
Abstract
Antidepressant drugs represent the principal form of treatment for major depressive disorder. While there are a plethora of medications available for this task, current drugs have many shortcomings. In the face of these deficiencies there is an ongoing search for new agents. The search has been guided, in part, by drug design based on existing agents and their putative mechanism of action. This has been less than fruitful in addressing inadequacies of existing medications as it has not produced compounds which are novel in terms of pharmacological mechanisms. Recent insights from molecular biological approaches hold promise for the discovery of novel compounds, in particular the so-called neurogenesis hypothesis suggests novel therapeutic approaches. Although significantly modified over the years, the monoamine hypothesis of depression and antidepressant drug action still remains an important driving force behind the development of new compounds. Several recently marketed agents and some in early-phase development tend to conform to these existing mechanistic hypotheses. Clearly the place of these agents in the treatment of depression is dependent on issues such as short- and long-term safety and efficacy. Duloxetine has been developed as a dual monoamine re-uptake inhibitor. Agomelatine is a compound with major effects on the circadian system as well as effects on subtypes of the serotonin receptor system. While the mechanism of action of this compound is not certain, recent evidence would suggest that the drug exerts its effects through antagonist actions at serotonin receptors. Compounds based on the hypothalamic pituitary adrenal axis, substance P antagonism and other neuropeptides have potential application for the treatment of depression but require further development before that potential is realized.
Keywords
Antidepressant medications remain at the forefront of the treatment of moderate to severe forms of major depressive disorder. Reputed generational changes in antidepressant drugs have taken place since the introduction of imipramine in 1957, including the launch of the selective serotonin re-uptake inhibitors and so-called dual acting agents. The shift to the new generation of antidepressants has not produced a quantum leap in depression treatment. Any unbiased assessment of the current status of antidepressants must admit that significant shortcomings in treatment remain [1]. Of particular concern has been the static improvement rates compared with older agents and more controversially a decrease in drug–placebo differences [2]. While newer medications may be better tolerated than the tricyclic antidepressant and monoamine oxidase inhibitors, leading to greater compliance, they nevertheless have different side effect profiles which in turn present unique long-term problems. To some extent these persistent shortcomings may be related to the prevailing paradigm concerning antidepressants' mechanisms of action, that is, the monoamine hypothesis has hindered the development of novel drug treatments. It might be expected that until there is a paradigm shift, antidepressant response rates will remain static and time to the onset of action slow. On the other hand, the inherent nature of depressive illness may of itself suggest that rapid therapeutic response will always be an unachievable goal. Increases in response rates may require a much greater degree of sophistication in the choice of drugs, perhaps based on pharmacogenetic principles [3]. Until there is a greater degree of understanding of the factors involved in the aetiology of depression, including the interaction between environment and genetics, it may be unrealistic to expect any significant change in response rates and onset of action. Despite this therapeutic nihilism, recent research on the mechanism of action of antidepressants has provided new insights with the potential for exploitation in the search for a new generation of antidepressant drugs [4].
The neurogenesis hypothesis
The traditional monoamine hypothesis has focused on the interaction between neurotransmitters and their cell surface receptors as critical for an understanding of antidepressant effects. By implication the changes induced by chronic drug administration are believed to reverse deficits present in depression, but evidence for transmitter and receptor changes in patients is equivocal at best [5]. On the other hand, magnetic resonance imaging studies have provided reasonably robust evidence for smaller hippocampal volumes in patients with depression compared with healthy controls [6]. Furthermore, hippocampal volume loss appears to be related to the length of time patients have spent depressed [7]. This loss may be caused by persistent elevations of glucocorticoids, particularly cortisol, in depressed patients as cortisol is known to be toxic to neurones, although the evidence for this is not particularly robust [6]. Hypercortisolaemia has long been recognized as a feature of at least some cases of depression, most prominently in patients with melancholic features. A hypothesis of neurodegeneration in depression has arisen with the notion that antidepressants may bring about their therapeutic effects by stimulating neurogenesis [4]. Recent studies in animal models have provided evidence for neurogenesis following chronic but not acute treatment with all classes of antidepressant treatments including ECT [8]. Neural cell proliferation and axonal growth have been shown to occur in the presence of antidepressants both in vitro and in vivo [9]. Neurogenesis in the dentate gyrus of the hippocampus has been demonstrated in animals following chronic antidepressant treatment [8]. These studies point to the importance of events beyond the receptor in mediating the therapeutic effects of antidepressants, rather than changes at the receptors themselves. Stimulation of receptors triggers an intracellular cascade of biochemical changes which increases the concentration of the protein CREB (cyclic AMP response element binding protein). In turn CREB binds to the gene for brain-derived neurotrophic factor (BDNF) a key protein for the proliferation and survival of neurones [10]. Several other mechanisms have also been identified including an increase in the anti-apoptotic protein Bcl-2 [5]. Together these biochemical changes produce conditions which are favourable to neurogenesis. As noted above, neurogenesis has been demonstrated in the hippocampus of animals treated with antidepressants. Furthermore, the effects of stress, which causes neuronal loss, can be prevented by pretreatment of animals with antidepressants [9]. Together these data suggest that any treatment which can increase neurotrophic factors (such as BDNF) or increase anti-apoptotic proteins may be a potential antidepressant therapy. Furthermore, neurotrophic factors themselves may offer a treatment of depression. Indeed it has been demonstrated in preclinical models that BDNF has antidepressant-like properties [11, 12]. Considerable practical difficulties would need to be overcome before a molecule such as BDNF could be used clinically (stability at gut pH, blood–brain barrier penetration). Nevertheless, an agonist of BDNF may reach clinical trials in the future. The prospect for the treatment of depression into the future is a generation of agents designed specifically to target appropriate intracellular messengers.
Monoamine-based treatments
Two newer agents developed on the basis of the monoamine hypothesis and marketed in some countries in recent times are duloxetine and agomelatine. Further compounds based on this approach or which combine agonism/antagonism at a particular receptor site with reuptake inhibition are in various stages of development [13].
Duloxetine
Duloxetine combines serotonin (5HT) and noradrenaline re-uptake inhibition with [14, 15] a low affinity for a range of serotonin, muscarinic, adrenergic and histaminergic receptors and does not significantly inhibit monoamine oxidase A or B activity [16]. Acute doses of duloxetine significantly enhance prefrontal cortical release of serotonin and noradrenaline in drug-naive rats [17]. On the other hand, in vitro studies in healthy volunteers suggest that at doses of 20 and 60 mg day−1 duloxetine was selective for serotonin re-uptake inhibition [18]. Nevertheless, serotonin and noradrenaline reuptake inhibition of duloxetine are likely to account for the antidepressant activity of the drug.
The efficacy of duloxetine in major depression was suggested in an open evaluation [19]. Patients received 20 mg of duloxetine once daily for 6 weeks and efficacy was evaluated with the HAM-D scale. Response based on a 50% or more reduction in the baseline depression score was achieved by 78.2% of subjects while remission (final HAMD score <6) was achieved by 60.3% of patients. The main side effects of the drug were nausea, insomnia, headache and diarrhoea. Subsequently a number of double-blind placebo-controlled clinical trials confirmed efficacy of duloxetine in the treatment of depression. In these studies duloxetine had superior efficacy to placebo in trials of 8–9 weeks' duration [20–25]. The drug was at least as effective as fluoxetine [20] and paroxetine [23, 24]. In these studies duloxetine appeared to be similar to other antidepressants in terms of the number of patients responsive to the medication. There was no indication that the rate of onset of action of duloxetine was faster than that of other agents. In a 52-week open evaluation duloxetine was effective in maintaining response in patients with DSM-IV major depressive disorder [26]. The effective dose of the drug was 40–60 mg twice daily. The drug appeared to be safe and well tolerated with no unexpected major adverse events. A summary of relevant trials is presented in Table 1.
Double-blind trials assessing the efficacy of duloxetine in major depression
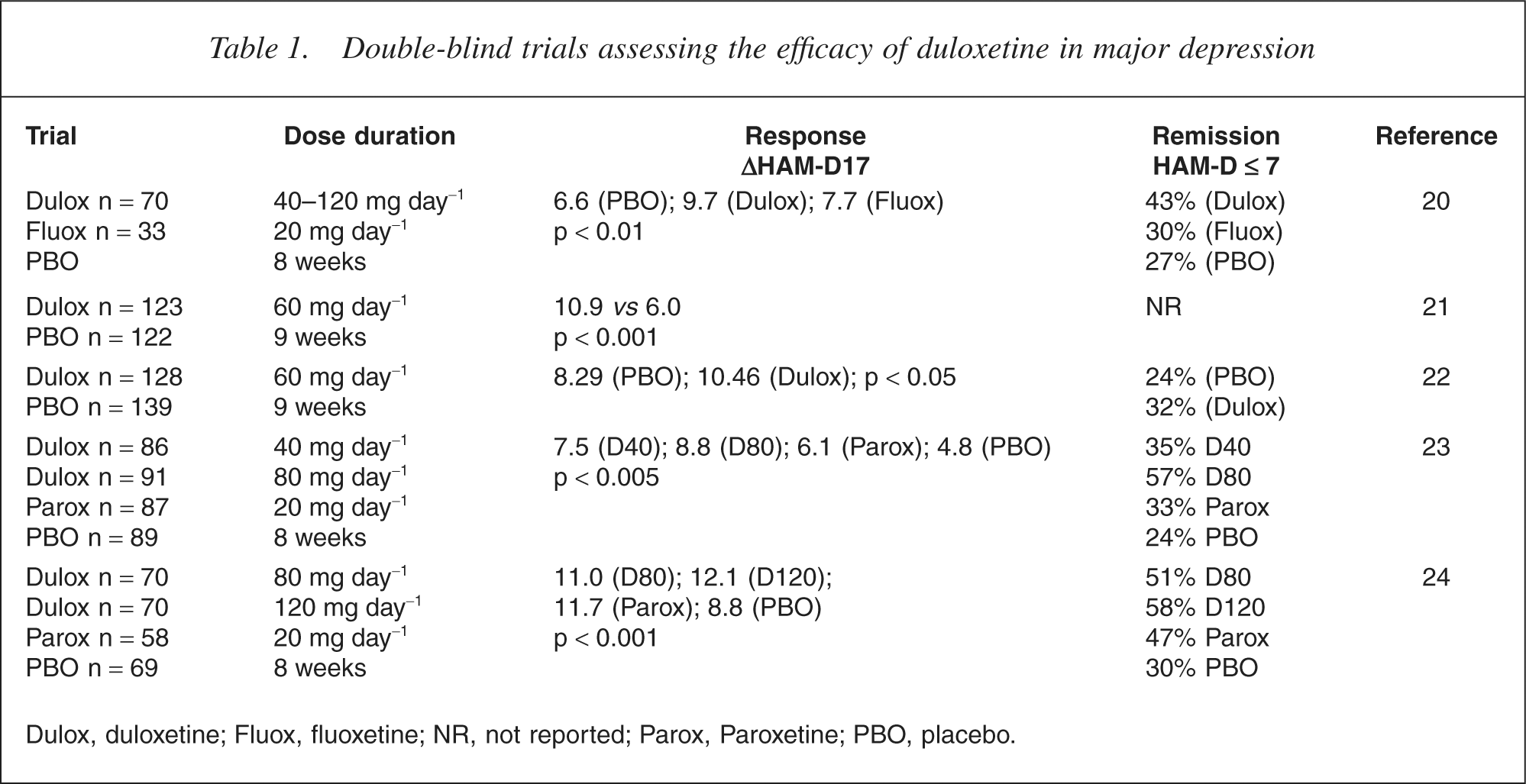
Dulox, duloxetine; Fluox, fluoxetine; NR, not reported; Parox, Paroxetine; PBO, placebo.
The safety and tolerability of duloxetine was available from a pooled analysis of eight placebo-controlled comparisons [27]. The data were from 1139 patients treated with duloxetine (40–120 mg day−1) for up to 34 weeks. Nausea, dry mouth, constipation, insomnia, dizziness and fatigue were the main side effects reported significantly more often than with placebo. Cardiovascular assessments showed small increases in heart rate, systolic, diastolic blood pressure compared with placebo, but were not considered to be clinically significant. The mean change in the QRS width of the ECG was also not regarded as clinically significant. The incidence of abnormal clinical laboratory parameters was similar to that of placebo-treated patients. There were some increases in liver enzymes values in patients treated with duloxetine but these were not likely to be clinically significant. There did not appear to be any withdrawal reaction following abrupt discontinuation of the drug but nevertheless a tapered drug withdrawal was recommended. Three deaths occurred in duloxetine-treated patients: one due to suicide, one due to cardiorespiratory arrest and the other resulted from non-cardiogenic pulmonary oedema.
Agomelatine
Agomelatine exhibits potent agonist effects at melatonin (MT1 and MT2) receptor sites with an affinity comparable to that of melatonin [28–30]. In an animal model of delayed phase sleep syndrome, agomelatine resynchronized circadian rhythms [31, 32]. Agomelatine possesses antagonist properties at serotonin 5HT2C receptors both in vivo and in vitro [33, 34]. At cloned human 5HT2C receptors agomelatine is an antagonist [34]. Consequently, agomelatine dose dependently increased noradrenaline and dopamine concentrations in the frontal cortex of rats [34], an effect not blocked by the selective melatonin antagonist S22153 [34]. In preclinical studies agomelatine is active in tests indicative of antidepressant activity: the forced swim test [35]; the chronic mild stress model [36]. These effects were attributed to 5HT2C antagonism as melatonin was inactive in the same tests. Agomelatine demonstrated anxiolytic activity in the Vogel conflict and social interaction tests [37]. Agomelatine may represent a novel approach to the treatment of depression, via both its chronobiotic activity and 5-HT2C antagonism.
Clinical trials have confirmed an antidepressant effect of agomelatine in humans [38–40]. In a double-blind randomized study agomelatine 5 and 100 mg day−1 were compared in patients with major depressive disorder for 4–8 weeks [38]. Total MADRS scores decreased from a mean of 30.7 at baseline to a mean of 14.8 in the 5 mg group and from 31.6 to 18.6 in the 100 mg group. There was no statistically significant difference between the groups for efficacy or safety, although the 5 mg dose was slightly more efficacious and better tolerated. A randomized, double-blind placebo-controlled trial compared fixed doses of agomelatine (1 mg, 5 mg or 25 mg in the evening) with paroxetine (20 mg) and placebo in 711 patients over an 8-week period [39]. Analysis of the HAM-D scores (LOCF dataset) showed a statistically significant difference between the three agomelatine doses and placebo (p < 0.05). Subsequent post hoc analysis showed that only the 25 mg dose was superior to placebo. Paroxetine was also significantly different from placebo at week 8. There were no discontinuation symptoms in patients abruptly ceasing 12 weeks' agomelatine treatment with 25 mg day−1 [40]. Further studies are necessary to assess the role of agomelatine in the treatment of depression.
Neuropeptide modulators
Hypothalamic pituitary adrenal (HPA) axis
The involvement of the HPA axis in depression has long been known [41]. Patients with major depression have elevated plasma cortisol [42, 43]. With recovery from depression there is normalization of hypercortisolaemia [43]. Circadian rhythm disturbances of cortisol as well as elevated concentrations of corticotropin releasing factor (CRF) have also been reported [44, 45]. Both hypersecretion of CRF and hypercortisolaemia are thought to arise from impaired feedback inhibition by endogenous corticosteroids. The failure of the synthetic glucocorticoid, dexamethasone, to suppress cortisol secretion has been taken as an indication of abnormal glucocorticoid receptors [46]. Secretion from the hypothalamus of CRF controls the release of ACTH which in turn stimulates the release of cortisol from the adrenal glands [43]. The treatment of depressive illness by the modulation of the activity of the HPA axis and/or CRF has therefore provided a target for the development of several potential therapeutic agents.
Raised concentrations of cortisol can be diminished by the administration of steroid synthesis inhibitors such as ketoconazole, metyrapone or aminogluthemide. These have been used as treatments for depression, but results from double-blind placebo-controlled studies have been inconsistent [47–49]. This strategy is not likely to be useful in all patients as elevated plasma cortisol is not a feature of all patients. For example, two double-blind placebo-controlled evaluations of ketoconazole in major depression showed a significant antidepressant effect of ketoconazole over placebo in one, but only in those patients with hypercortisolaemia at baseline [49], but not in another [50].
Initial clinical studies with the glucocorticoid receptor antagonist mifepristone have shown efficacy in psychotic depressive episodes [51, 52]. Further evaluations of this compound are necessary to evaluate its place in therapy.
The CRF family of neuropeptides (CRF, urocortin, urocortin II and urocortin III) bind with different affinities to the two major CRF G-protein coupled receptor subtypes. Several modulators of CRF receptor function have shown potential antidepressant and anxiolytic properties in animal models [53]. The CRF1 antagonist, DMP904 was shown to be anxiolytic in the elevated plus maze and defensive withdrawal paradigm in rats [54]. The compound blocked the stress-induced rise in plasma corticosterone. A further CRF1 antagonist, CP-154,526 demonstrated anxiolytic and antidepressant-like effects in some preclinical models [55, 56]. ORG 34517 showed significant antidepressant effects in a 4-week doubleblind study [57]. R121919, another CRF antagonist was investigated in 24 patients with major depression who were assigned to treatment with two different dose escalating regimens: 5–40 mg day−1 and 40–80 mg day−1 [58]. Ten patients completed the 30-day treatment period in each group. Statistically significant declines in HAMD scores from baseline were noted in both groups with a greater effect observed in the patients treated with the highest dose. This compound does not appear to have been developed any further. Clinical investigations for other drugs in this class are underway but none of the molecules has been marketed.
Neurokinin-1 (substance P) antagonists
Substance P belongs to the family of tachykinin neuropeptides which share a common C-terminal sequence. Biological effects are mediated through three G-protein coupled receptors designated NK1, NK2 and NK3 [59]. Substance P and its receptors are highly expressed in brain regions critical for the regulation of emotion and stress [60]. Antagonist effects are species dependent: NK1 antagonists at guinea pig and gerbil receptors also bind with similar affinity to the human receptor [59]. Clinical trials with aprepitant (MK869) showed it to be more effective than placebo and at least as effective as the SSRI paroxetine [61]. In a dose-finding study aprepitant (10, 30, 100 or 300 mg day−1) was as effective as fluoxetine 20 mg day−1 or placebo [62]. In this doubleblind study the HAM-D scale was used to evaluate the efficacy of the drug at 6 weeks. Improvements in depression ratings were similar in all arms of the study. A variant of the compound with higher oral bioavailability and greater brain penetration did not show separation from placebo [62]. Further development was terminated leaving the role of NK1 antagonists as antidepressants in limbo. Further compounds have been developed and the results of early phase clinical studies are awaited with interest.
Vasopressin
Gold et al. noted the similarities between the symptoms of depression and the effects of arginine vasopressin (AVP) and suggested a role in psychiatric disorders [63]. Subsequent investigations noted correlations between plasma and CSF concentrations of AVP and depressive symptomatology [64, 65], with AVP concentrations lowered by antidepressant treatment [66]. Advances in understanding the actions of AVP and its receptor subtypes-specific agents with putative antidepressant properties have been developed. Three subtypes of AVP receptor have been characterized in the central nervous system [67]. The V1 and V3 receptor in particular have been the targets for the development of potential antidepressants. The V1-antagonist d(CH2)5 Tyr(Me)AVP shows activity in the rat forced swim test when injected into the septum or amygdala [68, 69]. The V3 receptor antagonist SSR149415 is active in blocking stress and CRF induced increases in plasma ACTH, suggesting possible antidepressant activity [70]. Furthermore, V3 receptor knock-out mice exhibit marked reductions in AVP and stress induced ACTH release [71]. As overactivity of the HPA axis is a target of putative antidepressant drug development (vide supra) V3 antagonists may be antidepressant via an indirect effect on cortisol hypersecretion. In the chronic mild stress paradigm and the forced swim test SSR149415 produces dose-dependent antidepressant-like effects [72, 73]. Activity in the forced swim test was present in hypophysectomized animals, suggesting that the antidepressantlike effect is independent of the influence over the HPA axis [73]. The activity of V1 and V3 receptor antagonists in clinical trials is yet to be assessed.
Other compounds
Antidepressant activity in compounds not affecting traditional monoamine targets has been suggested from preclinical studies. Antidepressant agents, from different classes modulate the activity of the NMDA receptor [74, 75]. Thus NMDA receptor antagonists demonstrate antidepressant-like activity in preclinical models: 2-amino-7-phosphoheptanoic acid (AP-7), 1-aminocyclopropancarboxylic acid and memantine have been shown to be active [76]. Similarly compounds affecting the AMPA receptor such as LY392098 and LY451646 have also shown antidepressant effects in animal model studies [77]. The activity of these compounds awaits demonstration in methodologically sound clinical trials.
Modulation of neuropeptide Y (NPY) systems displays antidepressant-like activity in preclinical models [78]. Alterations in NPY activity, gene expression and function have been demonstrated in several putative animal models of depression and by chronic treatment with antidepressant drugs [79]. Available data suggest the NPY Y1 receptor subtype is the most likely target for the development of treatment strategies. Antidepressant-like activity for NPY has been demonstrated in the forced swim test [80]. Clearly the development of specific compounds affecting NPY receptors is necessary to evaluate the potential of this group of receptors to provide clinical treatments for depression.
The effects of melanin concentrating hormone (MCH), a component of the regulation of energy balance and body weight [81], are mediated through two subtype Gprotein coupled receptors in humans [82]. The distribution of the MCH-R1 receptor in the central nervous system is suggestive of a role mood and stress-related disorders [83]. In the rat forced swim test and the guinea pig ultrasonic vocalization test SNAP-7491, a MCH-R1 antagonist showed a dose-dependent, antidepressant-like effect [83]. Clinical testing of the compound does not appear to have taken place but the data suggest a potential therapeutic approach to treatment of depression.
Conclusions
It is unlikely that most of the compounds presented in this brief overview will reach clinical practice. Many compounds with promising preclinical profiles fail in the clinic because of toxicity or efficacy concerns. Other promising molecules are abandoned for reasons of formulation and bioavailability issues. Recent approaches to the treatment of depression have moved beyond the monoamine hypothesis to be based on a diversity of findings from preclinical and clinical studies. Nevertheless the monoamine hypothesis continues to drive drug discovery for the treatment of depression as the recent introduction of some compounds (e.g. duloxetine) attests. Other molecules based on neuropeptides are at once both novel (e.g. MCH antagonists) and old (e.g. modulation of the HPA axis). From preclinical studies of the effects of antidepressants has come the notion that modulation along crucial signal transduction pathways may provide a new generation of drugs for the treatment of depression. At present such approaches are necessarily theoretical but in the future molecules based on the hypothesis may resolve some of the current clinical shortcomings of the present generation of antidepressants.
Disclaimer: The author has been a member of advisory boards for Bristol Myers Squibb, Eli-Lilly, Lundbeck and Organon. He has received funding for research, lectures or attendance at conferences from Bristol Myers Squibb, Eli-Lilly, Lundbeck, Pfizer, Glaxo Smith Kline and Organon.