
Abstract
Introduction:
This article provides a strategy by which a manufacturing process with a Biosafety Level 2 (BL2) designation can be downgraded to Biosafety Level 1 (BL1). The principles of the downgrading process are based on the robust contamination controls in clinical and commercial manufacturing, which typically are not part of Research and Development processes. These strict requirements along with the application of current Good Manufacturing Practice (cGMP) principles provide a framework by which processes can be suitably managed and controlled to mitigate biohazard risk, specifically for cell lines that may be contaminated with human pathogenic viral agents.
Purpose:
We demonstrate how a risk assessment guide was used to define the risk profile of a theoretical process with a human cell line intended for clinical/commercial application. Based on the risk assessment, key BL2 elements were identified as suitable for downgrading, including facility containment controls, emergency spill response plans, and storage and shipping requirements. For various reasons, some aspects of the systems were deemed unsuitable for downgrading due to the severity of the control risk and, therefore, remained at BL2.
Summary and Conclusions:
We have used an established risk assessment guide to show how cGMP compliments and augments biosafety containment. We provide justification for downgrading from BL2 to BL1 for clinical and commercial cell and gene therapy manufacturing with human cell lines.
Introduction
The biopharmaceutical industry is expanding exponentially, with a focus of getting cell and gene therapy (CGT) products to market as quickly and safely as possible for the benefit of patients. One of the main impediments can be timelines around facility build, fit-out, and various certifications. If some operations are deemed acceptable to be performed in a Biosafety Level 1 (BL1) environment, it may allow retrofitting of existing facilities to facilitate quicker manufacturing timelines. In this article, we assess if biosafety level controls can be downgraded during technology transfer and scale-up from the Research and Development (R&D) setting to the Good Manufacturing Practice (GMP) clinical and commercial manufacturing environment.
As a global consortium of Environmental Health and Safety (EHS) and biosafety experts, our previous article 1 and publication 2 outlined best practices for biosafety risk profiling and controls in cell line expansion in R&D operations of CGT manufacturing. In this study, we use a hypothetical model of transitioning a CGT process utilizing a human cell bank from an R&D environment to a clinical or commercial manufacturing setting. We demonstrate the ancillary benefits of current Good Manufacturing Practice (cGMP) requirements and highlight the risk controls that enable us to downgrade the biosafety containment level without compromising employee safety.
Current industry practice for R&D and CGT manufacturing is to handle all human cell lines with Biosafety Level 2 (BL2)3,4 containment due to the potential risk for latent and adventitious agents. 5 The precedent in the United States has been to handle all human materials including human cell lines as potentially infectious under the Occupational Health and Safety Administration (OSHA) Bloodborne pathogen (BBP) standard. This practice for human cell lines is broadly in place, despite the 1994 OSHA interpretation letter 6 explaining that established human cell lines characterized to be free from recognized BBPs could be considered exempt from the BBP standard.
This is provided the determination was documented and kept on file for OSHA to review. In general, EHS professionals have maintained the requirement for BL2 containment for all human cell lines. However, in recent years, with advancements in technologies and medicines, there has been a call within the pharmaceutical clinical/commercial sector to reconsider the blanket BL2 policy for all human cell lines and to reassess based on the true risks of the materials being handled in these settings, which are highly regulated and designed to mitigate any product contaminations to protect patient safety.
The article explores the possibility of reclassifying a human cell line for pharmaceutical clinical/commercial production and how a company may document a comprehensive risk assessment that would ensure the safety of all employees, yet also allow for greater flexibility in certain areas such as facility design. The subtle changes from BL2 to BL1 containment can make a significant difference in enabling biopharmaceutical companies to deliver lifesaving or life-altering medicines quickly to the populations in need. Downgrading to BL1 would provide significant advantages for facility design, footprint, operations, cost savings, biosafety risk control, and more. In addition, downgrading to BL1 also would offer shared manufacturing access for processes of similar design but with different risk classifications. Although hypothetical, the scenario is representative of a realistic and relevant example commonly found in the field of biologics manufacturing.
Through the principles of risk assessment, the biosafety professionals who authored the article determined that a comprehensive panel for adventitious agents, as well as a full suite of microbial testing,6–9 provided protection for employee safety. Although no amount of testing will ever guarantee or prove a cell line to be free from all infectious agents or eliminate all risks, the authors believe the level of testing to utilize a cell line in a manufacturing process were robust and sufficient to allow for handling of the material with BL1 containment. Thorough analysis of the process ensured that employee health and safety would not be compromised by any of the changes from BL2 to BL1 containment. Note: The lowered risk profile of the tested and well-controlled cell lines does not eliminate the need for reporting of any exposures and providing adequate medical follow-up as determined by a qualified medical provider.
Background/Purpose
The reasons cGMP manufacturing usually is conducted at BL2 is to accommodate the risk group of the vectors produced, the potential use of infectious viruses in the manufacturing process and the use of human cells. The risk of human cell lines pertains to the potential of endogenous viruses (e.g., human papilloma virus in HeLa cell lines) or contamination with relevant adventitious human viruses requiring BL2 containment and practices for safe handling.
In the initial case study, the risk assessment guide1,2 focused on the manufacturing of a human cell line at scales exceeding 10 L in volume. Therefore, facilities and site controls supporting the initial case study were aligned to Biosafety Level 2 Large-Scale (BL2-LS) guidance.1,2,10 The continuation of this theoretical scenario progresses the human cell line manufacturing process from a research environment to the clinical/commercial manufacturing setting. In this setting, GMP compliance imposes requirements intended to improve process performance, product quality, and management of product contamination risk. For example, the human cell line is well characterized and tested for adventitious pathogens through a comprehensive panel of viral and microbial assays and the manufacturing processes are well developed, validated to defined operational limits, and monitored to established process specifications.
Other practices supporting contamination control include the use of industry-standard closed systems, single-use, sterile disposable supplies, raw materials free of animal origin, validated equipment, formal operator training, cleanroom facilities, and well-established site sanitization and segregation practices. Such practices and controls challenge whether this defined state of operations are adequate to mitigate the risk of (1) active and infectious endogenous virus being present in the manufacturing cell line and whether (2) processes are well managed to prevent viral contamination. Fundamentally, these are the two governing factors defining the risk profile of the process and, therefore, the biosafety control requirements.
Considering the robust cGMP requirements, we are interested in whether the previously developed risk assessment tool1,2 can adequately support biocontainment reclassification of a hypothetical clinical large-scale human cell line manufacturing process from BL2-LS to BL1-LS of the NIH guidance, 3 as previously outlined. We focus on the use of established and well-characterized human cell lines, as opposed to autologous and allogenic cell therapy processes. Furthermore, we do not include other process related functions that may influence the biosafety risk profile such as the use other biological reagents (e.g., bovine serum), process-dependent helper viruses or the product viral vectors.
The elements of the process and biosafety risk profile have consequences on the facility design and EHS safety systems, including occupational health practices and post-exposure medical surveillance requirements. Considerations for postsurveillance are predicated on the biosafety profile of the process, conformance of the manufacturing controls, and the employee health profile. As such, surveillance is not a predetermined response but should be managed case by case, based on all mitigating factors.
Risk Guide Overview
Table 1 provides the framework for risk assessment described in subsequent sections with corresponding implementation of appropriate measures commensurate with the defined risk factors of the process.
Risk assessment guide


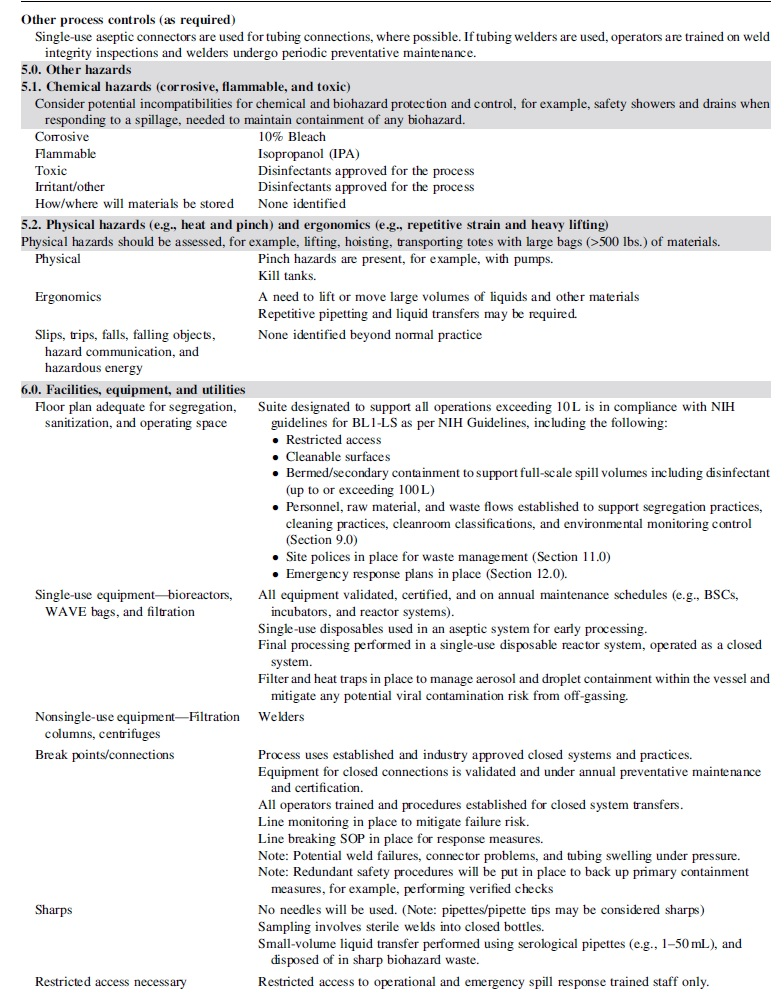
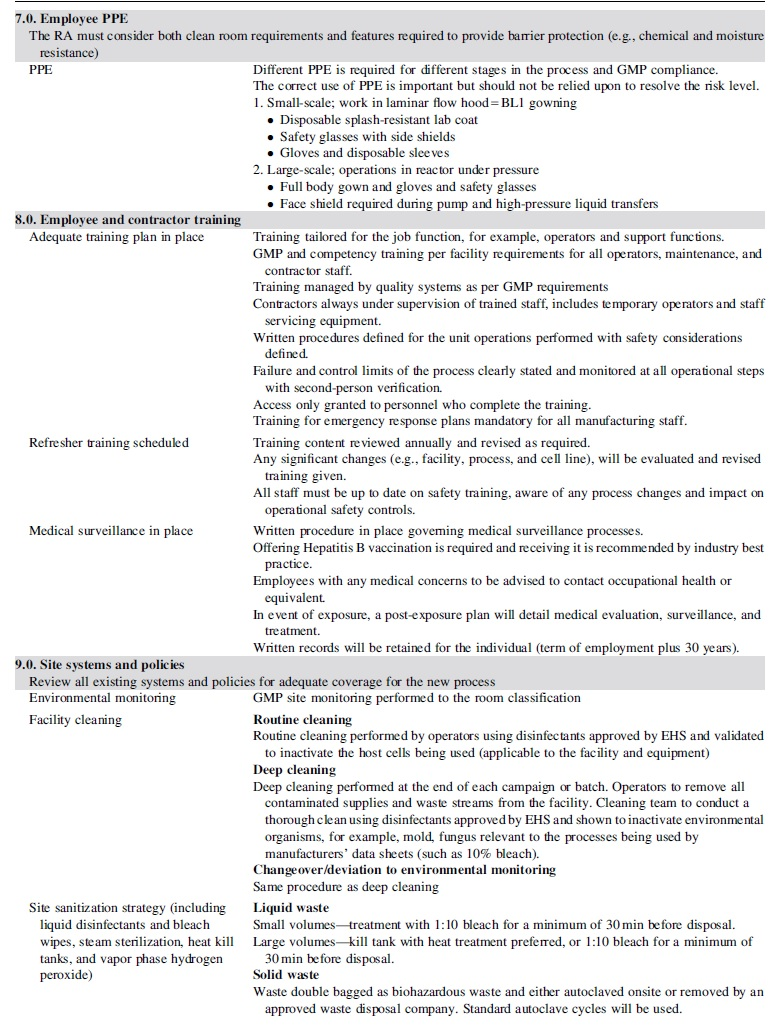
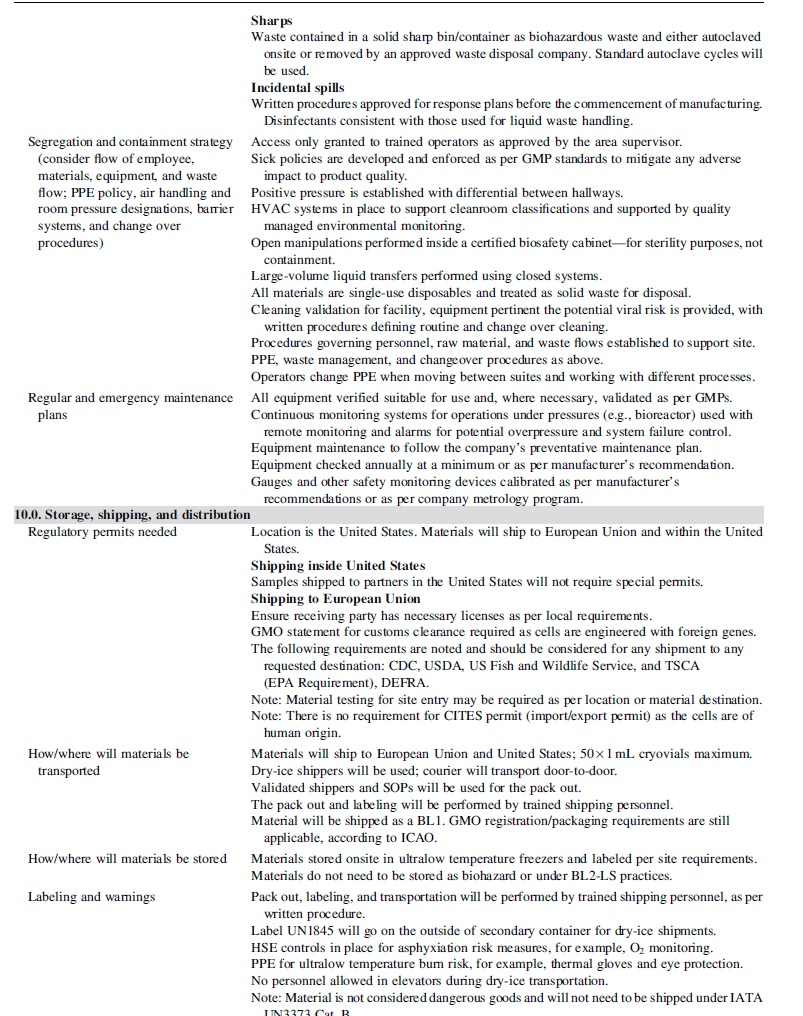
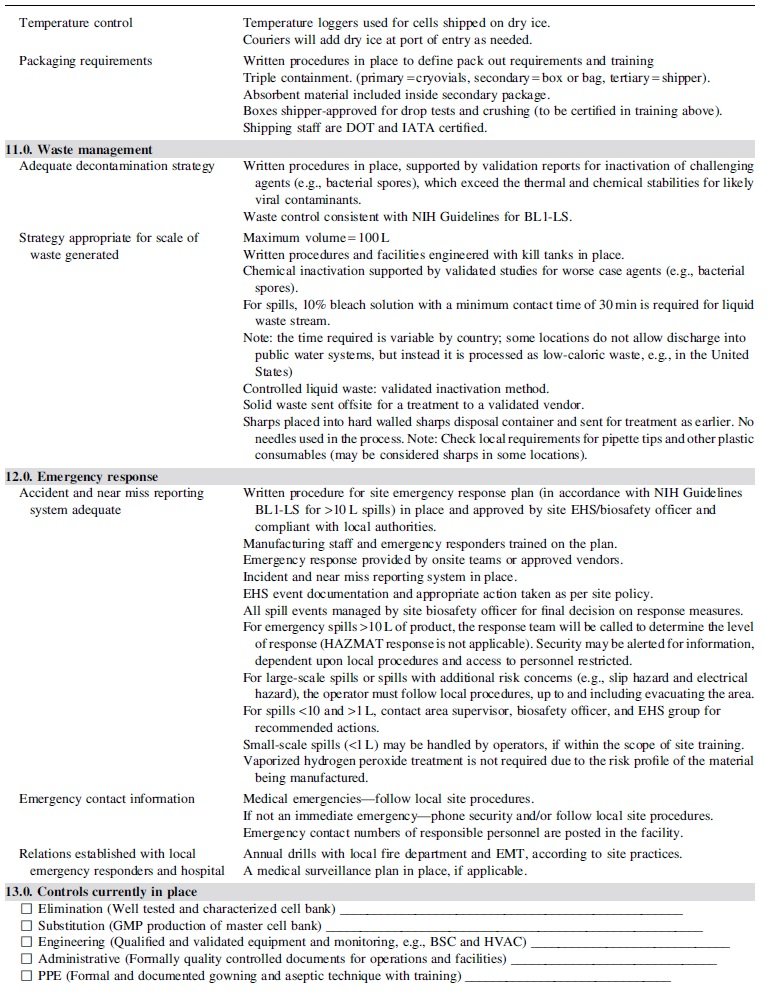
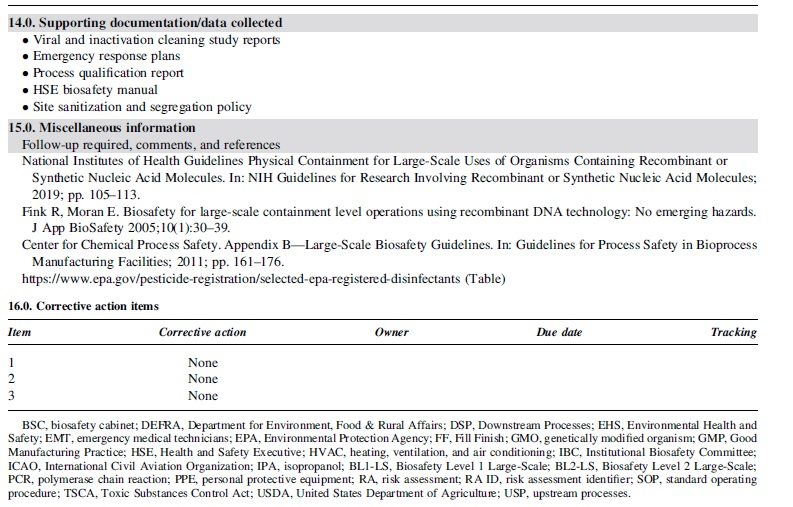
Sections 2 through 4 of the risk guide (Table 1) describe in detail the aspects of the theoretical process, as well as the controls supporting critical raw materials (e.g., manufacturing cell line) and operations.
The assumptions supporting the contamination control of the cell line and the process are summarized as follows:
The cell line is: Of human origin, well characterized, and managed under industry standard quality systems. Tested by a comprehensive panel of viral and microbial assays for adventitious agents and human viral polymerase chain reaction panel, as well as a full suite of microbial testing.6–9
Documented for its origins and traceability to show control of custody and prior exposure to materials of animal origin (e.g., bovine serum and TSE/BSE risk). The process is: Free of materials of animal origin, including growth media, which is serum-free and chemically defined. Uses single-use, sterile disposable supplies. Developed to regulatory standards and employs the use of open and closed manipulation up to the scale of 100 L. Performs open processing following aseptic practices. Supported and managed by a combination of operator training (e.g., operational, procedural, and gowning), engineering controls (e.g., biosafety cabinet, monitored to Grade A) and facility controls (e.g., clean room classification and environmental monitoring). Employs processing using industry standard closed systems for containment control and risk assurance. Monitors for contamination control by testing to an established sampling plan. At a manufacturing site with well-established sanitization and segregation systems in place compliant with applicable regulatory standards to mitigate the risk of contamination.
In summary, the control elements as listed earlier are relevant remediations applied to mitigate the risk of viral contamination to the process. Although the risk of viral contamination is not zero, the control measures are deemed to be appropriate to define and establish the biosafety profile of the process and, if adequately managed, would be justification for a BL1 classification.
Risk Guide Outcomes/Assessment
The robust cGMP requirements compliment biosafety containment controls and provide a strong justification that the process of transferring a human cell line to a GMP clinical manufacturing process can, in principle, be considered suitably controlled against viral contamination and, as such, managed under the principles of BL1-LS. 4 Based on this position, Sections 5.0 through 15.0 of Table 1 were assessed and the biosafety requirements defined. In some cases, the biosafety controls were aligned to the downgraded risk level; in other cases, the biosafety risk was considered appropriate, and the control level not altered. It is also relevant to note that in some cases, the necessary adherence to GMP and regulatory compliance were considered the driving factors of the control measures above the EHS requirements.
Examples of the changes where the downgrading of the risk level was supported by changes in site controls are as listed hereunder:
Post-exposure medical surveillance (Section 8.0) for any individual would be managed on a case-by-case basis and directed by site Occupational Health in consultation with qualified physicians. There is no universal solution, so although the likelihood of a viral pathogen is remote the individual health profile needs to be considered as well as other factors of the incident (e.g., chemical exposure and physical injury).
Site segregation practices (Section 9.0) supporting containment of aerosols for biosafety risk was downgraded from negative room pressure containment to positive room pressure. Risk of a virus contamination was deemed well managed and controlled by the process and supplemental testing of the cell bank. Any such contamination, although remote, would be managed by the containment systems of the process and the closed systems. Storage of cell banks was downgraded to BL1, allowing for a simplified storage solution for the site and eliminating the need for segregation.
Shipping practices (Section 10.0) were downgraded from dangerous/infectious agents 11 to regular biological shipments. No special permits for impact (e.g., CDC permits) were required.
Emergency response plans (Section 12.0) were aligned with NIH Guidelines, 4 where reporting of events and post-exposure medical surveillance was commensurate with the risk profile. Notably, post-spill facility cleaning response did not define vaporized hydrogen peroxide as a precaution, as the viral contamination risk was deemed low and supported by standard site cleaning practices.
The other sections did not have any discernible changes pertaining to the changed state of operations or were deemed not relevant to the adjusted biosafety classification. Waste inactivation was deemed prudent since the cell line was genetically manipulated and local site regulations may require treatment before disposal. Finally, adherence to cGMP principles as best practice for access control remained relevant, including site sick policy for employee health status for site entry.
The process risk profile does not define the requirement for hepatitis B vaccination against potential BBPs due to the testing and control of the cell line. However, such vaccinations must still be offered as per OSHA. 7 Personnel should be educated as to the hazards that could result to an exposure of the specific biological material to an immunocompromised individual, allowing individuals to consider consulting with an Occupational Health professional as to their suitability to conduct work, or if additional measures would be recommended.
Summary and Conclusions
Our team, a global consortium of EHS and biosafety experts, successfully leveraged an established risk guide to define the biosafety controls supporting a theoretical process of the transfer and scale-up of a human cell bank from an R&D environment to GMP manufacturing. We demonstrated through a comprehensive risk assessment success in downgrading the process classification from BL2-LS to BL1-LS and subsequently provided guidance on the appropriate technical operational controls and site systems. We also continue to advocate for retaining BL2 controls for other processes, such as waste management and access control. Post-exposure medical surveillance would always be on a case-by-case assessment based on any mitigating factors, not solely reliant on biosafety considerations but on other health hazards and employee health considerations.
In this theoretical case, our team determined the cGMP process afforded suitable controls for mitigating the risk of viral contamination and, therefore, provided a rational justification for a selective downgrade in containment level. Although we agree testing and operational controls are not sufficient to preclude absolute viral contamination, cGMP control measures do afford site EHS and biosafety personnel several crucial opportunities by defining appropriate risk-mitigation measures:
To leverage the GMP and regulatory requirements that echo and/or enhance the EHS and biosafety components of the facility design and control; the two need not be separated in the site controls as they work together to protect the product and patients; to control contamination; and to remediate biohazard risk to the operators, facilities, and environment.
To carry out a localized risk assessment and ensure the approach is aligned with the following parties to establish process-specific risk profiles:
Local regulatory requirements
GMP technical experts
As CGT manufacturing involving the use of human cell lines are traditionally performed following BL2 practices, the team offers the industry a risk-based strategy to establish suitable EHS site controls commensurate with the safety risk profile of the process and where justified proposals to downgrade BL2 requirements. This strategy builds on the current understanding for the safe use of well-characterized human cell lines for clinical and commercial products, and leverages enhanced biosafety analytical and operational controls supporting increasing regulatory expectations. This risk assessment guide, along with the input from a supporting cast of various interdisciplinary experts, is the foundation for a strategy with the potential for adoption across the industry. The authors hope to establish risk-based systems with opportunities to manage site costs, facility build, fit-out, and site certifications without compromising the biosafety control of the site, personnel, and environments.
Footnotes
Acknowledgments
Improvements were suggested by several subject matter experts who reviewed the article. Any recommendations and errors are our own and should not tarnish the reputations of these reviewers. Special thanks go to Jason Keaton (Acacia Safety Consulting, Inc.) for helpful insights and comments. The study was facilitated by BioPhorum, which since its inception in 2004 has become an open and trusted environment where senior leaders of the biopharma industry come together to openly share and discuss the emerging trends and challenges facing their industry. The BioPhorum Operations Group's mission is to create environments where the global biopharmaceutical industry can collaborate and accelerate their rate of progress, for the benefit of all. More information can be found at ![]() .
.
Authors' Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
![]() The cells risk assessment template can also be downloaded from the BioPhorum website at https://www.biophorum.com/download/biosafety-risk-assessment-template-for-manufacturing-and-processing-of-biological-materials/
The cells risk assessment template can also be downloaded from the BioPhorum website at https://www.biophorum.com/download/biosafety-risk-assessment-template-for-manufacturing-and-processing-of-biological-materials/
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.