
Abstract
The high incidence and serious nature of schizophrenia has led to extensive efforts to discover macro-or microscopic lesions in the brain that could cause the illness. Failure to identify such lesions [1] has lent support to a number of hypotheses that propose changes in the molecular architecture of the brain as the basis of schizophrenia. Both neuroimaging techniques and brain tissue obtained post-mortem have been used to gain direct evidence to support such hypotheses. It is now pertinent to review the outcomes of key studies using these two techniques and how they are influencing current thinking on the neuro-biology of schizophrenia. This review will not address the basic biochemistry of neurochemical systems which are extensively reviewed elsewhere [2].
The dopamine hypothesis of schizophrenia
The dopamine (DA) hypothesis of schizophrenia is a long-standing hypothesis that proposes that over-active dopaminergic neurones cause the psychoses associated with schizophrenia [3]. Support for the DA hypothesis comes from the effects of drugs that target the dopaminergic systems of the brain [4]. In particular, it has been observed that drugs which activate dopaminergic neurones may induce or exacerbate psychoses while those that reduce the activity of these neurones can lessen psychoses. Since the formulation of the DA hypothesis of schizophrenia there has been a concerted effort to obtain direct experimental evidence to prove its validity.
Initial attempts to confirm a role for DA in the pathology of schizophrenia were encouraging, with increased levels of DA being reported in the nucleus accumbens [5] and amygdala [6] from subjects with schizophrenia. Subsequently, findings on DA [7] and other neurotransmitters, as well as enzymes involved in the synthesis and degradation of neurotransmitters, have been inconsistent. It is now widely accepted that neurotransmitters and enzyme activity are not stable in the brain post-mortem [8]. Hence, it is likely the apparent variability in post-mortem studies measuring these variables could have been due to variability in tissue collection. This makes it difficult to interpret the early results on DA levels in the brains of subjects with schizophrenia.
The inconsistencies in results from measuring neurotransmitters and/or enzyme activity have increasingly led to the investigation of more stable components of the dopaminergic system, in particular the levels of DA receptors in the brain of subjects with schizophrenia. Initially it was reported that DA2 receptor was increased in the caudate nucleus from subjects with schizophrenia [9]. These results led to the proposal that there was an overabundance of DA2 receptors in the brain of subjects with schizophrenia and, in the presence of normal or increased levels of DA, resulted in a super-sensitive state and over-activation of DA responsive neurones. However, it has been reported that DA2 receptors are not altered in schizophrenic subjects never treated with antipsychotic drugs [10]. This suggested that the increase in density of DA2 receptors in tissue from subjects with schizophrenia was due to antipsychotic drug treatment during life.
Positron emission tomography (PET) has also been used to measure DA2 receptors in the human brain. As with post-mortem studies, some studies using PET have reported an increase in the density of DA2 receptors in the caudate nucleus of subjects with schizophrenia, whether or not they were naive to anti-psychotic drugs [9]. These data appeared to support the argument that an increase in DA2 receptors in the caudate-putamen is involved in the pathology of schizophrenia. However, later PET studies could not confirm an increase in DA2 receptors in drug naive subjects with schizophrenia. Hence, the use of PET has not fully resolved whether there is an increase in DA2 receptors in the brain of subjects with schizophrenia [9].
Early studies on DA receptors based their conclusions on the hypothesis that there were two DA receptors [11]. One of these receptors, the DA2 receptor, was partly defined as the site that bound antipsychotic drugs with high affinity. Cloning studies have now shown that the pharmacologically defined DA2 receptor encompasses a family of receptors, now termed the DA2-like receptors [12]. This family of receptors is made up of the D2, D3 and D4 receptors, each being a separate gene product. Moreover, the D2 receptor undergoes post-transcriptional modification producing a long (D2L) and short (D2S) form of the receptor. Pharmacological agents that are totally specific to a single DA receptor have yet to be developed. Thus, the study of each individual DA receptor in schizophrenia has involved measuring levels of messenger RNA (mRNA) encoding each receptor in brain tissue obtained postmortem. One such study reported increased levels of mRNA for the D2 receptor in the ventral orbital gyrus, inferior orbital gyrus and caudate nucleus from subjects with schizophrenia [13]. This would, at least, suggest that any increase in DA2 receptor protein in the brain from subjects with schizophrenia has resulted from an increase in the rate of expression of that receptor. The development of radioactive ligands and/or effective receptor antibodies that are selective for the D2 receptor will be required to pursue this hypothesis.
Using an indirect pharmacological subtraction technique, a number of studies have reported a relatively high concentration of the D4 receptor in the human caudate-putamen and an increase of that receptor in that brain region in schizophrenia [9]. This was taken as being highly significant as it had been suggested that clozapine binds with high affinity to the D4 receptor [14] and may therefore have some of its unique therapeutic effects [15] by selectively blocking that receptor. However, concerns about the validity of these results have arisen because there appears to be no mRNA for the D4 receptor in the caudate-putamen [16] making it unlikely this region would contain high levels of D4 receptor, as suggested by the subtraction technique. In addition, more direct pharmacological approaches to measuring the D4 receptor failed to show differences in schizophrenia [9] and methodological problems associated with the use of the subtraction technique suggest that method would not accurately measure the density of D4 receptors in schizophrenia [17]. Significantly, mRNA encoding the D4 receptor is present in the frontal cortex and has been reported as decreased [16,18] or unchanged [19] in schizophrenia. Hence, current data on mRNA for the D4 receptor favour a change in that receptor in the frontal cortex in schizophrenia.
Decreases in mRNA for the D3 receptor have been reported in Brodmann's areas 1 through 5 [20] and 11 [16] of the frontal cortex, but not in the striatum or nucleus accumbens [16], from subjects with schizophrenia. This is a particularly intriguing finding as there is growing evidence that suggests the D3 receptor is the presynaptic autoreceptor in certain areas of the brain [21,22]. The role of the DA autoreceptor is to regulate DA synthesis and release [23]. Hence, alterations in the D3 autoreceptor in the brain of subjects with schizophrenia is likely to result in an aberrant control of DA release. Significantly, two functional neuroimaging studies have reported an increase in the release of DA following a standardised amphetamine challenge [24,25]. These data further support the hypothesis that there is an increase in DA release in subjects with schizophrenia which results in an increase in levels of extra-neuronal DA, a finding consistent with some early studies using tissue obtained post-mortem [5,6].
While the DA hypothesis of schizophrenia still requires total validation, studies using brain tissue obtained at autopsy and PET suggest there are changes in the functioning of the dopaminergic systems in subjects with schizophrenia. Most intriguingly, recent data could indicate that the major problem in subjects with schizophrenia is presynaptic and involves the control of DA release. If proven, this would represent a major shift in the focus of research into abnormalities in dopaminergic function in schizophrenia.
The dopamine/serotonin hypothesis of schizophrenia
Changes in the serotonergic systems of the brain in schizophrenia are also supported by neuropharmacological observations [5]. At the centre of this hypothesis is the tenet that certain indoles, such as the serotonin (5HT) receptor agonist lysergic acid (LSD), induce or exacerbate hallucinations. In addition, it is increasingly accepted that antipsychotic drugs which antagonise both DA and 5HT receptors have a therapeutic advantage over drugs that more selectively target DA receptors [26]. These data suggest it is over-active serotonergic neurones that are involved in schizophrenia.
In addition to neuropharmacological evidence, there has been a significant amount of data obtained from studies using tissue obtained post-mortem to support a role for changes in the serotonergic system in the pathology of schizophrenia. An early study reported a decrease in [3H]-LSD binding to 5HT receptors in the frontal cortex from subjects with schizophrenia [27]. However, in a follow-up study, [3H]-LSD binding was reported to be increased in tissue from subjects who had not received anti-psychotic drugs close to death [28]. These authors suggested the decrease in 5HT receptors in the earlier study was an effect of antipsychotic drug treatment prior to death.
[3H]-LSD binds to a number of 5HT receptors. Thus, in an attempt to better understand the nature of the changes in 5HT receptors in schizophrenia, more recent studies used radioactive ligands that were more selective for specific 5HT receptors. Several of these studies have now reported a decrease in 5HT2A/2C receptors in the frontal cortex of subjects with schizophrenia [29–32]. Moreover, the changes in the density of 5HT2A/2C receptors were independent of the antipsychotic drug received by the schizophrenic subjects close to death [30,31] and may be associated with decreases in levels of mRNA encoding for the 5HT2A receptor [32]. It is now widely accepted that the 5HT2A/2C receptor is decreased in regions of the cortex from subjects with schizophrenia, but it is remains to be established whether this is due to a pathological process.
Positron emission tomography has also been used to examine the density of 5HT2 receptors in the cortex. These studies did not show a difference in the density of cortical 5HT2 receptors in schizophrenia [33,34], results which differed from the majority of those obtained using post-mortem tissue. It is important to note that both PET studies used [18F]-setoperone as the radioligand to measure the 5HT2 receptors. Given that the use of [3H]-LSD in postmortem tissue led to an inconsistency of results, it could be that the use of a different radioligand in PET and post-mortem studies has led to inconsistencies in results on 5HT2 receptors in schizophrenia.
An increase in the density of the 5HT1A receptor has been reported in the prefrontal cortex [32,35–37], orbital frontal cortex [37,38], temporal cortex [34], posterior cingulate, motor cortex and hippocampus [39] from subjects with schizophrenia. This increase in 5HT1A receptor density does not appear to be associated with a change in mRNA encoding for the receptor [32]. Significantly, the 5HT2A/2C and the 5HT1A receptor are differentially distributed in the frontal cortex [31,40] (Fig. 1). This has led to the suggestion that the changes in these two receptors in the frontal cortex were ‘normalising’ some abnormal drive in the cortex of subjects with schizophrenia [41]. It would seem most likely that such an abnormal drive would involve changes in innervating serotonergic neurones.
The distribution of serotonin2A/2C (A) and serotonin1A (B) receptors in Brodmann's area 9 of the frontal cortex. In these images, the density of radioligand binding and hence receptor is proportional to the ‘greyness’ of the image: the darker the image, the more receptor.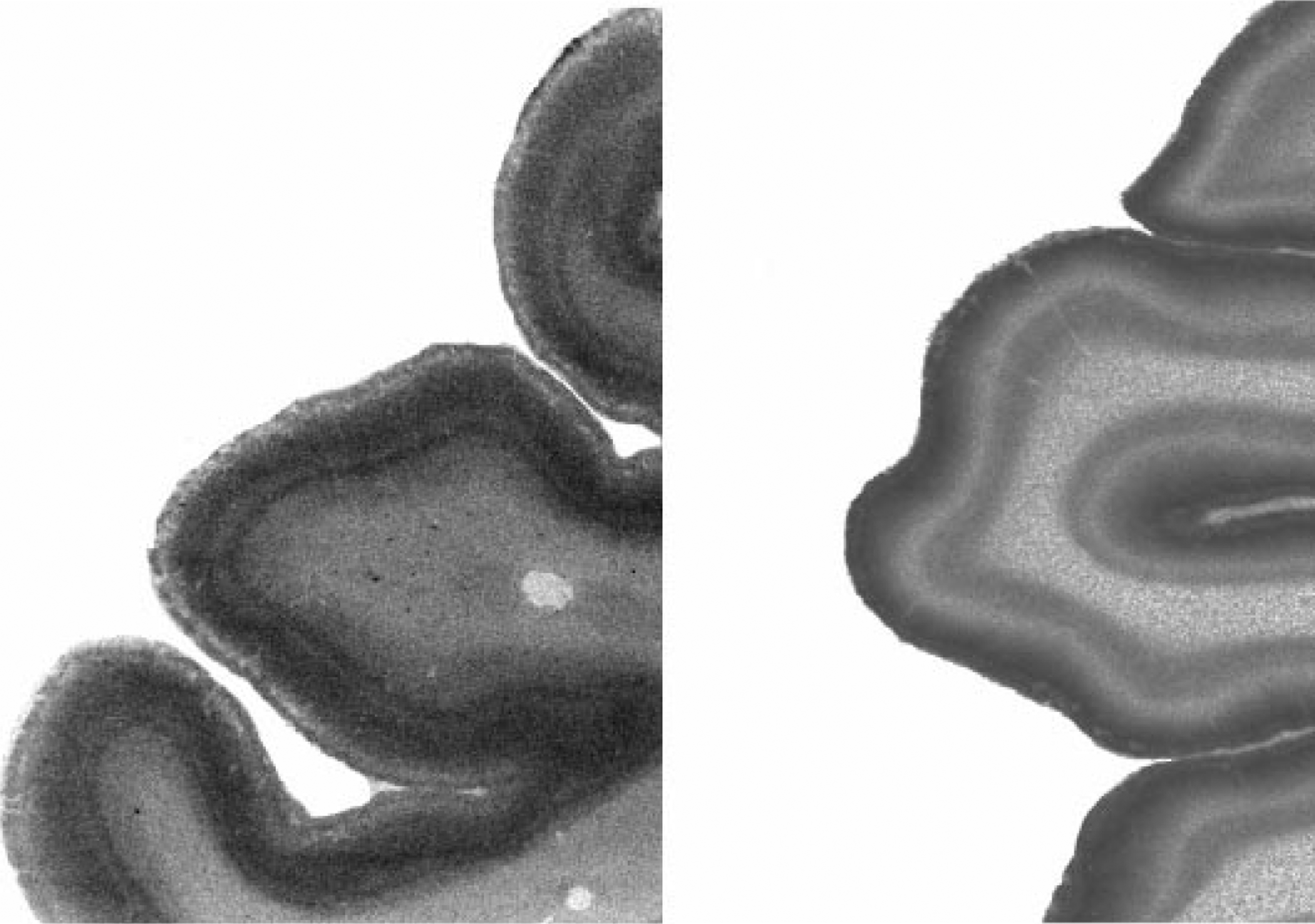
There have been two reports that have suggested there is a decrease in the density of the serotonin transporter in the prefrontal cortex of subjects with schizophrenia [36,39]. Such differences would be expected to change 5HT levels in that brain region. Moreover, this change in levels of 5HT could be the cause of the changes in 5HT receptors reported in a number of studies. It has also been suggested that there is a conformational change in the 5HT transporter in the hippocampus from subjects with schizophrenia [42,43]. This conformational change is not associated with any particular polymorphism in the 5HT transporter gene promoter region [44], but manifests as a change in the affinity of [3H]-paroxetine binding [43]. Thus, changes in the 5HT transporter in schizophrenia could also be important in the hippocampus as well as in the frontal cortex. Moreover, in both brain regions, it could be by antagonising 5HT receptors that atypical drugs counteract the ‘flow-on’ effects of changes in the serotonin transporter and hence give improved clinical outcomes [26].
The glutamate hypothesis of schizophrenia
The glutamate hypothesis of schizophrenia is mainly based on the observation that drugs such as phencyclidine and SKF 10,047, which block the ion channel of the N-methyl-D-aspartate (NMDA) receptor, precipitate psychoses [45]. Moreover, it has been proposed that these NMDA receptor ion channel blockers are the only pharmacological agents that cause the deficit symptoms associated with schizophrenia [46]. These observations have led to the proposal that deficiencies in glutamate systems provide the best model of the symptoms of schizophrenia.
Studies using tissue obtained post-mortem have subsequently examined the density of the binding site for phencyclidine on the NMDA receptor. One of these studies reported an increase in the binding of N-(1-[2-thienyl] cyclohexyl) 3,4-[3H] piperidine ([3H]-TCP), a phencyclidine derivative, to orbital frontal cortex from subjects with schizophrenia [47]. [3H]-TCP binding has also been reported to be decreased in the Cornu Ammonis (CA3) region of the hippocampus from subjects with schizophrenia [48]. Other radioactive drugs that bind to the NMDA receptor have been used to study these receptors in brain tissue obtained post-mortem. These studies have shown an increase in the density of the NMDA receptors in the putamen [49,50], caudate nucleus [51] and superior temporal cortex [52] from subjects with schizophrenia. Hence, many studies on the NMDA receptor suggest this receptor is changed in the brain of subjects with schizophrenia, but that these changes are not consistent throughout the brain.
Glutamate has also been shown to have effects through a number of non-NMDA receptors including kainate and amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors [53]. The density of kainate receptors has been reported to be increased in the frontal [54] and orbital [55] cortex, as well as in the striatum [56] from subjects with schizophrenia. By contrast, the level of kainate receptors has been reported to be decreased in the CA4/CA3 regions of the hippocampus from subjects with schizophrenia [57]. These data, added to those on the NMDA receptor, suggest that changes in glutamate function in the hippocampus from subjects with schizophrenia differ to those in other brain regions. Finally, it has been shown that there are no significant changes in AMPA receptors in the cortex of subjects with schizophrenia [58].
The NMDA, kainate and AMPA receptors have been shown to be made up of subunits, all of which are separate gene products [59]. Using data from cloning studies it has been possible to construct probes to measure levels of mRNA for the different subunits of the glutamate receptors in tissue from subjects with schizophrenia. In addition, by translating the genetic codes for the subunits into amino acid sequences it has been possible to produce subunit specific antibodies that have been used to measure levels and distribution of receptor subunits in human brain tissue. Studies on levels of mRNA showed a reduction in the expression of subunits of the NMDA receptor in the superior temporal cortex [60] as well as frontal cortex [61] of ‘neuroleptic-free’ subjects with schizophrenia. By contrast, it has been reported that only levels of mRNA for the NR2 subunit family of the NMDA receptors is increased in the frontal cortex of subjects with schizophrenia [62].
Messenger RNA for the non-NMDA kainate/AMPA receptor was initially reported to be reduced in the CA3 region of the hippocampus from subjects with schizophrenia [63]. More recently, reduced expression of a subunit of the kainate receptor (KA2) has been measured in the dentate gyrus, CA1 and CA2 regions of the hippocampus from subjects with schizophrenia [64]. In addition, in contrast to the lack of change observed in radioligand studies, low levels of mRNA for the two spliced variants of the GluR2 subunit of the AMPA receptor were observed in the hippocampal formation from subjects with schizophrenia [65]. These data were supported by a immuno-autoradiographic study which showed a decrease in GluR1 subunit in parahippocampal gyrus and a decrease in GluR2/3 subunits in the CA4 region of the hippocampus from subjects with schizophrenia [66].
Data from studies using brain tissue obtained postmortem seem to indicate that there is disruption to the cortical and hippocampal glutamatergic systems in subjects with schizophrenia. This hypothesis has been tested in a nuclear magnetic resonance (NMR) study which reported an increase in glutamine levels without a change in glutamate levels in the frontal cortex from subjects with schizophrenia [67]. This result was taken as revealing an abnormality in the conversion of glutamine to glutamate by glutamatergic neurones. This raises the possibility that, as for dopamine and serotonin, aberrant control of the levels of available neurotransmitter by presynaptic neurones maybe involved in the pathology of schizophrenia. In addition, a PET study has shown that ketamine, a non-competitive NMDA receptor antagonist, increases cerebral blood flow in the anterior cingulate cortex in subjects with schizophrenia but reduces blood flow in the hippocampus and primary visual cortex [68]. This is further evidence that the changes in glutamate function in the hippocampus differ from that in certain cortical regions of subjects with schizophrenia.
Finally, two studies have reported that glutamate uptake sites are reduced in the striatum from subjects with schizophrenia [69,70]. Intuitively, a reduction of glutamate uptake sites would most likely to be associated with a reduction in glutamate uptake leading to increased levels of glutamate and hence increased glutamate activity. However, the reduction in uptake sites could also be an appropriate physiological response in an attempt to rectify the decrease in glutamate synthesis which has been suggested by the NMR finding on cortical glutamate in schizophrenia.
The γ-aminobutyric acid/dopamine hypothesis of schizophrenia
Unlike other hypotheses of schizophrenia, the γ-aminobutyric acid (GABA) hypothesis is not primarily based on drug effects in humans, but on data from studies using animals. Such studies suggest that if the GABA system, which has an inhibitory drive on dopaminergic systems of the brain, is under-active, this would result in an over-active dopaminergic system and hence induce psychoses [71].
Studies using tissue obtained post-mortem have attempted to confirm a role for a decrease in GABA functioning in the pathology of schizophrenia. Initially, the finding that GABA was decreased in the nucleus accumbens and thalamus of subjects with schizophrenia [72] seemed to lend support for a decreased GABA inhibitory drive onto the dopaminergic system in schizophrenia. However, in a similar paradigm to that which occurred with findings on DA, the inability to replicate this result led to the study of GABA receptors in the brain of subjects with schizophrenia. The most abundant GABA receptor in the brain is the GABAA receptor and it has been shown that levels of GABAA receptors are inversely related to levels of GABA [73]. Significantly, increases in the density of the GABAA receptor, as measured by [3H]-muscimol binding, have been reported in the superficial layers of the cingulate cortex [74] as well as in the prefrontal cortex [75,76], the dentata, CA4, CA3 of the hippocampal formation, the subiculum and presubiculum [77] from subjects with schizophrenia. Importantly, the increase in the density of the GABAA receptor is thought to be a response to a decrease in activation by GABA rather than a primary deficit relating to the pathology of schizophrenia.
The GABAA receptor contains a number of binding sites for various centrally active compounds. [3H]-muscimol is a GABAA receptor agonist that binds to the GABA binding site of the receptor. Another binding site on the GABAA receptor is the benzodiazepine binding site, which is where benzodiazepines exert their anxiolytic effects. The presence of this site on the GABAA receptor means that radioactive benzo-diazepines, like [3H]-muscimol, can be used to measure the density of GABAA receptors in the brain. Such studies have reported an increase in benzo-diazepine binding in the superior temporal gyrus, but not medial frontal cortex, orbitofrontal cortex, medial and inferior temporal gyri, CA1–3 or putamen from subjects with schizophrenia [78]. By contrast, other studies have reported that benzodiazepine binding is not changed in the hippocampal formation [79] or frontal cortex [80] from subjects with schizophrenia. Finally, one study has reported a decrease in benzo-diazepine binding in the anterior cingulate cortex, hippocampus, somatomotor cortex, cerebellar cortex and globus pallidus from subjects with schizophrenia [81]. These studies, plus the finding that mRNA for the subunits of the GABAA receptor is not altered in prefrontal cortex from subjects [82,83], make the case for an increase in GABAA receptors in the brain of subjects with schizophrenia less compelling.
Benzodiazepine binding in schizophrenia has also been studied using single photon emission computed tomography (SPECT). These studies were unable to find changes in benzodiazepine binding in subjects with schizophrenia [84–86] and therefore do not support a role for changes in GABAA receptors in schizophrenia. A study on the binding of [3H]-muscimol and radioactive benzodiazepine in tissue from the same individuals would seem essential to help reconcile differences in existing data on GABAA receptors in schizophrenia.
As with other neurotransmitter systems, the uptake system for GABA has been studied in schizophrenia. These studies have reported a decrease in GABA uptake sites in the hippocampus [87,88], striatum [69] and frontal cortex [89] of subjects with schizophrenia. Finally, a decrease in glutamic acid decarboxylase (GAD) has been reported in the frontal cortex of subjects with schizophrenia [90]. Glutamic acid decarboxylase is the rate-limiting enzyme in the synthesis of GABA and is present in GABA-producing neurones. These data could indicate either a decrease in GABA-producing neurones in subjects with schizophrenia, or a decrease in GABA uptake sites on an unchanged number of neurones. This is an important issue that needs to be resolved.
The cholinergic hypothesis of schizophrenia
The cholinergic hypothesis of schizophrenia has been developed from many lines of evidence [91]. These include evidence that the modulation of the cholinergic system can affect both positive and negative symptoms of schizophrenia [91]. In addition, it is becoming apparent that the cholinergic system is central to processing of sensory stimuli and in ensuring that the higher functions of the brain operate optimally [92]. A derangement of these functions could well result in the symptoms of schizophrenia. In addition, atypical antipsychotic drugs such as clozapine and olanzapine, that appear to have improved therapeutic outcomes [15,93], bind with high affinity to muscarinic receptors [94]. Finally, anticholinergic agents have proven useful in reducing extrapyramidal side-effects of typical antipsychotic drugs.
Direct evidence to support a role for changes in the muscarinic system in schizophrenia came from the study of a critical enzyme in the synthesis of acetylcholine. The first evidence to suggest that the cholinergic system was altered in the brains of subjects with schizophrenia was the demonstration that the activity of choline acetyltransferase (ChAT) was increased in the hippocampus, striatum and nucleus accumbens from subjects with schizophrenia [95]. Differences in the activity of ChAT were not detected in any of the other 47 regions studied. These findings were challenged by another study that showed that ChAT was decreased in the septal area and caudate head from subjects with schizophrenia [96]. ChAT was not altered in any of the other 13 regions studied, including the pons. Finally, a study looking at levels of protein rather than enzyme activity showed that levels of ChAT was reduced in the pons of subjects with schizophrenia [97]. As with other neurotransmitter-related enzymes, the differences between these studies could be due to problems in the stability of enzymatic activity post-mortem.
Studies on muscarinic receptors are limited and contradictory. A radioligand-binding study that measured the summed density of all muscarinic receptors showed an increase in receptor density in the orbito-frontal cortex and caudate from subjects treated with antipsychotics up until death [98]. However, using a more selective radioligand, reduction in density of muscarinic receptors M1 + M4 has been reported in the caudate-putamen not related to antipsychotics [99]. Moreover, this was not accompanied by a change in the levels of M1 receptor mRNA in the caudate-putamen [100]. The availability of increasingly specific radioligands for muscarinic receptors, receptor specific antibodies and measurement of levels of mRNA for each receptor will make it possible to determine which receptors are altered in schizophrenia. This will allow the precise nature of changes in muscarinic receptors to be determined.
Conclusions
Studies using neuroimaging techniques or tissue obtained post-mortem have revealed extensive changes in neurochemical markers in different regions of schizophrenic patients. Moreover, it is clear these changes would affect many neurotransmitter systems and could, therefore, precipitate diverse symptoms [101]. It would seem logical that the atypical antipsychotics affect a greater number of the systems altered in schizophrenia [94].
Interpreting data from post-mortem and neuro-imaging studies is difficult. Growing evidence supports the hypothesis that changes in functioning of dopa-minergic neurones is central to the genesis of schizophrenic psychoses and that changes in serotonergic and glutamatergic neurones are important in the pathology. Further evidence is required to implicate changes in GABAergic and cholinergic systems.
Data from both neuroimaging and post-mortem studies have identified changes in the presynaptic proteins and/or function in the brains of schizophrenic patients: in dopamine release and glutamate synthesis, in the dopamine autoreceptor, and the serotonin, glutamate and GABA transporters. Changes in functioning of presynaptic processes could be a component of the pathology. As molecules in these processes are isolated and their function understood [102], it should be possible to determine if changes in a single protein common to all affected presynaptic processes is the cause of schizophrenia. Alternatively, it results from changes in different proteins such as the serotonin, glutamate and GABA transporters. Whichever proves to be the case, if the ‘presynaptic hypotheses’ is proven, then current antipsychotics probably act by blocking the ‘flow-on’ effects of abnormal neurotransmitter levels by antagonising post-synaptic receptors. It would, therefore, seem that the focus of future drugs should change from blockade of the post-synaptic receptor to modulation of presynaptic function, that is, ‘modulate signal transmission’ rather than ‘reduce signal reception’.
Footnotes
Acknowledgements
I thank Elizabeth Scarr and Geoffrey Pavey for their suggestions, scientists in the Rebecca L. Cooper Research Laboratories for their commitent to our research, and the National Alliance for Research into Schizophrenia and Depression, The Rebecca L. Cooper Medical Research Foundation and the Stanley Foundation for supporting that research.