
Abstract
Sepsis-related multiple organ dysfunction syndrome is a leading cause of death in intensive care units. There is overwhelming evidence that oxidative stress plays a significant role in the pathogenesis of sepsis-associated multiple organ failure; however, reactive oxygen species (ROS)–associated biomarkers and/or diagnostics that define mortality or predict survival in sepsis are lacking. Lung or peripheral blood gene expression analysis has gained increasing recognition as a potential prognostic and/or diagnostic tool. The objective of this study was to identify ROS-associated biomarkers predictive of survival in patients with sepsis. In-silico analyses of expression profiles allowed the identification of a 21-gene ROS-associated molecular signature that predicts survival in sepsis patients. Importantly, this signature performed well in a validation cohort consisting of sepsis patients aggregated from distinct patient populations recruited from different sites. Our signature outperforms randomly generated signatures of the same signature gene size. Our findings further validate the critical role of ROSs in the pathogenesis of sepsis and provide a novel gene signature that predicts survival in sepsis patients. These results also highlight the utility of peripheral blood molecular signatures as biomarkers for predicting mortality risk in patients with sepsis, which could facilitate the development of personalized therapies.
Acute overwhelming sepsis is the leading cause of morbidity and mortality among critically ill patients in intensive care units (ICUs). The sepsis syndrome manifests as a spectrum of conditions, including systemic inflammatory response syndrome (SIRS), sepsis, severe sepsis, septic shock, and multiple organ system dysfunction. The overall mortality rate ranges from 26% in patients with SIRS to 82% in patients with septic shock.1,2 Currently, there are no available biomarkers that are predictive of survival in patients with sepsis. 3 Traditional acute-phase markers such as C-reactive protein, interleukin 6, or procalcitonin do not predict survival.3–5 Procalcitonin has been used to indicate the presence of bacterial infection, but its use a prognostic biomarker has not established.
Over the past 2 decades, major advances have been made in our understanding of the underlying biologic features of sepsis. However, these findings have not been successfully translated to effective therapies. One potential barrier to the development of effective therapies and improved survival outcomes is the paucity of reliable biomarkers for diagnosis, prognosis, and responses to therapy. Biomarker studies are often limited by the number of candidate biomarkers, cohort size, and lack of replication in a validation cohort. 6 Technological advancements have enabled the utility of expression microarrays providing analysis of genome-wide profiles capable of generating molecular signatures, from either disease tissues7–14 or peripheral blood mononuclear cells (PBMCs).15–17We have previously employed this strategy to identify diagnostic and/or prognostic biomarkers for several respiratory diseases, such as sarcoidosis, 18 idio-pathic pulmonary fibrosis, 15 asthma, 19 and lung cancers.20,21
Interestingly, there is overwhelming evidence implicating an underlying role of oxidative stress in the pathogenesis of sepsis.22–25 Under normal physiologic conditions, redox balance exists through a complex interplay of genes that mediate oxidant generation and antioxidant responses. An imbalance between the production of reactive oxygen species (ROS) and the capacity for detoxification of their reactive intermediates results in oxidative stress. Numerous studies demonstrate an association between sepsis and elevated oxidative-stress levels.23,25,26 It is likely that sepsis triggers the alteration of the activity of various genes that mediate both oxidant generation and/or antioxidant capacity. Here, we analyzed previously generated microarray gene expression data from PBMCs of sepsis patients and constructed the first ROS gene expression–based signature capable of predicting the odds of patients' outcome. In-silico analyses of expression profiles and phenotypic data allowed the generation of a 21-gene ROS-associated molecular signature that predicts survival, in both the discovery and validation cohorts. These results demonstrate the feasibility of using peripheral blood to identify molecular signatures that can serve as relevant biomarkers for predicting survival and its potential for facilitating individualized therapies.
METHODS
Subjects and blood samples
For the discovery cohort (GEO [Gene Expression Omnibus] accession: GSE54514), whole-blood samples were collected from a total of 35 patients with confirmed sepsis at day 1 in the ICU. The diagnosis of sepsis was based on documented bacterial infection, meeting SIRS criteria, and consensus by the consulting physician that sepsis was the cause of the patients ICU stay. The validation cohort (GEO accession: GSE63042) included patients who met at least two SIRS criteria and had a suspected or known acute infection. 15 Peripheral blood samples were collected from 106 patients. We segregated the sepsis patients into two groups: survivors and nonsurvivors. Specifically, we utilized data from 35 patients (9 nonsurvivors and 26 survivors) as the discovery cohort and data from 106 patients (28 nonsurvivors and 78 survivors) as the validation cohort (Table 1). For the samples in the discovery cohort, the whole-genome gene expression pattern was profiled by Illumina HumanHT-12 V3.0 expression beadchip, while the gene expression data in the validation cohort were obtained by high-throughput sequencing using Illumina Genome Analyzer II. The study was approved by the Institutional Review Boards of each institution, with written informed consent obtained from the patients or their relatives. 27,28
Characteristics of individuals included in analyses
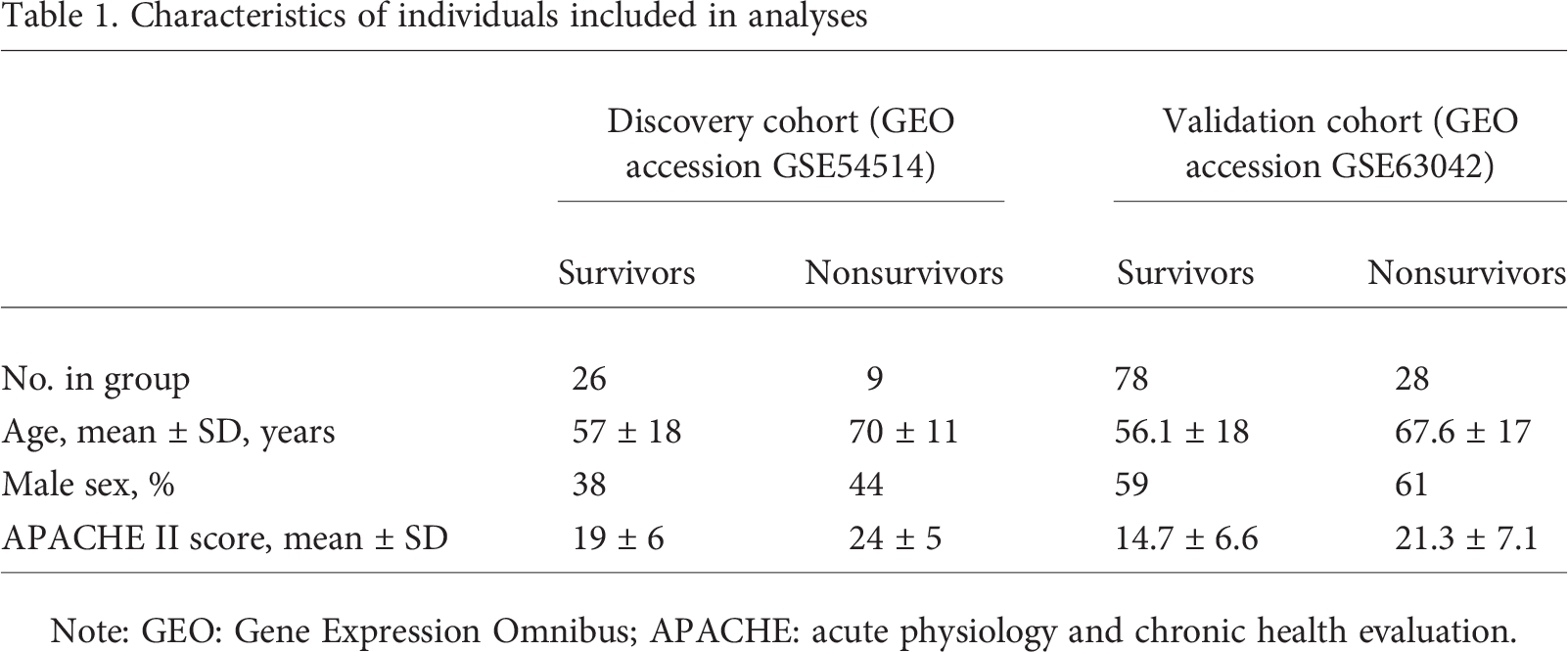
Note: GEO: Gene Expression Omnibus; APACHE: acute physiology and chronic health evaluation.
Risk score
A risk score was calculated for each patient using a linear combination of expression values of genes in the signature.14,20,21 The formula is
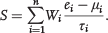
Here, S is the risk score of the patient; n is the number of genes in the signature; Wi denotes the weight of gene i (as shown in Table 2), which indicates the direction of deregulation for gene i (1 or −1); ei denotes the expression level of gene i; and μi and τi are the mean and standard deviation of the gene expression values for gene i across all samples, respectively.
21-gene signature
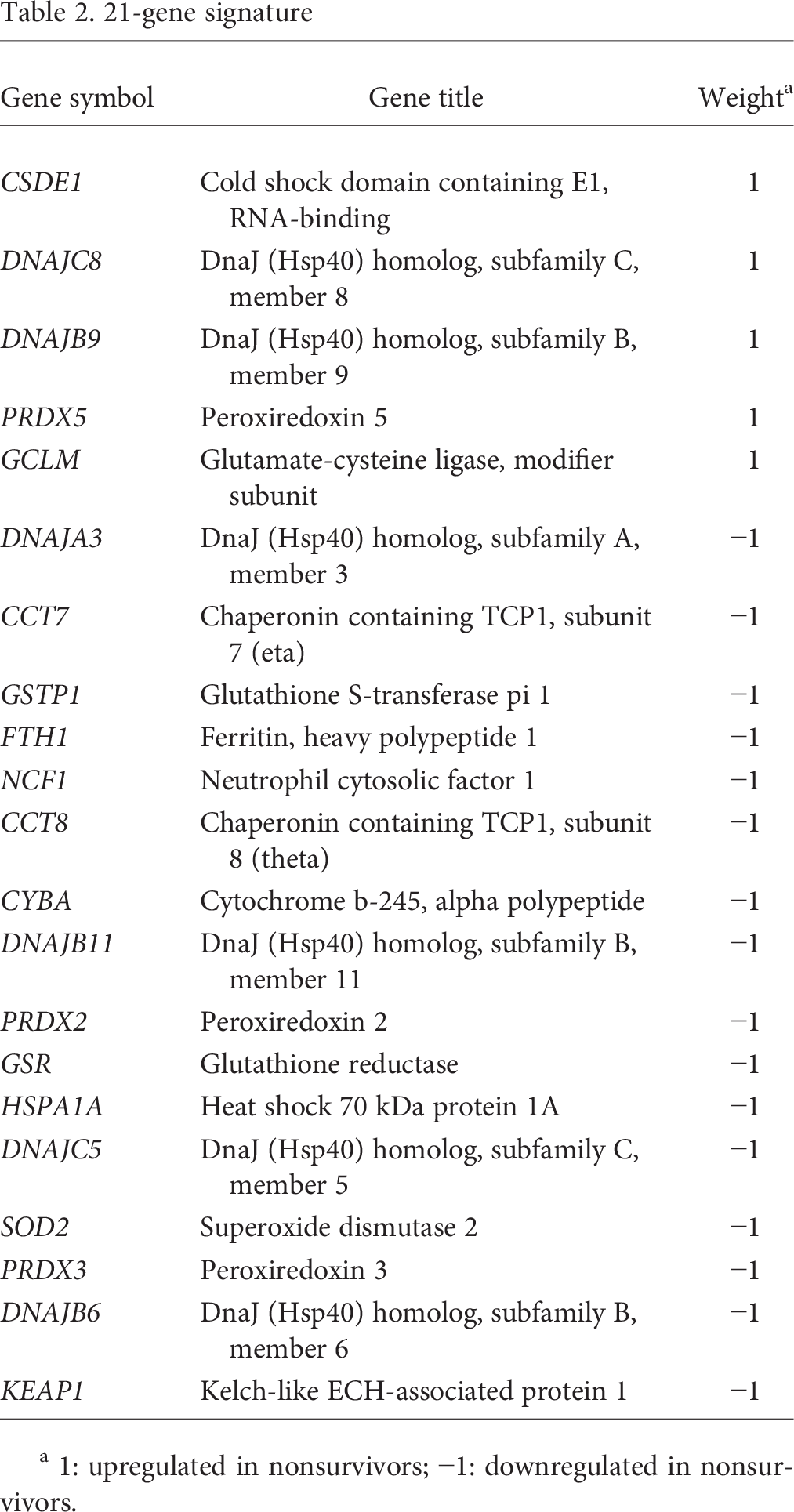
1: upregulated in nonsurvivors; −1: downregulated in nonsurvivors.
RESULTS
Patient characteristics
Our in-silico analyses focused on 35 patients (9 nonsurvivors and 26 survivors) as the discovery cohort and 106 patients (28 nonsurvivors and 78 survivors) as the validation cohort. The characteristics of individuals included in our analyses for survivors and nonsurvivors within the discovery and validation cohorts are presented in Table 1.
Identification of differentially expressed genes in blood associated with survival of sepsis patients
To determine differentially expressed protein-coding genes in PBMCs associated with survival of sepsis patients, we first analyzed gene expression patterns in the discovery cohort. We then linked gene expression levels with survival. We identified 450 genes upregulated and 1,027 genes downregulated in nonsurvivors compared to survivors (Fig. 1).
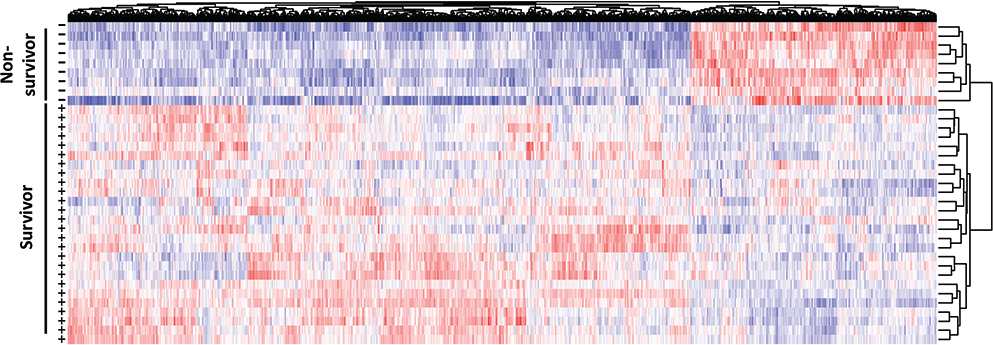
Differentially expressed genes associated with survival (+) and nonsurvival (−) in sepsis patients. The heat map is generated on the basis of peripheral blood mononuclear cell gene expression; red represents increased expression, while blue represents decreased expression.
We searched for any enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) 29 physiological pathways among the differentially expressed genes, using the National Instituted of Health DAVID (Database for Annotation, Visualization, and Integrated Discovery).30,31 Our analyses revealed 19 KEGG pathways associated with differentially expressed genes (P < 0.05); the top KEGG pathways significantly associated with differentially expressed genes were identified, with “oxidative phosphorylation” as the most significantly altered KEGG pathway (Fig. 2). Gene ontology analysis on Gene Ontology Consortium biological-process terms 32 also confirmed that oxidative phosphorylation is significantly associated with the genes expressed differentially between survivors and nonsurvivors (Fig. 3).
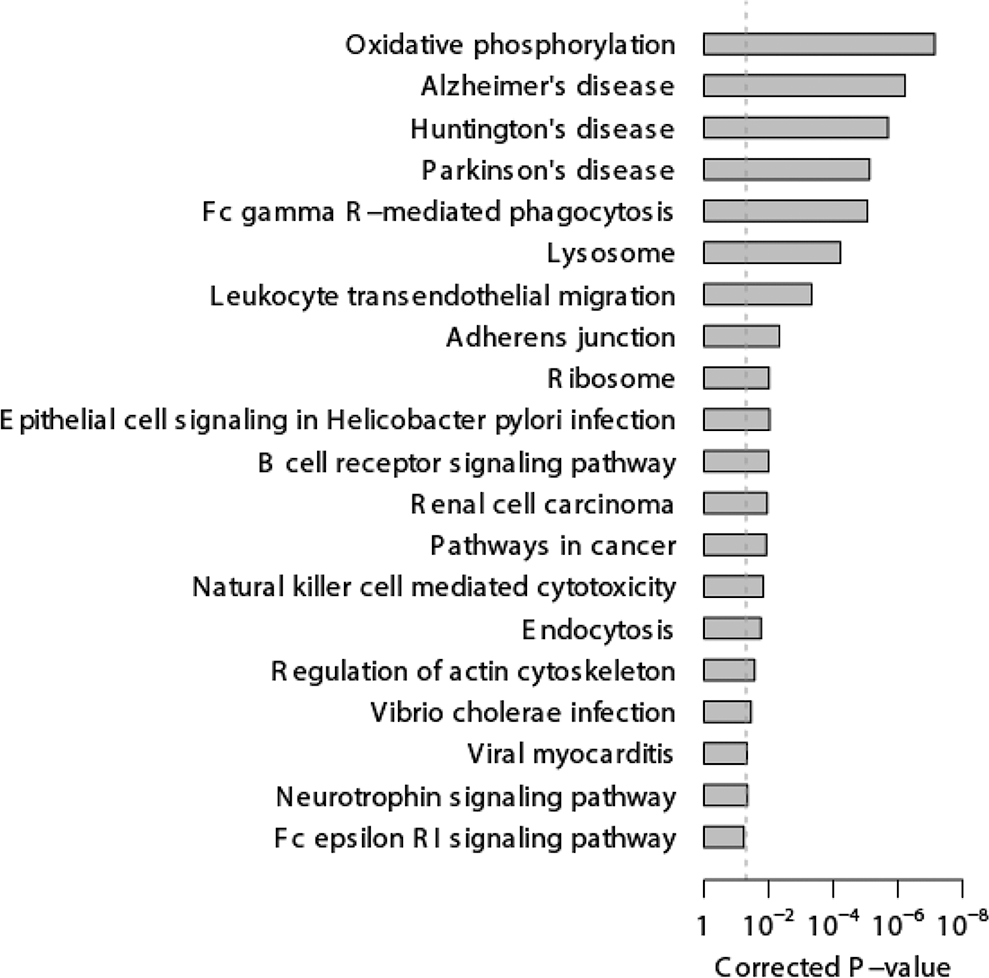
Enriched pathways among the genes expressed differentially between survivor and nonsurvivor sepsis patients. The 20 top-ranked Kyoto Encyclopedia of Genes and Genomes pathways are listed. The dotted line indicates the cutoff of significance (0.05). The Fisher exact test was used to calculate the P values.
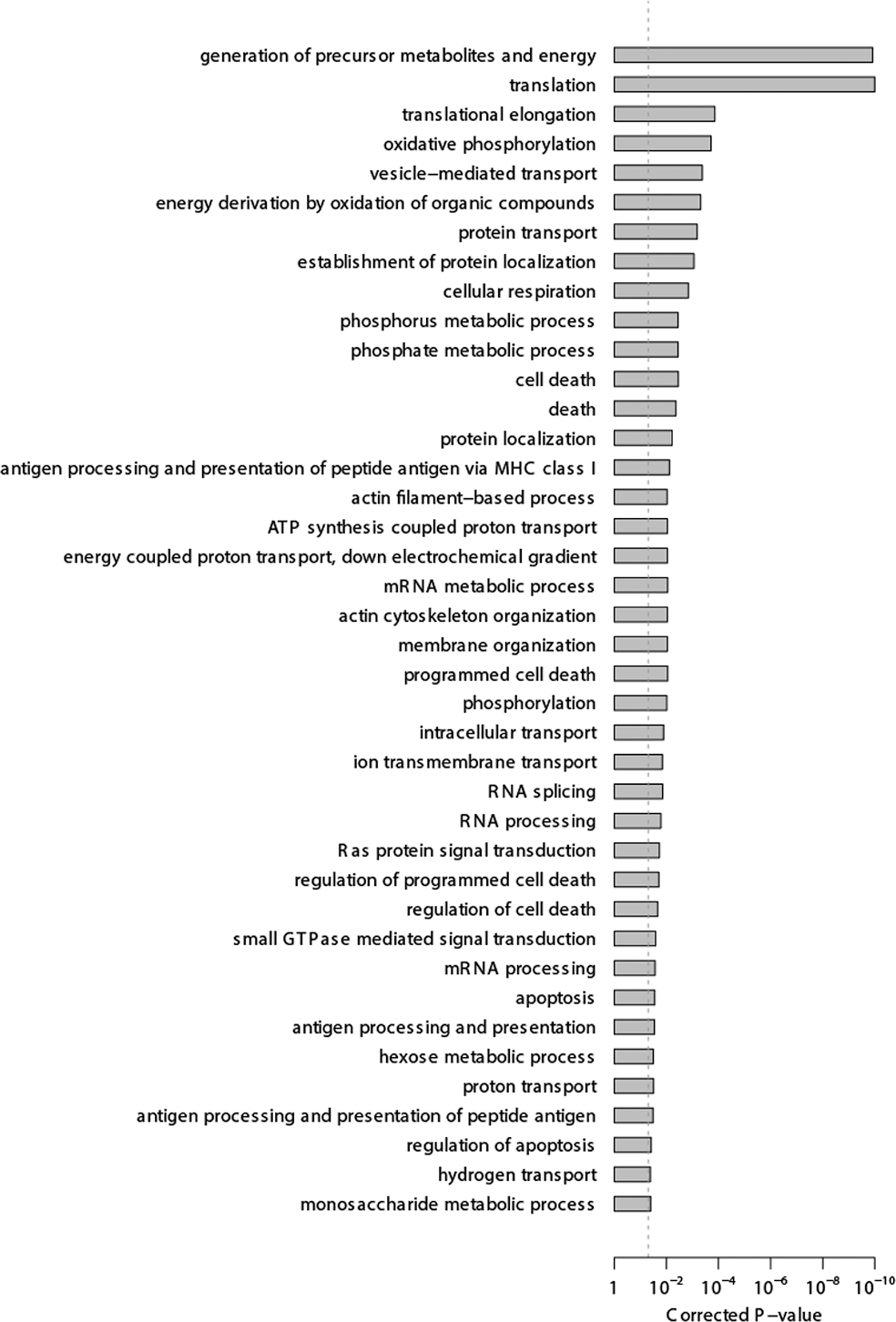
The 40 top Gene Ontology Consortium 32 biological-process terms associated with the genes expressed differentially between survivors and nonsurvivors. The dotted line indicates the cutoff of significance (0.05). The Fisher exact test was used to calculate the P values.
ROS gene signature predicts survival in the discovery cohort
We identified a group of 137 genes that are known to play a role in mediating ROS levels (Table S1, available online). This list includes genes involved in oxidant generation (such as NADPH oxidases), metabolism (superoxide dismutases), and/or antioxidant response genes (Nrf2 glutathione peroxidases, peroxiredoxins). Interestingly, we confirmed that these deregulated genes are significantly enriched in sepsis survival–related genes (cumulative hypergeometric test: P = 0.012), suggesting a statistically significant overlap (21 genes) between ROS genes and sepsis survival–related genes (Fig. 4A). We designated the genes within the overlap a 21-gene signature (Table 2; Fig. 4B). The weights listed in Table 2 indicate the direction of differential gene expression in nonsurvivors.
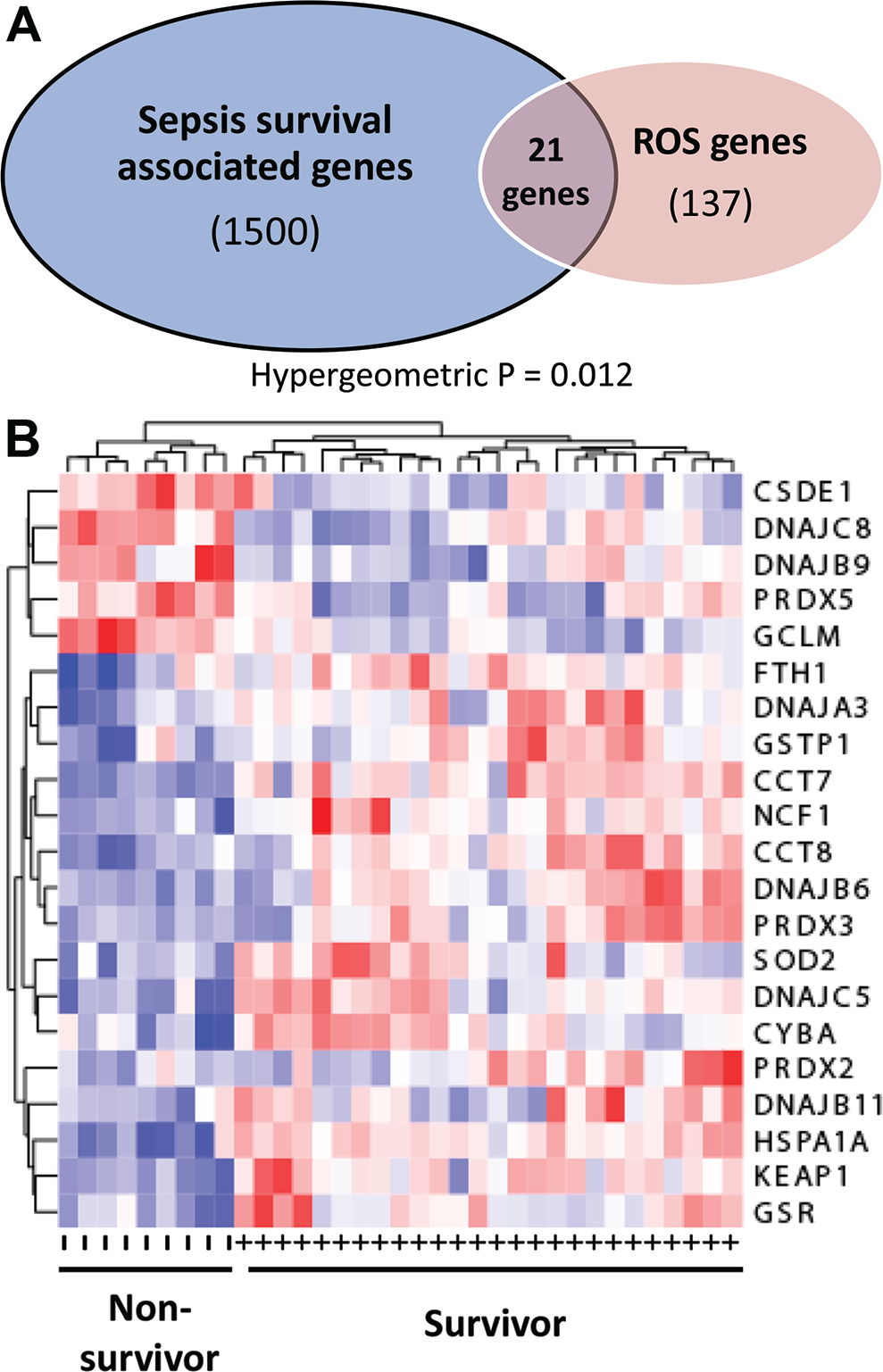
Reactive oxygen species (ROS)–associated gene signature in sepsis patients. A, Schematic diagram representing genome-wide and ROS-associated overlapping genes. Twenty-one overlapping genes were identified, which is statistically significant (hypergeometric test: P = 0.012). B, Heat map representing the 21 differentially expressed ROS-associated genes. Red represents increased gene expression, while blue represents decreased expression. Patients are characterized as survivors (+) or nonsurvivors (−).
On the basis of the 21-gene signature, we constructed a scoring system to assign each subject a risk score, representing a linear combination of the 21 gene expression values, weighted by the direction of differential expression: 1 for the upregulated and −1 for the downregulated genes in nonsurvivor (see “Methods” for details). A higher risk score suggests a poorer clinical outcome. Our in-silico analyses focused on 35 patients (9 nonsurvivors and 26 survivors) as the discovery cohort (Table 1). As expected, the risk scores of nonsurvivors were significantly higher than those of survivors in the discovery cohort (t test: P = 6.45 × 10−13; Fig. 5A). The area under the receiver operating characteristic curve (AUC) was 1.000 (Fig. 5B).
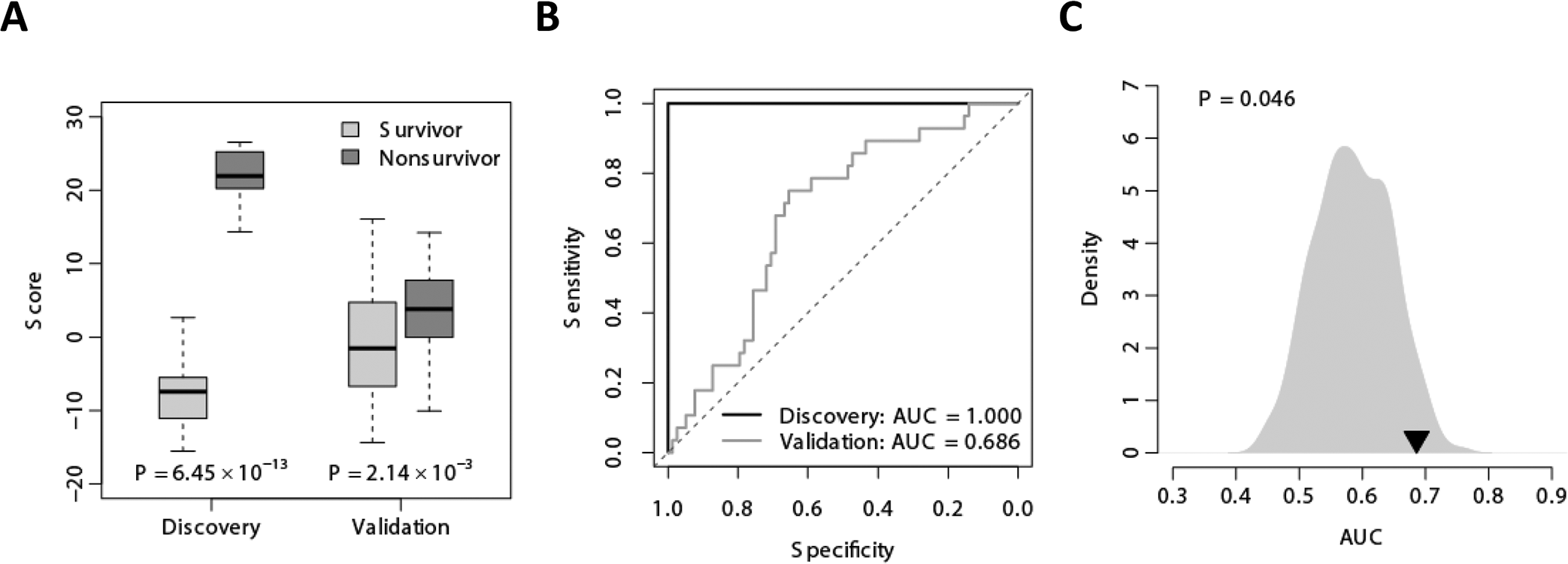
The 21-gene signature–based risk score. A, The 21-gene signature–based risk score differentiates the nonsurvivors from the survivors in both the discovery and validation cohorts. The box plot indicates the distribution of the risk score in each category. B, The receiver operating characteristic (ROC) curves of the 21-gene signature in distinguishing nonsurvivors from survivors in both the discovery and validation cohorts. C, Superior predictive power of the 21-gene signature–based risk score compared with random gene signature. The gray area shows the area under the ROC curve (AUC) for 1,000 resampled gene signatures picked up from human genome with sizes identical to that of the 21-gene signature. The black triangle stands for the AUC value of our 21-gene signature. The right-tailed P value of the sampling distribution was calculated.
Validation cohort confirmed the predictive power of the 21-ROS-associated-gene signature
We utilized a second data set as validation cohort (Table 1), where a unique risk score based on the expression of the 21-gene signature was assigned to each patient. This signature performed well in predicting the patients' survival, as the 21 gene–based risk scores of nonsurvivors were significantly higher than those of survivors in the validation cohort (t test: P = 2.14 × 10−3; Fig. 5A). The AUC was 0.686 (Fig. 5B).
We also conducted a resampling test to confirm the predictive power of the 21-gene signature. We obtained 1,000 random gene signatures by randomly selecting 21 genes from the human genome and calculated the AUC for each random gene signature. It is of interest that our signature significantly outperformed randomly generated signatures composed of the same number of genes (right-tailed P = 0.046), highlighting the presence of a valid biological signal (Fig. 5C).
DISCUSSION
Sepsis syndromes remain a major cause of morbidity and mortality in ICUs. Oxidative stress plays an important role in the pathogenesis of sepsis and is associated with decreased survival,23,25,26 yet reliable ROS-associated biomarkers for predicting survival and assessing response to therapy in sepsis are lacking. Here we report the first ROS-associated, PBMC-derived novel gene expression signature for predicting survival in humans with sepsis. This 21-gene signature was identified from approximately 1,500 differentially expressed genes in PBMCs of sepsis nonsurvivors in a discovery cohort and 137 preselected ROS-associated genes. This signature was validated in an independent cohort to predict survival in sepsis patients.
The strengths of our analysis include (1) a robust methodology for identification of the differentially expressed genes in PBMCs that are associated with survival in patients with sepsis, (2) emphasis on ROS-associated genes, (3) use of survival as the outcome, and (4) confirmation of this signature as a significant predictor of survival in sepsis in an independent validation cohort. Here, we demonstrate that the 21-gene signature outperformed randomly selected genes from the human genome in both cohorts, suggesting that we are capturing a true effect.
We have identified some potential limitations of this study. First, we were not able to account for the timing of blood sample collection, particularly given that the sepsis syndrome evolves rapidly and that these cohorts were evaluated at different sites. It is possible that gene expression profiles may differ according to the time of sample collection. Future studies could be designed to standardize the timing of sample collection. We also noted that the mean age of nonsurvivors was higher than that of survivors, although these differences were not statistically significant. We recognize that elevated ROS levels are also associated with increased age; therefore, our ROS-associated gene signature might be capturing the effect of age on sepsis-related mortality. Future studies should focus on evaluating this gene signature in a larger cohort of sepsis patients while adjusting for age.
In the past 2 decades, our group and others have used microarray-based gene expression profiling to identify transcriptional signatures that predict clinical outcome or response to specific therapies, thus enabling the translation of gene expression research to bedside clinical practice.15,18–21 This approach has worked well for diseases that evolve slowly and where tissue samples are available for gene expression analyses, such as cancer or chronic inflammatory diseases such as inflammatory bowel disease. It is much more difficult in diseases that evolve rapidly, such as sepsis, resulting in organ failure and mortality, and without the ability to sample specific tissues for gene expression analysis. These studies demonstrate further proof of concept that PBMCs, which can be obtained immediately, can be used as a diagnostic or prognostic marker in severe acute diseases such as sepsis. It would be interesting to determine whether this gene signature predicts survival for other disease states with rapid onset and progression, such as acute respiratory distress syndrome. The use of genetic biomarkers from peripheral blood holds promise for improved personalized medicine in the critical-care setting.