogen) and individual volatile species if they are sufficiently abundant. Once the state of the sample tube seals has been determined, sample headspace gas should be retrieved. The retrieval process should recover all available gas originally contained within one individual sample tube volume stored in a respective container each that allows future allocation. The remaining headspace should be back-filled with an inert curation-grade gas. Small aliquots of the gas should be used to assess contamination by terrestrial atmosphere, and the remaining larger aliquots may be sterilized (see Velbel et al., 2022) and sealed in preparation for continued analysis outside biocontainment. Because mineral-bound volatiles will be subject to degassing when the seal is breached, the sample tube headspace extraction procedure should facilitate multiple attempts at extracting headspace gas. This will minimize the escape of mineral-bound volatiles to the headspace and facilitate their characterization. The extraction and handling of sample headspace gas will involve specialized equipment including custom-made gas transfer tubes constructed of suitable material, vacuum pumps, inert carrier gas (i.e., He) and/or cold traps to concentrate and transfer gaseous species.
The composition of major and minor gaseous species present in relatively small amounts of sample headspace gas could be achieved by using gas chromatography-isotope ratio mass spectrometry (GC-IRMS). In addition, GC-IRMS can provide stable isotopic composition of C and N in the gas phase at the expected quantities. However, in addition to this, spectroscopic methods such as cavity ringdown spectroscopy (CRDS) can provide higher precision measurements of C-isotopes and characterization of CO2 isotopologues and triple oxygen systematics, which are all important tracers of atmospheric evolution. Here, either analytical capability could be recommended to facilitate complete chemical and isotopic characterization of sample headspace gas, and a final decision on the suitability of a particular method for low volume and molar quantities expected for returned samples must await further technique development.
Goal 2: Characterize organic material of potential biological origin (including volatile hydrocarbons)
To meet this goal, the time-sensitive focus group recommends the following:
Prior to analysis, and during sample handling and preparation, exposure of samples to aqueous conditions and/or oxidants should be minimized.
If early analysis suggests the potential for biology, samples should be preserved in such a way as to prevent further deterioration. This should include sealing a small aliquot of the sample core in a hermetically sealed container stored at low temperature (i.e., <-20°C).
If volatile hydrocarbons and/or reactive O-containing species are detected above background levels, a portion of the remaining sample core should be hermetically sealed and stored at -20°C or lower to prevent additional volatile loss and chemical reactions at room temperature.
Time-sensitive analyses targeting trace volatile species present in the sample tube headspace gas should be performed using non-destructive techniques. In addition, non-destructive measurements of reactive O-containing species of the solid sample should be performed prior to exposure of the samples to solvents or elevated temperatures and humidity.
Time-sensitive analyses targeting molecular / genetic material should include instrumentation that permits (1) high resolution in situ chemical imaging to distinguish organic material from inorganic material, (2) the determination of stable isotope ratios of elements such as C, H, and N in complex mixtures of organic material, (3) the extraction and identification of organic molecules and polymers, and (4) analysis and characterization of nucleic acids, proteins, lipids, and metabolites.
Instruments that (in some combination) fulfill these requirements include, for example, confocal laser scanning microscopy equipped with Raman and UV fluorescence analysis, as well as gas chromatography isotope ratio mass spectrometry (GC-IRMS), ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS), electrospray ionization mass spectrometry (ESI-MS), and matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF-MS) of samples/extracts suspected of containing biological material. In addition, a DNA sequencer is required to fully characterize the genetic sequence of nucleic acids.
Goal 3: Characterize mineral-bound volatiles
To meet this goal, the time-sensitive focus group recommends the following:
Time-sensitive analyses targeting hydrous sulfate minerals, poorly crystalline or X-ray amorphous materials, clay minerals, and hydrous carbonate minerals should aim to characterize, in detail, the structure and chemistry of the materials, which in many cases may change over time as volatiles are exchanged with ambient surroundings (see Table 4 and Section 4.3).
Suitable analytical capabilities include a combination of powder X-ray diffraction, total X-ray scattering and pair distribution function analysis (which is now possible with commercial lab-based XRD instruments provided a high energy X-ray source and suitable detector are used), Fourier transform infrared and Raman spectroscopy, field emission scanning electron microscopy with energy dispersive X-ray spectroscopy, and BET surface area analysis, which should be utilized to monitor the evolution of poorly crystalline or X-ray amorphous materials in response to ripening/recrystallization reactions.
Goal 4: Characterize solid-phase volatile hosts
To meet this goal, the time-sensitive focus group recommends the following:
Time-sensitive analyses that target mineral-bound volatiles should characterize the concentration, speciation, and isotopic composition of volatiles in question (i.e., H2O, SO2, CO2, etc.).
Suitable analytical capabilities include temperature conversion/elemental analysis-isotope ratio mass spectrometry, a technique that utilizes combustion and isotope ratio mass spectrometry to determine the stable isotopic compositions of components in bulk samples. Although other analytical techniques have been developed that target volatiles hosted in specific minerals (such as coupled thermogravimetry/differential scanning calorimetry and evolved gas analysis via cavity ringdown spectroscopy), these techniques are prone to ambiguity with complex mineral mixtures of more than one or two phases, which together may release multiple components that may react at high temperature and alter the isotopic composition of the evolved gas phase (e.g., Ming et al., 2014). For these reasons, bulk methods such as thermal combustion elemental analysis-isotope ratio mass spectrometry (TC/EA-IRMS) are recommended here, although samples may be subject to physical processing (i.e., particle size separation) to prepare and/or concentrate mineral separates before analysis.
Time-sensitive analytical instruments that target the identity and speciation of volatiles adsorbed on mineral surfaces (e.g., electron paramagnetic resonance spectroscopy, EPR); should be used along with instruments capable of analyzing the chemical reactivity of materials in aqueous solution (an investigation supporting iMOST Objective 7; Table 1). Methods such as ICP-OES and ion chromatography should be utilized to permit analysis of major and trace cation and anion chemistry, respectively.
Recommendations for Future Work
As highlighted in Section 5, some degree of uncertainty surrounds the exact timescales over which time-sensitive processes may influence the scientific utility of specific investigations. Table 4 highlights uncertainty in two main areas: the timescales over which organic material may degrade under various conditions, and the timescales over which volatile-bearing solid phases may exchange with ambient atmospheric conditions. Regarding the former, it is currently not possible to anticipate a priori how rapidly organic material, if detected, may degrade, which is in part a function of chemistry, sample homogeneity, and several other factors. Regarding the latter process of mineral-volatile exchange, it is important to note that currently available studies of the timescales of mineral-volatile exchange are mainly limited to single phase or simple mineral mixtures that utilize laboratory studies of finely powdered, high surface area (and therefore high reactivity) samples. Minerals bound in lithified or otherwise cemented materials are expected to exchange volatiles more slowly as the timescale is likely limited by sample porosity/permeability and grain boundary diffusion. To that end, future work should prioritize constraining the timescales over which analogous samples (e.g., cores of lithified sedimentary rocks) exchange volatiles with ambient surroundings. This should involve, for example, monitoring chemical/mineralogical changes on analog cores.
A final consideration for estimating the loss of scientific information as a function of time involves the degree to which the degradation timescales of a specific investigation target can be extended through mitigation strategies or special storage conditions. For instance, studies of mineral-volatile exchange among hydrous sulfate minerals, hydrous carbonate minerals, and clay minerals have all shown that the rates of exchange depend strongly on RH (Section 4.3). In fact, many studies have shown that storage of a specific mineral within a RH buffered environment (i.e., achievable with a sealed vessel containing a saturated salt solution of a known water activity and T) can greatly extend the window of time over which volatile transfer (e.g., Jerz and Rimstidt, 2003; Chou et al., 2013) and complex interactions between hydrous minerals (e.g., Wilson and Bish, 2012) might occur. Similarly, freezing or vacuum sealing within impermeable materials have each been noted to stabilize or mitigate volatile loss and irreversible mineral transformations among hydrous minerals (e.g., Konrad et al., 2016). Investigation targets for which supporting literature indicates that extension of the timescale might be possible through a mitigation or storage strategy are indicated in Table 4.
Footnotes
Acknowledgments
We would like to thank the entire MSPG-2 committee for their input and discussions during our bi-weekly meetings, especially Kim Tait and Michael Velbel and for their input on this document. We are grateful for the generous input from subject matter experts Paul Niles (NASA-JSC) and Matthew Brady (University of Cambridge) on evolved gas analysis of volatile-bearing minerals, and to Lisa Mayhew, Karim Benzerara, and Ralph Milliken for comments and feedback which improved the clarity of the report.
The decision to implement Mars Sample Return will not be finalized until NASA's completion of the National Environmental Policy Act (NEPA) process. This document is being made available for planning and information purposes only.
Disclosure Statement
No competing interests.
Funding Information
A portion of this work was funded by the National Aeronautics and Space Administration (NASA) and the European Space Agency (ESA).
A portion of this work was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration (80NM0018D0004).
This work has partly (H. B.) been carried out within the framework of the NCCR PlanetS supported by the Swiss National Science Foundation. M.A.V.'s participation in MSPG2 was supported in part by a sabbatical leave-of-absence from Michigan State University. M.-P.Z. was supported by projects PID2019-104205GB-C21 of Ministry of Science and Innovation and MDM-2017-0737 Unidad de Excelencia ‘Maria de Maeztu’–Centro de Astrobiología (CSIC-INTA) (Spain).
Acronyms Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.