
Abstract
Pulmonary hypertension (PH) is characterized by severe exercise limitation mainly attributed to the impairment of right ventricular function resulting from a concomitant elevation of pulmonary vascular resistance and pressure. The unquestioned cornerstone in the management of patients with pulmonary arterial hypertension (PAH) is specific vasoactive medical therapy to improve pulmonary hemodynamics and strengthen right ventricular function. Nevertheless, evidence for a beneficial effect of exercise training (ET) on pulmonary hemodynamics and functional capacity in patients with PH has been growing during the past decade. Beneficial effects of ET on regulating factors, inflammation, and metabolism have also been described. Small case-control studies and randomized clinical trials in larger populations of patients with PH demonstrated substantial improvements in functional capacity after ET. These findings were accompanied by several studies that suggested an effect of ET on inflammation, although a direct link between this effect and the therapeutic benefit of ET in PH has not yet been demonstrated. On this background, the aim of the present review is to describe current concepts regarding the effects of exercise on the pulmonary circulation and pathophysiological limitations, as well as the clinical and mechanistic effects of exercise in patients with PH.
Introduction
Pulmonary hypertension (PH) is characterized by an elevated pulmonary arterial pressure (PAP) and an increased pulmonary vascular resistance due to remodeling of the pulmonary arteries. 1 If left untreated, right ventricular (RV) maladaptation and RV failure ensue as a consequence of prolonged exposure to excessive afterload. 2 Maladaptive hypertrophy and/or dilatation represent central characteristics of the pathophysiological RV response. 2 Moreover, the maladaptive process affecting the RV is described as a key factor in determining the occurrence of relevant clinical symptoms and overall survival. 3 A further key characteristic of PH pathobiology is chronic inflammation which has been detected in the airways as well as the systemic circulation and which contributes in particular to vascular remodeling. 4
For a long time, it was believed that exercise would critically enhance RV stress by substantially increasing RV afterload, and it was assumed that this stress would result in a worsening of RV failure rather than having beneficial effects.
5
As a central consequence, a limitation of physical activity and exercise was recommended during the late 1990s for patients with PH (Fig. 1).
5
However, evidence is growing for positive effects of exercise training (ET) on pulmonary hemodynamics and exercise capacity. Anti-inflammatory effects have also been observed, although these have not yet been directly linked to the therapeutic benefit of ET in PH, and the precise mechanism by which ET positively influences RV function, the pulmonary vascular system, and/or immunity in patients with PH is still unknown.
6
The present review aims to summarize the current status of ET in PH by describing current knowledge of RV and exercise physiology and discussing the available data regarding the effects of ET on the cardio-pulmonary and immune systems with possible transition into the clinical setting.
Timeline of clinical evidence for exercise training in PH.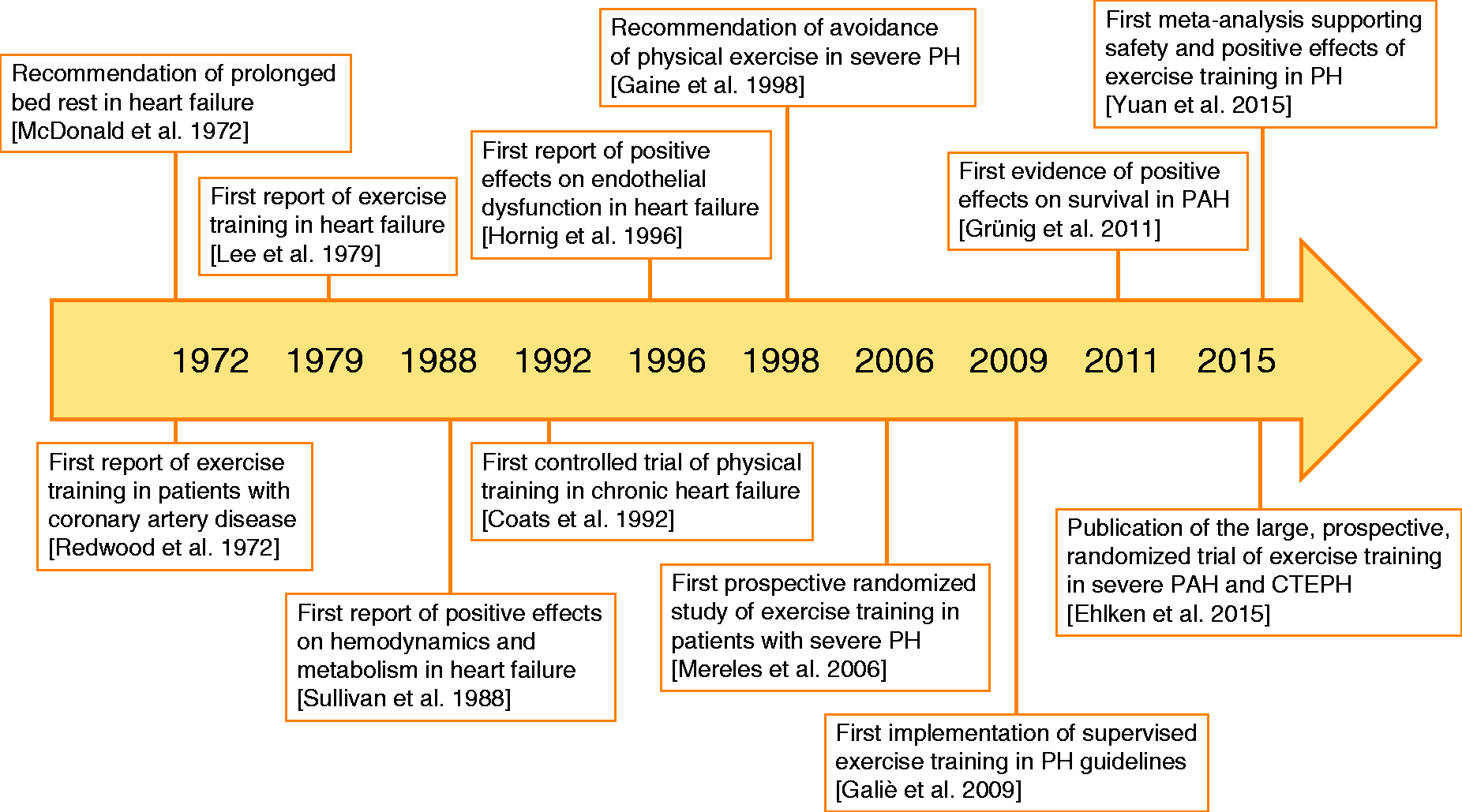
The healthy pulmonary circulation under exercise
Physiologically, the pulmonary circulation resembles a low-resistance and high-compliance system. 7 The response to exercise of the crescent-shaped right ventricle differs dramatically from the response of the left ventricle. 7 During moderate or extensive exercise, both the pulmonary and the systemic circulation have to adapt to manage increased cardiac outputs (CO), which can reach up to 35–40 L/min in trained athletes. 8 Moreover, healthy individuals show a slight (age dependent) rise in mean PAP and pulmonary arterial wedge pressure during exercise.9,10 The healthy pulmonary circulation has several mechanisms to compensate for such a rise in CO, pressures, and RV afterload. Healthy individuals show a slight reduction in pulmonary vascular resistance to allow the increased CO to pass the pulmonary vessels during exercise. 11 Interestingly, the reduction in pulmonary vascular resistance during exercise depends on body position; in the supine position (which allows complete pulmonary vascular recruitment), only a slight decrease in pulmonary vascular resistance is observed. 7 Various invasive hemodynamic studies in healthy volunteers have shown that mean PAP and CO increase in a specific physiological relationship. It is widely accepted that the pressure/flow relationship can be estimated with a linear model (despite the distension of the pulmonary vessels resulting in a slight curvilinearity). 12 Therefore, during exercise, healthy individuals show a mean PAP/CO slope of 0.5–3.0 mmHg/L/min. The flattened slope indicates that even during extensive exercise with a concomitant rise in CO, only a moderate increase in mean PAP is evident. The RV itself compensates for the increased CO demand and the challenge of an elevated afterload by increasing contractility, heart rate, diastolic function, and RV–arterial coupling. 13 As a noteworthy secondary effect, ET subsequently leads to an increase in myocardial mass with concomitant RV hypertrophy and dilatation. 14 Within this framework, the ability of the healthy RV to adapt to ET and to an extensively increased afterload is an intensively discussed issue. Extensive exercise in healthy individuals challenges the RV with a disproportionately high afterload and a greater increase in wall stress compared with the left ventricle. 14 The stress from extreme and prolonged exercise is suspected to result in RV dysfunction with cardiac injury due to myocardial inflammation, substrate deficiency, and oxidative stress. 15 The occurrence of ventricular arrhythmias in athletes has been associated with mild structural and functional RV abnormalities. 16 Whether moderate/normal ET also results in RV dysfunction or an increased risk of RV failure in healthy individuals remains unknown. 11
Exercise limitation in pulmonary hypertension
Even in mild PH, numerous relevant pathological alterations contribute substantially to exercise limitation. The multifactorial pathophysiology of exercise limitation in PH includes impairment of the circulatory, respiratory, and peripheral muscle systems (Fig. 2).
Major pathophysiological hallmarks of exercise limitation in patients with PH. Alterations within the pulmonary circulation, combined with maladaptive responses of the right and partly the left ventricle, influence the respiratory and peripheral muscle systems as well as contributing directly to exercise limitation. In addition, alterations within the respiratory system such as increased dead space ventilation and ventilation/perfusion mismatch result in exercise-induced hypoxemia and thus exaggerate exercise limitation and the sensation of dyspnea. Moreover, reduced peripheral and respiratory muscle strength might lead to excessive muscle fatigability, increased ventilatory drive, and increased perception of effort.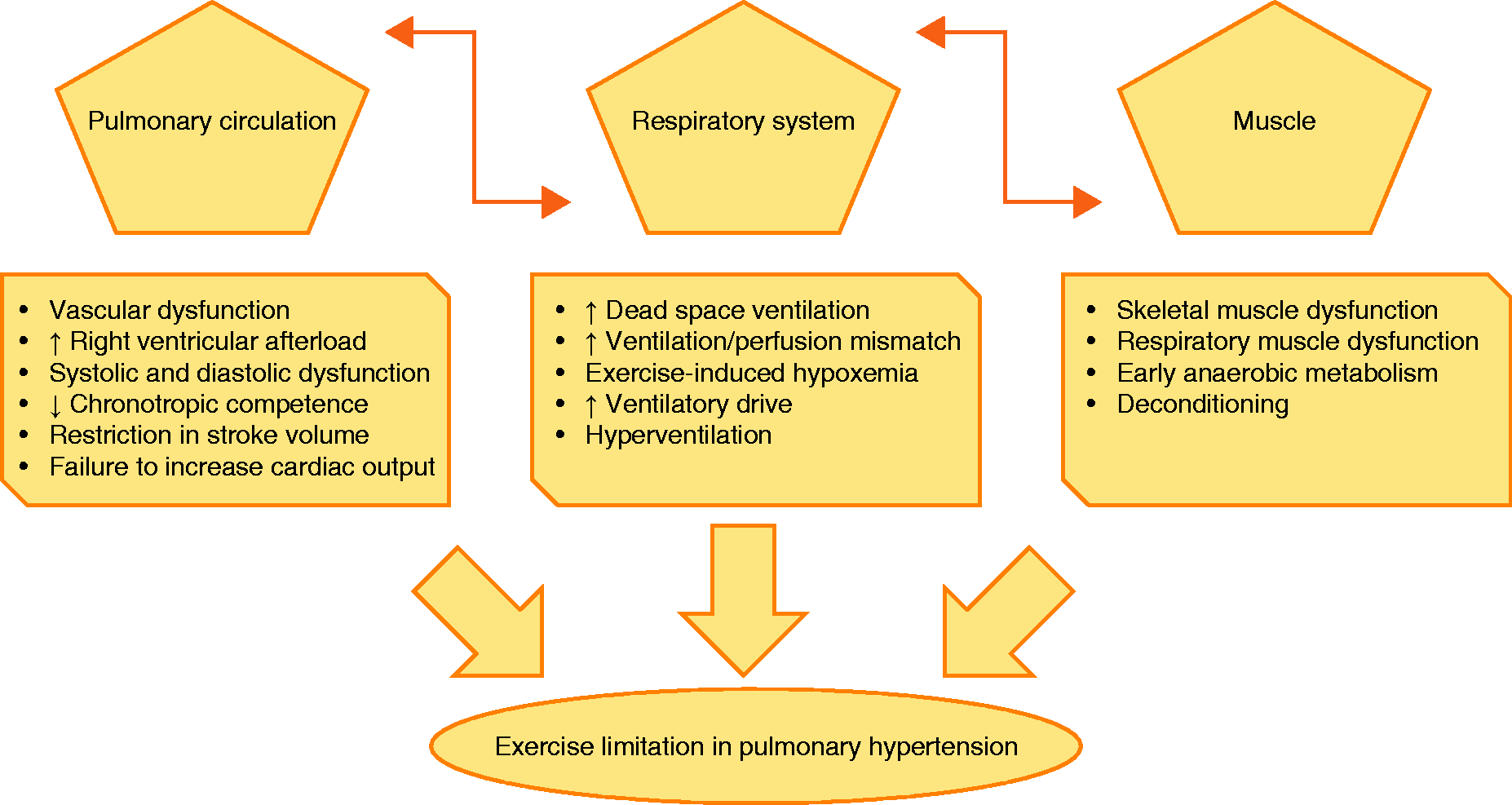
Hemodynamic hallmarks
A key contributing factor to the pathogenesis of PH is the reduced elasticity and patency of the pulmonary vascular system, characterized by an imbalance of vasoconstrictive / vasodilatory mediators and increased proliferation of cells within the pulmonary arterial and capillary vessel walls and extracellular matrix.17,18 These changes lead to an increased RV afterload, 19 and the initial adaptive response of the right ventricle to maintain CO is a rapidly commencing RV hypertrophy. 20 Interestingly, this initial compensated response is characterized by a concentric pattern of hypertrophy, enhanced contractility, preserved ejection fraction, and absence of biomarkers of cardiac dilatation.20,21 In PH, this compensated status can deteriorate into a maladaptive response characterized by an eccentric pattern of RV hypertrophy, a decreasing contractility, RV dilatation with markedly reduced ejection fraction, release of biomarkers of cardiac dilatation, and a secondary neuro-hormonal activation.20,21 The imbalance between the RV oxygen demand (which is increased owing to the increased RV myocardial mass) and the delivered oxygen (which is insufficient owing to insufficient capillarization) is considered to be the main cause of the associated right heart failure.22,23 With the progression of the disease and the aggravation of contractile dysfunction, diastolic dysfunction develops, resulting in a further increase of filling pressures and leading to RV output failure. 24 This, in turn, leads to a depletion of left ventricular preload, 25 which combines with the increased RV pressure and the accompanying paradoxical leftward shift of the inter-ventricular septum to lead to a compression of the left ventricle, 26 resulting in a decreased left ventricular output and depleted systemic oxygen supply at rest and during exercise.27,28 Moreover, exercise limitation in patients with pulmonary arterial hypertension (PAH) is partly attributed to impaired chronotropic competence 29 (evident in cardiopulmonary exercise testing [CPET] as a low oxygen pulse [oxygen uptake (VO2)/heart rate]) 30 as well as a restriction in stroke volume. This phenomenon is attributed to a downregulation of ß-adrenoreceptor activity in the RV myocardial mass 31 and is associated with disease severity.29,32 The combination of these two negative effects prevents an adequate rise of CO and systemic blood pressure during exercise. Animal models have shown that there is a close relationship between RV and right atrial pressure and the ventilatory response. Pressure-related stimulation of mechanoreceptors in the right atrium and right ventricle results in an aggravated sensation of dyspnea that increases ventilation.33–35 Moreover, the right atrial pressure has a strong negative association with exercise capacity 36 and correlates with survival in PH. 37
Respiratory system
Further relevant mechanisms besides the hemodynamic alterations contribute to exercise limitation in PH. Patients suffering from moderate to severe PH show a decrease in oxygen saturation during exercise. 30 This decrease has been associated with the impaired CO response described above, which leads to insufficient oxygen delivery to the peripheral tissue accompanied by a compensatory rise in peripheral extraction. 38 Impaired diffusing capacity 39 combined with ventilation/perfusion mismatch 40 also results in relevant hypoxemia during exercise in patients with PH. Within this framework, reduction of diffusing capacity for carbon monoxide is a common finding in PH.39,41 The reduction is a result of impaired pulmonary membrane diffusing capacity and, to some degree, reduced pulmonary capillary blood flow.39,41 Ventilation-perfusion mismatch (indicated in CPET by an elevated ventilatory equivalent for CO2 [VE/VCO2], steep VE/VCO2-slope, and reduced end-tidal CO2 tension)42,43 is caused by an obstruction of the small pulmonary vessels, non-efficient ventilation, and hyperventilation. 30 The reduction in ventilatory efficiency is partly attributed to impaired blood flow and reduced pulmonary vascular perfusion, 44 which lead to increased dead space ventilation;30,44 the elevated VE/VCO2 is primarily attributed to increased dead space ventilation and is influenced by alterations in ventilatory response (e.g. hyperventilation). 45 The exaggeration of hypoxemia during exercise is associated with stimulation of central and peripheral chemoreceptors, a pronounced sensation of dyspnea, hyperventilation, and substantially increased respiratory demand.45,46 The imbalance between the increase in oxygen demand and the insufficient oxygen supply within the skeletal muscle cells during exercise leads to the early onset of anaerobic metabolism, resulting in a low VO2/workload ratio in CPET (the VO2/workload ratio may not show any alteration until the anaerobic threshold is reached). 45 These changes lead to stimulation of intracellular and extracellular chemoreceptors and thus, via the so-called ergoreflex, increase ventilation.47,48
Muscle dysfunction
During the past decade, numerous studies have focused on the impact of muscle dysfunction within the complex pathophysiology of exercise limitation in patients with PH. In this context skeletal and respiratory muscle dysfunction have been reported mostly in patients with PAH.49–53 It is assumed that reduced peripheral and respiratory muscle strength might contribute to exercise limitation in patients with PAH by causing excessive muscle fatigability, increased ventilatory drive, and increased perception of effort.45,54,55 Moreover, muscle dysfunction might be associated with early anaerobic metabolism which could exaggerate early peripheral muscle fatigue and make a substantial contribution to exercise limitation. 56
It is believed that the muscle dysfunction is caused by reductions in the proportion of type I muscle fibers, capillary to fiber ratio, and aerobic enzyme activity, impaired mitochondrial biogenesis/increased muscle protein degradation mediated by the ubiquitin–proteasome system, and altered excitation–contraction coupling.51,53,57–59 The origin of these multifactorial causes is still under investigation. Systemic inflammation has been suggested to contribute to muscle dysfunction, because pro-inflammatory cytokines have detrimental effects on striated muscle, damaging the function of contractile proteins and stimulating their proteolysis. However, contributory roles have also been proposed for peripheral endothelial dysfunction, impaired anabolic signaling, chronic hypoxemia, and abnormalities of mitochondrial function. The precise mechanism by which skeletal muscle dysfunction interacts with circulatory, inflammatory, and neuronal pathways involved in the exercise pathophysiology of PAH remains unknown. 60
Emerging concepts in hemodynamic measurement at rest and under exercise in pulmonary hypertension
A recent study by Spruijt et al. showed that patients with PH (in contrast to non-PH controls) were unable to increase their ventricular elastance (Ees) during exercise. 61 Ees is considered to be the gold standard for the assessment of load-independent myocardial contractility. 62 RV afterload can be evaluated via the measurement of pulmonary arterial elastance (Ea) and calculation of the Ees/Ea ratio reflects RV–arterial coupling. 62 In patients with PH, only limited data exist regarding the response to exercise of Ees, Ea, and Ees/Ea derived from pressure–volume curves. 63 This is because of the complexity of direct measurement of these parameters: the maximum end systolic pressure from pressure–volume curves is combined with the maximum isovolumic pressure obtained by the so called single-beat-method.62,64,65 Although simplified formulas exist to calculate Ees without pressure–volume curves, for example from cardiac magnetic resonance imaging (MRI),66,67 accurate and reliable assessment of Ees requires conductance catheter technology. 68
Analogous to the findings of Spruijt et al.,
61
Hsu et al. observed a blunted response of Ees to exercise in patients with PAH associated with systemic sclerosis indicating an impaired contractility.
63
Ea increased significantly during exercise while RV–arterial coupling decreased in PAH associated with systemic sclerosis.
63
However, Hsu et al. also showed a contradictory Ees and RV–arterial coupling response in patients with idiopathic PAH.
63
It has to be noted that in PH the rest-to-exercise response in load-independent measures of RV contractility and RV–arterial coupling has only been studied in small cohorts (the studies from Spruijt et al.
61
and Hsu et al.
63
each included only 24 participants). However, the given preliminary data indicate that the impaired rest-to-exercise response in Ees, the increase in Ea, and the deterioration in RV–arterial coupling are important contributors to exercise limitation in PH beyond the increased pressure and resistance of the pulmonary circulation. Fig. 3 shows a conductance pressure–volume loop measurement at rest and during exercise in a patient with PH due to congenital heart disease. The observed right shift of the averaged pressure–volume loops indicates a concomitant increase in RV volumes and pressures. Nevertheless, the derived single-beat measurement of Ees in our patient indicated an increase in contractility and thus RV–arterial coupling during exercise.
Pressure–volume loops from a patient with PH due to congenital heart disease at rest (a) and during maximal exercise (b). The observed right shift of the averaged pressure–volume loops indicates a concomitant increase in RV volumes and pressures. The derived single-beat measurement of Ees in our patient indicated an increase in contractility and thus RV–arterial coupling during exercise. Placement of the conductance catheter and calibration of the RV volume by cardiac MRI were done as reported previously.63,65 Approximately 10 pressure–volume loops were averaged.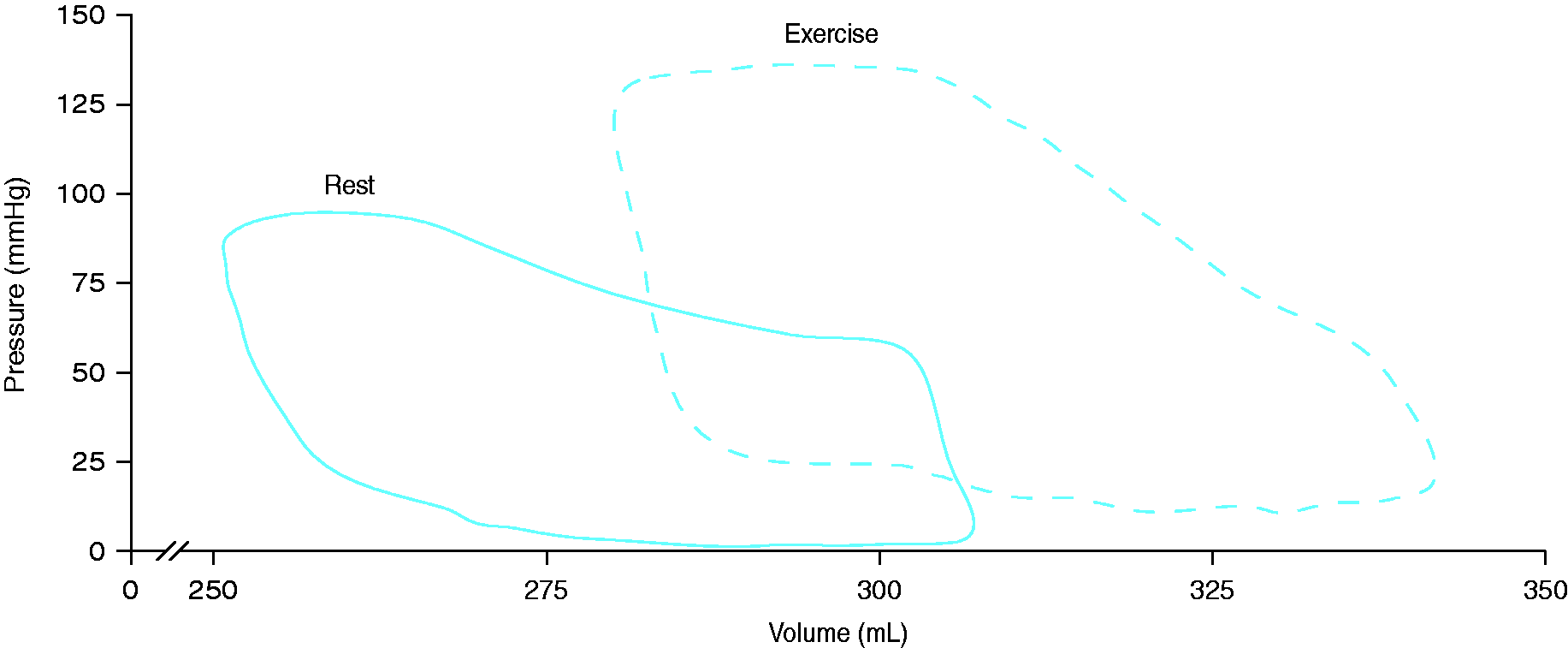
Role of inflammation in the pathogenesis of pulmonary hypertension
There is increasing evidence that inflammation plays a key role in PH pathobiology. 69 For example, pathologic specimens from patients with PAH show an accumulation of perivascular inflammatory cells such as macrophages, dendritic cells, T and B lymphocytes, and mast cells; 70 interestingly, pulmonary arteries from patients with idiopathic PAH show tertiary (ectopic) lymphoid tissues often adjacent to areas of vascular remodeling. 71 In addition to the local inflammation, systemic circulating levels of certain cytokines and chemokines are elevated, and these correlate partly with a poor clinical outcome.70,72 Furthermore, certain inflammatory conditions such as connective tissue diseases are associated with an increased incidence of PAH. Although to date there is a lack of data showing a precise causal or mechanistic relationship between inflammation and PAH pathology, 70 the emerging focus on inflammation provides a new perspective in understanding (and potentially treating) PAH. 73
Despite the lack of mechanistic data, the role of specific cytokines in the initiation and progression of PAH has been intensively discussed. 74 In particular, levels of inflammatory proteins such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-10 have been found to be slightly but chronically increased in patients with PAH. 75 Some of these cytokines have been shown to modulate vascular function or represent risk factors for cardiovascular diseases.
One of these cytokines is the C-reactive protein (CRP) which is known to be associated with systemic arterial hypertension. It was shown that this acute phase protein modulates endothelial cell function by reducing endothelial nitric oxide synthase expression and bioactivity, 76 and by increasing endothelin-1 release. 77 While some studies have found associations between increases in PAP and CRP levels in patients with chronic obstructive pulmonary disease (COPD), 4 a direct causal relationship between CRP and PH pathogenesis has not yet been demonstrated.
IL-6 has a potential role in severe primary PH and PH associated with connective tissue diseases. Hypoxia induces upregulation of hypoxia-inducible factor 1α which is followed by an increase in IL-6 expression. 78 Overexpression of IL-6 induces PAH and vascular remodeling in rodents and further augments hypoxia-driven PH. There are several indications that IL-6 modulates smooth muscle and endothelial cell function leading to vascular remodeling.79,80 On a molecular level, overexpression of IL-6 induces vascular endothelial growth factor resulting in increased proliferation. In parallel, IL-6 upregulates Bcl proteins, the inhibitors of apoptosis, leading to a decrease in apoptotic cell death. 79
TNF-α is a proinflammatory cytokine with potent modulatory effects on the pulmonary circulation. In murine studies, TNF-α was shown to potentiate pulmonary vasoconstriction and increase pulmonary vascular reactivity. In transgenic mice overexpressing TNF-α, severe PH developed. 81 In contrast, TNF receptor-deficient mice were protected against PH. It is suggested that TNF-α signaling in PAH is related to the increased production of reactive oxygen species (ROS). ROS are suggested to play direct and indirect roles in vascular remodeling. NADPH oxidases are important internal sources of ROS and TNF-α is known to be an important regulator of NADPH oxidases in vascular cells. Therefore, it is assumed that increased levels of TNF-α induce ROS production by NADPH oxidases.82,83 However, human studies of the potential direct link between TNF-α and the pulmonary circulation have yielded inconsistent results, which emphasizes the need for further mechanistic studies.
Therapeutic effects of exercise
Clinical effects of exercise training and physical activity on PH
It is widely accepted that regular physical activity, among other lifestyle factors, protects against a series of chronic diseases and disorders. 84 Consequently, various international health associations and institutes such as the American College of Sports Medicine, the American Heart Association, and the World Health Organization have published exercise and physical activity recommendations for adults and older people.85,86
Summary of all major interventional studies of exercise training in pulmonary hypertension.

6MWD, 6-minute walking distance; 6MWT, 6-minute walking test; AHA, American Heart Association; C, controls; CAMPHOR, Cambridge Pulmonary Hypertension Outcome Review; CPET, cardio-pulmonary exercise testing; Ex, exercise; FC, functional class; HR, heart rate; NT-proBNP, N-terminal pro-brain natriuretic peptide; NYHA, New York Heart Association; PASP, pulmonary arterial systolic pressure; QoL, quality of life; SF-36, 36-item Short Form Health Survey; VO2, oxygen uptake; WHO, World Health Organization.
Since the development of the 2015 PH guidelines, further interventional studies addressing the effects of exercise training have been published and the outcomes of these and previous studies have been analyzed in several systematic reviews.104–107 One main conclusion was that improvements in 6MWD varied with different exercise modalities, favoring a combination of aerobic resistance and respiratory muscle training. 105 Further evidence comes from two systematic reviews with meta-analyses which included controlled interventional studies published up to 2013 10 6 and prospective interventional studies published up to 2015. 107 The meta-analyses demonstrated that ET led to a significant increase in 6MWD with a mean improvement of 72 m versus controls and 53 m versus baseline, respectively, accompanied by slight increases in peak VO2 (2.2 mL/kg/min versus controls and 1.8 mL/kg/min versus baseline, respectively).106,107 The highest mean increase in peak VO2/kg was demonstrated in a recent study by Ehlken et al. (3.1 mL/min/kg vs baseline); 6 overall, published data suggest that patients with severe PH who undergo ET increase their peak VO2/kg by about 15–25%. In addition, Ehlken et al. observed substantial improvements in pulmonary hemodynamics for the first time in a prospective randomized study. 6
It has been suggested that the lower improvements of endurance capacity observed in some studies are due to lower intensities and lower frequencies of ET. Since exercise-related improvements of peak VO2 depend on several factors such as lung diffusion, stroke volume, blood volume, and oxygen supply to the skeletal muscle, some studies have focused on changes in muscle structure after training. In this regard, Mainguy et al. found that improvement in 6MWD following ET was associated with a decrease in the proportion of type IIx fibers in patients with idiopathic PAH, indicating a shift of muscle fibers to a more oxidative phenotype. 90
As RV afterload is increased in patients with PH, it has been discussed if exercise-induced increases in pulmonary artery pressures could exceed the RV contractile reserve in these patients. However, all evidence to date indicates that negative effects of exercise on the right ventricle are transient and that function normalizes within days. It is further known that regular ET in healthy individuals promotes healthy physiological remodeling of the heart, as long as the exercise is not too strenuous and prolonged. 11 Therefore, more studies are needed to investigate whether an acute bout of exercise or regular ET may have a negative or positive impact on RV function in patients with PH. On this background, a recent meta-analysis by Pandey et al. demonstrated a slight reduction in resting pulmonary arterial systolic pressure of −3.7 mmHg and an increase in peak exercise heart rate of 10 bpm after ET. 107 Overall, it was reported that exercise was tolerated well with low dropout rates and no serious adverse events related to ET.105-107
Effects of exercise on inflammation
Several studies have demonstrated that both acute and chronic ET affect systemic and local inflammation,108,109 and data from a recent study have shown for the first time that a single bout of exercise may induce an immune response in patients with idiopathic PAH. 73 Up to now, a direct connection between the anti-inflammatory effects of exercise and the therapeutic benefits of exercise in PAH has not been shown. However, our experimental data provide some support for this therapeutic link, showing that regular exercise training downregulates phosphodiesterase-5 in lungs from mice with hypoxia-induced PH. 110
In general, data derived from other studies focusing on cardiovascular, metabolic, or pulmonary diseases have shown that regular physical activity lowers the levels of various proinflammatory cytokines. More precisely, several longitudinal studies of the immunologic effects of ET demonstrated that regular ET resulted in a reduction of systemic CRP and TNF-α levels in patients with chronic low grade inflammation. 111 Systemic CRP levels in the US general population were found to be significantly lower among physically active individuals when compared with their inactive peers, 112 and a recent meta-analysis of interventional studies demonstrated that ET is associated with a decrease in CRP levels regardless of the age or sex of the individual. 113 Regarding TNF-α, it was shown that exercise inhibits the endotoxin-induced increase in circulating levels of TNF-α in healthy individuals. 114 A major mechanism suggested to underlie this phenomenon is the release of myokines, which are cytokines with mainly anti-inflammatory properties released by muscular tissue. In this regard, it was shown that contracting muscle releases IL-6 as a response to glucose depletion during exercise. Although IL-6 has negative effects during chronic disease states, the exercise-induced periodic release of IL-6 is followed by the appearance in blood of IL-1RA (which inhibits the pro-inflammatory actions of IL-1β) and IL-10 (which downregulates the adaptive immune response). It was also demonstrated that IL-6 exerts inhibitory effects on TNF-α and IL-1 production.115,116
Another exercise-related mechanism that might affect inflammatory status during disease is the release of adrenal hormones. Exercise is known to activate the hypothalamic–pituitary–adrenal axis and the sympathetic nervous system, which is followed by increased secretion of cortisol, epinephrine, and norepinephrine. Cortisol is known to elicit potent anti-inflammatory effects. Catecholamines have been shown to downregulate the lipopolysaccharide-induced production of cytokines such as TNF and IL-1β. Therefore, it is assumed that both hormones and myokines contribute to the anti-inflammatory effect of exercise (Fig. 4).
117
However, it remains to be shown in future experimental studies if ET exerts its therapeutic effect in PAH via a reduction of proinflammatory cytokines.
Proposed role of systemic inflammatory effects in the pathobiology of PAH and the hypothetical potential of exercise to counteract vascular remodeling. PAH is induced by various pathological mechanisms, with inflammatory and autoimmune processes contributing to the increased proliferation and decreased apoptosis of pulmonary vascular smooth muscle cells (vascular remodeling). Increased NADPH oxidase activity increases oxidative stress and induces inflammatory pathways via expression of TNF-α, which in turn stimulates NADPH oxidase. Increased CRP levels decrease eNOS activity, leading to pulmonary vasoconstriction. Exercise affects inflammation and redox status, and could thus potentially counteract vascular remodeling, though this proposed mechanistic link remains to be demonstrated in experimental studies. In particular, exercise training decreases CRP levels and increases eNOS activity, which could lead to improved vascular compliance. It further stimulates anti-oxidative enzyme activity and inhibits NADPH oxidase activity, leading to an overall reduction of ROS. Finally, exercise stimulates the release of myokines such as IL-6 from the contracting muscle followed by an increase of IL-10 and IL-1RA, which exert anti-inflammatory effects.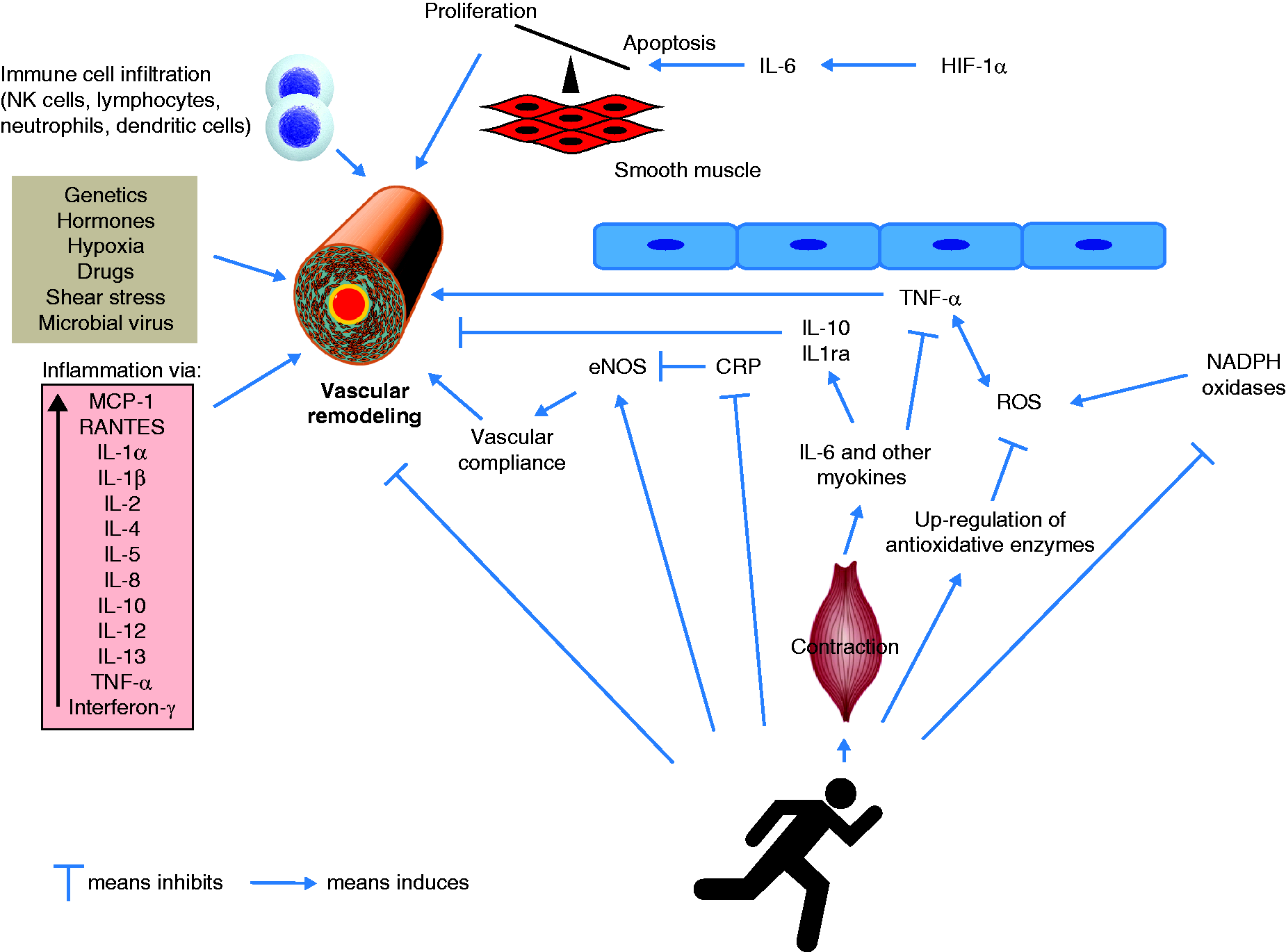
Anti-oxidative effects of exercise
Given the involvement of ROS in inflammation and vascular remodeling, the relationship between exercise and oxidative stress must also be discussed. During acute exercise, increased amounts of free radicals are generated, which are known to modulate muscle contraction, antioxidant protection, and oxidative damage repair. Furthermore, exercise-induced ROS formation is suggested to mediate upregulation of antioxidant molecules, as reflected by increased glutathione reductase or superoxide dismutase levels in response to regular ET. These effects are currently explained by the hormesis theory, in which an agent that is detrimental at high doses can induce an adaptive beneficial effect in the cells or the organism at low doses.118,119 Therefore, it is concluded that exercise training seems to induce an antioxidant effect. On this background, it can be suggested that patients who exercise regularly benefit due to an improved balance of their redox status. However, the direct benefit patients with PAH gain from the exercise-induced changes in redox status is still unclear.
Perspectives and future directions
Since 2006 it has been repeatedly demonstrated that an exercise program is safe and effective in improving exercise and functional capacity as well as quality of life in patients with PH. Indeed, there is still a need for further investigations to titrate the most effective exercise variables in this patient group. For example, it would be interesting to evaluate the value of eccentric strength training, which induces a lower drive to breathe during high workloads than concentric training and might therefore be more feasible for patients with PAH. Similarly, recent studies demonstrated that high-intensity interval training (using an intensity calculated relative to patients’ exercise capacity) could also be successfully used for exercise therapy.11,120 However, no data are available to demonstrate if this also applies to patients with PAH.
Many interventional studies have investigated individual, home-based exercise rehabilitation programs. For most chronic diseases, it was shown that group-based and supervised exercise programs are more effective than home-based training. On this background, we suggest that the regular participation of patients with PAH in specific ambulatory training groups might be a favorable therapeutic approach. Programs like this are supervised by a professional instructor and offer – beyond targeting the main symptoms – psychosocial and educational support.
Recommendation for current concepts of different exercise training protocols in PH.
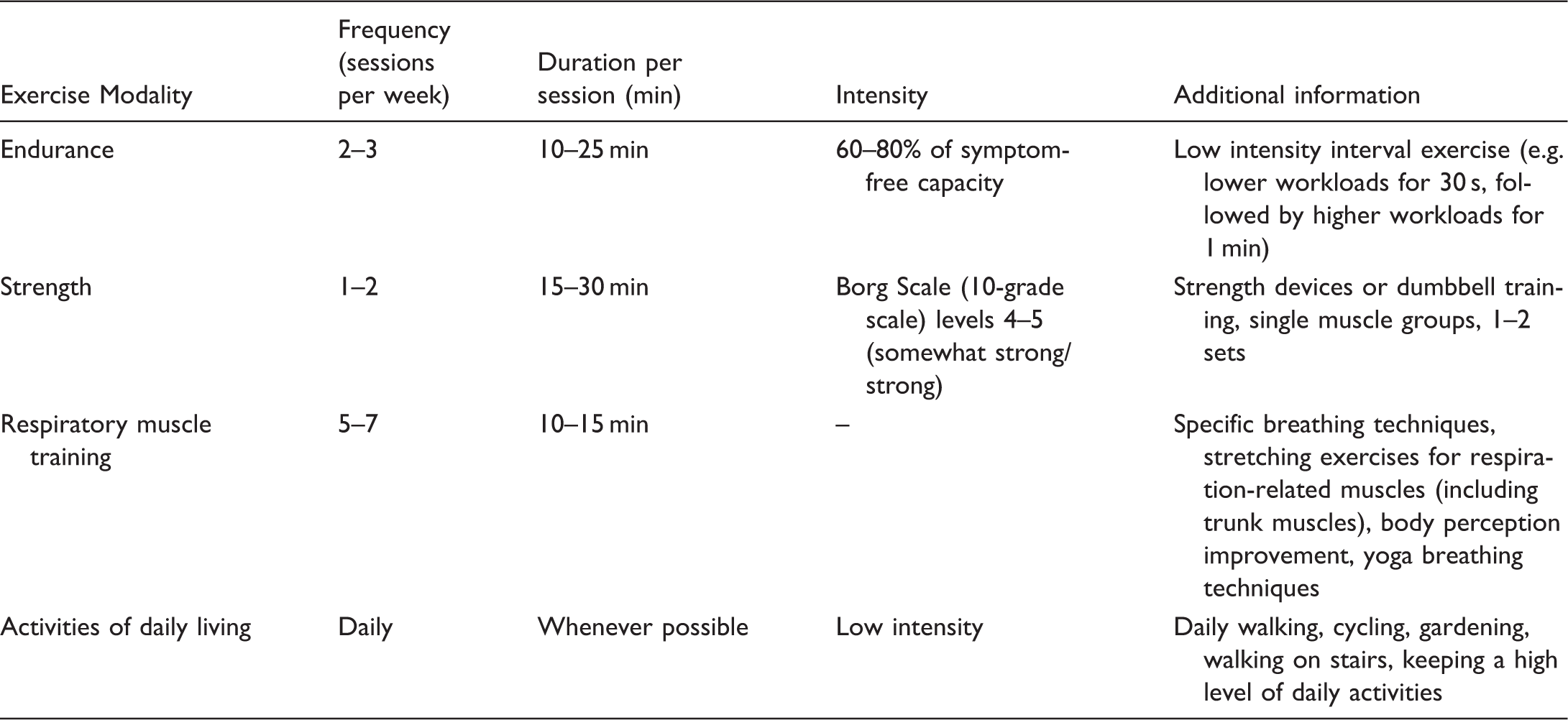
Summary
In conclusion, ET is emerging as a promising additional therapy option for patients with PH. Besides the impact of ET on functional capacity and pulmonary hemodynamics, recent studies have suggested that ET has anti-inflammatory effects, although it is not yet known if these effects contribute to the therapeutic benefits of ET in PH. Despite the emerging evidence from various controlled trials, the actual mechanistic link between ET and improvements of major pathophysiological PH features remains unknown. Whether ET directly influences RV maladaptation or improves pulmonary arterial remodeling has to be investigated in the future.
Footnotes
Acknowledgments
The authors thank Claire Mulligan, PhD (Beacon Medical Communications Ltd, Brighton, UK) for editorial support. We thank Jens Axmann for providing data of the pressure-volumen loops as part of his doctoral thesis.
*
Equal contributors.