
Abstract
Not long after the introduction of neuroleptic drugs, it was reported in the German literature that the use of these drugs could result in the development of a movement disorder [1]. Reports gradually accumulated in the 1960s with the use of descriptive terms such as ‘bucco-linguo-masticatory syndrome’, ‘orofacial dyskinesia’, ‘terminal extrapyramidal insufficiency syndrome’ and others. The term ‘tardive dyskinesia’ (TD) was introduced by Faurbye and Rasch [2] to emphasise the delayed or tardive onset of the syndrome secondary to neuroleptic drug use. Interest in TD increased in the 1970s and 1980s when it became apparent that it was a common and potentially irreversible side-effect that could lead to considerable suffering and disability. The introduction of ‘atypical’ antipsychotic drugs in the 1990s has, for the first time, opened up the possibility of treating chronic psychotic illnesses with drugs that have a low or negligible risk of TD.
Clinical features
Nature of the movement disorder
The term dyskinesia (literally ‘abnormal movement’) is a generic term that refers to a range of movement abnormalities. In the case of TD, the movements are either choreiform (rapid, jerky and non-repetitive, involving particularly the proximal muscles), athetoid (slow, sinuous or writhing, and involving distal muscles), dystonic (slow and sustained muscle contractions) or stereotypic (rhythmic and repetitive) or a combination of these [3].
The movements most commonly involve the orobuccal, lingual and facial muscles, especially in older individuals. The lingual involvement in the form of fine vermicular movements of the tongue while it is sitting at the base of the oral cavity is a common and early feature. With worsening, writhing of the tongue develops, and the tongue may thrust out of the mouth irregularly (‘fly catching’), repeatedly lick the lips or press against the cheek to produce a bulge (‘bonbon sign’). Dyskinetic blinking may be another early sign of TD. Lip smacking, puckering or pouting, chewing, jaw clenching or mouth opening, facial grimacing, blowing, blepharospasm and frowning are also common features. The limbs and trunk are often involved and the involvement of the respiratory muscles has been uncommonly reported [4]. The fingers may display stereotypic movements, especially when held in extension, as if the patient is ‘piano playing’. There may be irregular, jerky flexion or extension movements of the upper limbs. Involvement of the lower limbs manifests in the form of irregular movements of the toes, foot inversion or eversion and irregular flexion or extension at the ankle or knee. Stereotypic leg movements are often present. Truncal movements are in the form of lateral, posterior or irregular neck movements, shoulder shrugging, twisting, flexion or extension of the trunk and pelvic rotation or thrusting. Respiratory muscle involvement manifests in the form of irregular breathing, belching and grunting sounds, whistling or sucking and aerophagia. Abdominal and pharyngeal muscles may be rarely involved.
The oro-buccal-lingual-facial (OBLF) musculature is involved in three-quarters of affected individuals, the limbs in one-half and the trunk in up to one-quarter, with all three groups being affected in about 10% [5]. While the OBLF involvement is typical of the elderly, limb-truncal (LT) involvement is more likely in the young. This kind of involvement of respiratory, pharyngeal or abdominal muscles rarely occurs without OBLF or LT dyskinesia.
While the movements described above are choreoathetoid, which are recognised to be characteristic of TD, neuroleptic-induced tardive movements may sometimes be dystonic, akathisic, tic-like, myoclonic or tremorous in character. This has led to a debate between the ‘lumpers’ and ‘splitters’, i.e. those who include all these different movements within the rubric of TD, and those who make a distinction between TD and other tardive syndromes such as tardive dystonia (TDt), tardive akathisia, tardive tics or Tourette's syndrome and others (Table 1). There are some empirical observations, other than the descriptions of the movements themselves, that favour the splitters. To take the example of TDt, there is evidence that the risk factors (most cases have been reported in young individuals, with a male excess) and the pharmacological profile and treatment response may be different from TD (e.g. improvement of TDt with anticholinergic drugs) [6,7]. For a more detailed discussion, the reader is referred to a recent review of TDt [8]. Differences have also been reported for the relatively common syndrome of tardive akathisia which often overlaps with TD [9].
Neuroleptic-induced tardive subsyndromes

The status of the behavioural syndromes is uncertain.
The movements of TD typically fluctuate in intensity over time, increase with emotional arousal, decrease with relaxation and disappear during sleep. The spontaneous fluctuations may occur from day to day, and may even be apparent over hours or minutes.
They also decrease when the affected muscles are used for voluntary activity. Distracting tasks, such as finger tapping or mental arithmetic, tend to exaggerate the movements and bring out movements that may otherwise be latent. Walking may bring out movements in upper extremities. Poor dental status may exaggerate oral movements in some patients. In mild cases, the patient may be unaware of the movements.
The movements of TD have a variable response to medication. Neuroleptics, with the possible exception of clozapine, are recognised as suppressing the movements. Increasing the dose of the offending drugs may therefore suppress the movements. Anticholinergic drugs, on the other hand, usually aggravate TD. These responses are, however, often unpredictable.
Associated features
Some neurological, behavioural and cognitive features have been described as associated with TD. These include saccadic eye movement abnormalities [10], neurological soft signs and ventricular enlargement [11]. Tardive dyskinesia, when it occurs in patients with schizophrenia, is more likely to be associated with negative symptoms and cognitive impairment [9,11]. It has also been suggested that, analogous to TD, patients on chronic neuroleptic treatment may develop behavioural or cognitive changes, and the syndromes of supersensitivity psychosis [12], tardive dysmentia [13] and tardive dysbehaviour [14] have been described by a few investigators. Empirical support for these behavioural analogues of TD is weak as no large, systematic data sets exist to support or refute their existence.
Onset
Tardive dyskinesia develops after a person has been on neuroleptics for months to years. The DSM-IV (American Psychiatric Association, Table 2) requires exposure of at least 3 months, but this may be as brief as a month in elderly individuals. The onset may be either while the patient is still on neuroleptics or within a few weeks of their withdrawal. The latter (withdrawal-emergent dyskinesia) may remit spontaneously or go on to become persistent. The onset is usually gradual and the disorder is mild except for withdrawal dyskinesias which can be severe from the early stages.
DSM-IV research diagnostic criteria for tardive dyskinesia

Source: American Psychiatric Association, 1994.
Diagnosis
The nature of the movement disorder as described above and its occurrence following exposure to neuroleptic drugs help to diagnose the disorder. In order to distinguish from mild dyskinetic movements that may appear transiently in some patients, a duration of 4 weeks is arbitrarily required for a DSM-IV research diagnosis (see Table 2 for research criteria). Tardive dyskinesia is considered ‘persistent’ if it lasts for longer than 3 months, and chronic if it lasts beyond 6 months, according to the most commonly used research diagnostic criteria for TD [15]. These authors [15] suggest making a definite research diagnosis of TD only if movements of moderate severity are present in one or more body parts, or of mild severity if in two or more body parts. As the disorder fluctuates, patients with minimal or mild movements should be reassessed within a week to confirm their presence.
The examination of a patient for the assessment of TD should be systematic and standardised, and the use of a multi-item rating scale is recommended to evaluate the severity of the symptoms. The most commonly used scale is the Abnormal Involuntary Movements Scale (AIMS) [16] which rates dyskinetic movements in seven body regions, each on a five-point scale, and also includes a global severity rating of movements, the incapacity they cause and the patient's awareness of them. The AIMS comes with instructions for a systematic examination procedure with which all clinicians should be familiar. Rating scales are not, however, diagnostic instruments and the diagnosis should be based on a complete neuropsychiatric history and examination. Although the diagnosis is easy in most cases, and laboratory investigations (Table 3) to rule out other disorders are only occasionally necessary, the list of disorders that may resemble TD is long indeed.
Investigations for the differential diagnosis of tardive dyskinesia and their rationale

CT, computed tomography; CVA; cerebrovascular accident; MRI, magnetic resonance imaging; SLE, systemic lupus erythematosis.
Differential diagnosis
Acute neuroleptic-induced dyskinesia
Dyskinesias may occur as an acute effect of neuroleptic treatment; in these cases they are dose-dependant, remit with dose reduction or cessation, and respond to anticholinergic or antihistaminergic drugs. A prevalence rate of 2.3% has been reported [17]. A rare subacute extrapyramidal syndrome is the ‘rabbit syndrome’ characterised by a tremor of the lips and perioral region imitating the chewing movements of a rabbit. This is a restricted form of neuroleptic-induced parkinsonism which responds to anticholinergic treatment, and should not be confused with TD [18].
Dyskinesias due to other drugs
Some drugs reported to be associated with acute dyskinesia are anticonvulsants (phenytoin, carbamazepine), oral contraceptives, chloroquine-based antimalarials, stimulants, dopamine agonists, lithium and tricyclic antidepressants. These are reversible upon dose reduction or discontinuation of the offending drug.
A delayed-onset or ‘true’ tardive dyskinesia may result from other dopamine antagonists that are not used as antipsychotics (e.g. metoclopramide, prochlorperazine),
Dyskinesia secondary to other neurological and systemic disorders
Some disorders to consider in the differential diagnosis are Huntington's disease, Wilson's disease, Sydenham's chorea, Tourette's syndrome and rare disorders such as Hallervorden-Spatz disease and Fahr's syndrome. Huntington's disease may pose a diagnostic problem because its psychiatric manifestations may precede the movement disorder by years, and in this period the patient might have been treated with antipsychotic drugs. Tumours of the basal ganglia or infarcts may also produce dyskinesia. Immune disorders such as systemic lupus and antiphospholipid syndrome can have dyskinesia as a feature if central nervous system involvement has occurred. Endocrine disorders such as hyperthyroidism and hypoparathyroidism also need to be considered.
Spontaneous dyskinesias
Dyskinesias, in particular orofacial movements, have been reported to occur in some individuals with none of the above causes. They are more likely in the elderly, with one study reporting prevalence rates of 0.8%, 6.0% and 7.8% in the 6th, 7th and 8th decades of life of otherwise healthy subjects [21]. Kane et al. [22] reported a rate of 4.0% in healthy, elderly (mean age 73 years) subjects. The prevalence is higher in psychogeriatric patients and those in institutions who have not received neuroleptics, and is particularly so in dementia [23]. Ill-fitting dentures also increase the prevalence [3].
Is dyskinesia a feature of schizophrenia?
The occurrence of ‘peculiar, sprawling, irregular, choreiform, outspreading movements’ in schizophrenic patients were noted by Kraepelin [24]. Many studies have investigated neuroleptic-naive schizophrenic patients and reported rates of dyskinesia that vary from nil [25] to as high as 53% [26]. These movements are usually described as grimacing, ticlike or stereotypic, but orofacial dyskinesias and choreiform movements are less commonly observed. The studies show an increasing prevalence with age and the severity of illness. In a study of first-episode schizophreniform psychosis, Chatterjee et al. [27] reported dyskinesias in 1% of untreated patients. A more recent study [28] reported spontaneous dyskinesias in 7.6% of 79 first-episode patients. The movements in these reports were mainly orofacial or involving the upper limbs and similar in severity to those seen in TD patients. These findings have raised the argument that dyskinesias may be intrinsic to the pathophysiology of schizophrenia, and that neuroleptics serve to enhance the process.
Epidemiology
The prevalence of TD has been extensively investigated and 76 published studies were reviewed by Yassa and Jeste [29]. The rates ranged from 3% to 70%, with a median figure of about 24% in patients on chronic neuroleptic treatment. The higher rates were likely to be reported in the elderly. The wide variation in rates can be explained on the basis of the fact that different populations were studied, and there are many methodological issues: diagnostic criteria used, role of antipsychotic drugs masking the disorder, role of anticholinergic and other drugs, fluctuation in symptoms, duration of neuroleptic exposure and others. The majority of TD is fortunately mild. The study by Woerner et al. [30] is noteworthy for its attempt to address some of the methodological issues. The overall prevalence in neuroleptic-treated individuals was 23.4%, of which 3.8% had another neuromedical illness that might have had an aetiological role, thus giving a conservative prevalence rate of 19.6%. The rate varied with the setting, being 13.3% at a voluntary hospital with a young population, 23% in a Veterans Administration hospital and 36% in a state hospital. In the same study, when a group of patients with no evidence of TD was withdrawn from neuroleptic drugs and examined weekly for 3 weeks, 34% developed emergent dyskinesia.
Prevalence of TD has been examined in some special populations. TD has been reported in children and adolescents, but adequate epidemiological studies are few [31]. Rates of withdrawal-emergent dyskinesia of 8% [32] and 51% [33] have been reported in two studies of children on long-term neuroleptics. High rates have almost invariably been reported in elderly patients on neuroleptics [34,35]. High rates have also been reported in neuroleptic-treated individuals with mental retardation [36,37].
Given the difficulties inherent in prevalence estimates, it has been much more revealing to examine the incidence of TD in newly medicated patients followed longitudinally. The salient studies are summarised in Table 4. These studies suggest that the cumulative incidence of TD increases with increasing duration of neuroleptic treatment, at the rate of about 3–5% per year for the first several years, to reach a plateau at about 20–25%, but new cases continue to occur many years after drug initiation. It is difficult to identify a point of time after which the risk decreases. The incidence is many times higher in elderly individuals [38].
Summary of longitudinal studies of incidence of tardive dyskinesia in schizophrenia patients
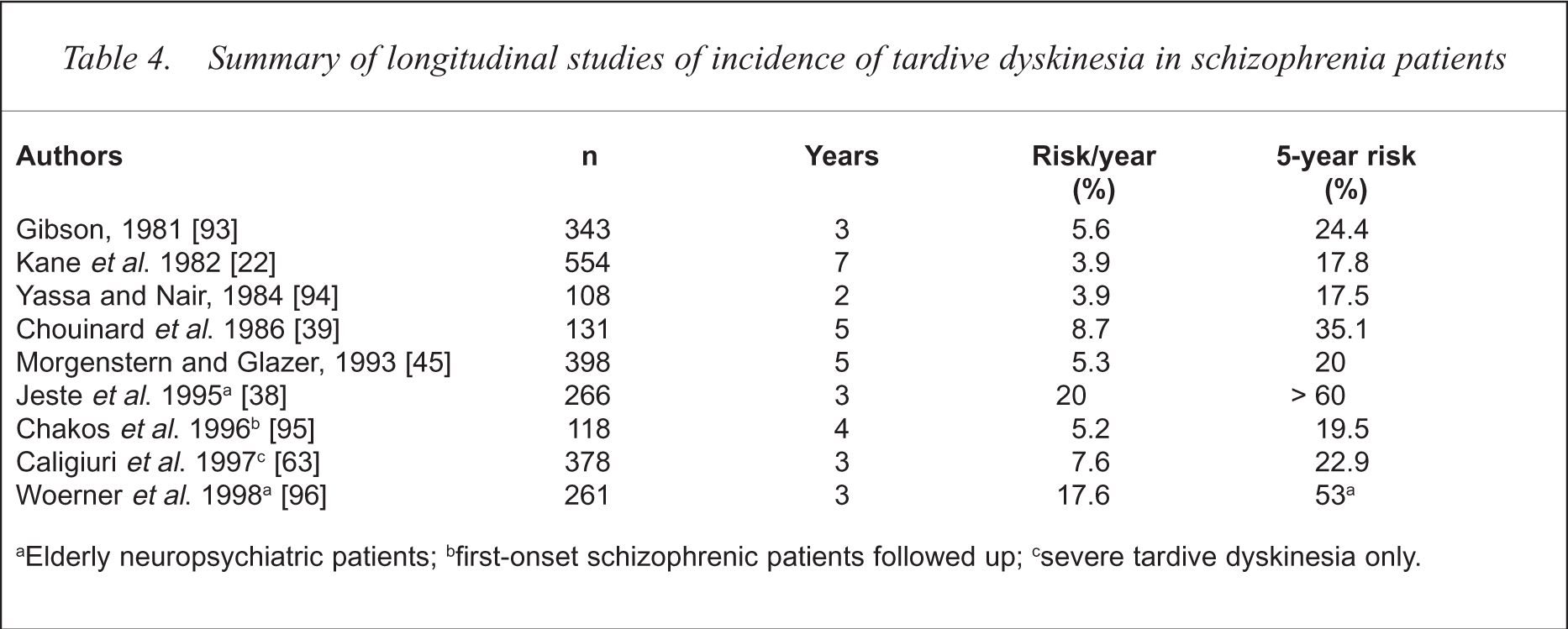
Elderly neuropsychiatric patients;
first-onset schizophrenic patients followed up;
severe tardive dyskinesia only.
Natural history
A number of studies have examined the course of TD once it has been diagnosed in a group of patients [39–41]. The overall conclusion from these studies is that TD does not become progressively worse except in a minority, and generally tends to show a fluctuating course with some spontaneous remissions. The remission rates do not tend to be high in the first year, but 5-year data are more favourable. In a 5-year follow-up study of Bergen et al. [41], 24% showed a fluctuating course, 11% improved while 7% worsened. From 5 to 10 years, about 50% of patients demonstrated a reduction in symptoms of at least 50%. The outcome is more favourable in the young, and if drug treatment can be stopped [42]. Improvement can be expected to continue for many years after neuroleptics have been ceased. The prognosis of withdrawal-emergent dyskinesia is more favourable, with over three-quarters showing improvement in 3 months [5].
Complications
The majority of TD patients have a mild disorder and may in fact be unaware of its presence. In these cases, TD is largely an aesthetic problem, but may impair social relationships and be an impediment to employment. About 5–10% of TD patients suffer impairment from the dyskinesia. Orofacial movements may lead to difficulty in eating or retaining dentures, and weight loss or cachexia may develop. Pharyngeal involvement may rarely cause aspiration pneumonia. Involvement of the limbs and trunk may interfere with ambulation. Diaphragmatic involvement may lead to respiratory dysfunction and even failure, and speech abnormalities are not uncommonly reported [4]. A rise in creatine kinase levels may occur because of continuous muscle activity, but malignant rhabdomyolysis has not been reported. In one study, the mortality rate of schizophrenics with TD was reported to be twice that of those without TD, but the reasons for this were unclear [43].
Risk factors
Sociodemographic factors
Advancing age is the most consistently established risk factor for TD, and there appears to be a linear correlation between age and both the prevalence and the severity of TD [44]. Not only is TD more common in the elderly, it is more likely to be severe, develop early in the course of neuroleptic treatment and be irreversible. The elderly are also more likely to have the orofacial manifestations of TD.
While earlier studies suggested that TD was more likely in women, the female gender has not consistently emerged as a risk factor in recent studies [45]. There may, however, be a female excess in elderly TD sufferers, suggesting a gender and age interaction [46].
Some ethnic differences, with higher rates in African Americans and lower rates in Chinese and other Asian populations, have been reported, the basis for which is not clearly understood [45,47].
Psychiatric disorder
Neuroleptics are used for a number of neuropsychiatric disorders, and sometimes for other indications such as vomiting, belching, agitation and sleeplessness. The dosage and duration of use of drugs for different indications vary considerably and it is difficult to assess the relative risk that the psychiatric condition itself presents. Since TD has been reported after neuroleptic use for non-psychiatric conditions [48], the presence of a psychiatric disorder is clearly not necessary. A number of investigators have commented on a higher relative incidence of TD in patients with affective disorder treated long-term with neuroleptics [49,50], but this finding is inconsistent, and may apply to early and not late-onset TD [45]. Kane et al. [51] reported incidence figures of 26% for affective and schizoaffective disorders and 18% for schizophrenia. In schizophrenic patients, a family history of affective illness is reported to increase the risk. Depression may, furthermore, produce a state-dependent exacerbation of TD [52] while mania may lead to the reverse [53].
Whether schizophrenia increases or decreases the risk for TD is not known. The similarity between the spontaneous dyskinesias of schizophrenia and TD has been suggested as evidence for schizophrenia as a risk factor, but the opposite has also been suggested [54]. Within schizophrenia, those with the negative syndrome, or evidence of cognitive impairment and neurological deficits, are reported to be more at risk [11], and the presence of TD indicates a poorer prognosis for schizophrenia.
The presence of brain damage (as evidenced by epilepsy, head trauma, dementia etc.) has been suggested as a risk factor, but the evidence is inconsistent. TD is known to develop in Tourette's syndrome patients treated with neuroleptics, but the prevalence rate, though not well studied, is likely to be lower than that seen in schizophrenia, possibly because of the youth of the patients and the small neuroleptic doses used [55].
Other patient-related factors
There has been recent interest in variables such as smoking, alcohol abuse and diabetes as risk factors. A recent study found that alcohol or other drug abuse increased the risk of TD by threefold [56]. Diabetics have an increased risk of both spontaneous dyskinetic movements (21% in the study by Ganzini et al. [57]) and TD (79% in diabetics vs 53% in non-diabetics [57]). Woerner et al. [58] reported a risk ratio of 2.3 for diabetics exposed to neuroleptic compared with non-diabetics, with the risk greater in aged diabetics. Alcoholics and smokers have an increased prevalence of spontaneous dyskinesias as well as TD [59].
It has been suggested that patients who are more prone to develop acute extrapyramidal side-effects with neuroleptics are also at a higher risk of TD [60]. The basis of this sensitivity to basal ganglia disease is not understood. Genetic factors have been studied. An intriguing finding is that heterozygous carriers of mutated alleles of the CYP2D6 gene have an increased susceptibility to TD [61]. An association between TD and a dopamine D3 receptor gene variant has also been reported [62].
Drug-related factors
Neuroleptic drugs
While there has always been a suspicion that neuroleptic dose was important for the risk of TD, empirical evidence for this was lacking. Recent longitudinal studies have provided that empirical support [45,63]. It is possible that in previous studies this dose effect was masked by the high current doses the subjects were receiving, and became apparent when patients treated with low-effective doses were studied [64]. The dose effect may, moreover, be apparent in the first few years of exposure to drugs. Longitudinal studies also suggest that the prevalence increases with duration of exposure as new cases are added, but this may reach a plateau after about 5 years of exposure. New cases continue to occur after that, but since others remit, the overall prevalence may not go up considerably after the first 5 years or so, although definite data to confirm this are lacking [51,60,65]. Taken together, the total neuroleptic load can be considered to be a risk factor and an attempt should be made to minimise this. The data in relation to neuroleptic blood level and the onset of TD are inconsistent [5].
Whether drug type is important has remained controversial. There is no convincing evidence that, once drug dosage has been accounted for, any of the conventional neuroleptics presents a differentially smaller risk; nor is there empirical evidence that depot neuroleptics are more likely to cause TD [5]. The evidence in relation to the newer atypical drugs is still preliminary, but suggests that these drugs may present a lower risk for TD. While TD has been reported with risperidone and olanzapine, there is no convincing report of TD with clozapine monotherapy and this may indeed be the safest drug [5]. Clozapine is only partially successful in suppressing TD, is considered to permit the remission of TD [66] and has been proposed as a treatment for TD and TDt [67]. In an analysis of seven 1-year trials of risperidone, the incidence of TD at relatively high doses (mean = 7.6–9.4 mg/d), was estimated at 0.3% [68]. In a comparative study of olanzapine and haloperidol, haloperidol had a fourfold (4.8%) higher incidence of TD in the last two visits after 7 months of treatment compared with olanzapine (1%) [69]. More experience with these drugs is necessary before their lower risk can be generally accepted.
The practice of drug holidays, which was once advocated to reduce the total neuroleptic load in an individual, is now believed to in fact increase the risk of TD [70]. Brief, intermittent or targeted treatment may, however, offer an advantage, but it should be further investigated and balanced against any increased risk of schizophrenic relapse [71].
Other drugs
Anticholinergic drugs are known to exaggerate TD or make latent TD become manifest, but there is no convincing evidence they are risk factors for TD per se [5]. The concurrent administration of lithium in affective disorder has been suggested to possibly reduce the risk of TD. This is supported by animal data [72], but the clinical evidence is tentative [60].
Pathophysiology
The dopamine supersensitivity hypothesis
The pathophysiology of TD is not completely understood. Much of the traditional conceptualisation of the disorder has been guided by the causative role of antipsychotic and other dopamine (DA) antagonists. This resulted in the proposal of the dopamine supersensitivity hypothesis of TD [73], which dominated debate in the 1970s and 1980s. Put simply, the hypothesis states that the chronic administration of DA antagonists leads to the development of post-synaptic DA receptor super-sensitivity, thereby producing the hyperkinetic state of TD. This hypothesis is compatible with many clinical and laboratory observations. Dopamine agonists such as
This hypothesis has a number of limitations. Observations of the animal models do not parallel clinical observations, as DA supersensitivity in rats develops rapidly (often after a single injection) and persists for only a short period after cessation of the DA antagonist drug. Supersensitivity in animals declines after ‘desensitisation’ with DA agonists, but these drugs are not effective in treating TD. Moreover, supersensitivity in animals is almost invariable whereas TD develops only in a fraction of patients. Dopamine supersensitive rats do not exhibit a behavioural change which is brought out by challenging them with dopaminomimetic drugs. Rats that do develop an analogue of TD continue to show the movements after DA supersensitivity has disappeared. There is no direct evidence of DA super-sensitivity in TD patients, either from post-mortem studies, cerebrospinal fluid studies or radioligand imaging studies using positron emission tomography. There is no difference between dopamine-mediated endocrine function in TD subjects and that in non-TD controls. The effects of DA agonists and antagonists are also inconsistent among TD patients as shown by clinical observations and challenge studies. Moreover, the DA hypothesis does not explain the spontaneous occurrence of dyskinesia in many schizophrenic and healthy subjects, and the increased risk with age and many other host factors.
The DA hypothesis, therefore, explains only some aspects of TD and must be considered to be inadequate. Some modifications have been suggested. One such suggestion is that patients vulnerable to TD develop a greater and more persistent increase in DA turnover in response to DA blockade [75]. Evidence for this is lacking in humans. Another postulate is that TD is the consequence of an imbalance between D1 and D2 subtypes of DA receptors [76]. The latter is consistent with the observation of an increase in D1 and a reduction in D2 receptor density in ageing brains, with age being the major risk factor for TD.
The γ-aminobutyric acid insufficiency hypothesis
The inadequacy of the DA hypothesis has led to the consideration of many other pathophysiological models. In relation to other neurotransmitters that are affected by neuroleptics, some attention has been given to changes in norepinephrine (NE), serotonin (5HT) and acetylcholine (ACh), but changes in γ-aminobutyric acid (GABA) are considered to be the most salient for the development of TD [76]. Reduced activity in a subgroup of striatal GABA neurones has been suggested as the basis of TD, and this is supported by evidence from animal and human work. Studies of rats [76,77] indicate that chronic neuroleptic treatment decreases GABA turnover and increases GABA receptors, which can be explained on the basis of reduced GABA release or a loss of GABA terminals. A reduction in glutamic acid decarboxylase (GAD), a rate-limiting enzyme in the synthesis of GABA, has been demonstrated in neuroleptic-treated animals, with a correlation with the development of dyskinesia [77]. A GABA antagonist, bicuculine, injected directly into the substantia nigra causes dyskinesia in rats [77]. Some human studies have supported the GABA hypothesis. A reduction of GAD in the subthalamic nuclei of TD patients was shown in a small study [78], and reduced GABA levels in the cerebrospinal fluid were demonstrated in TD patients in another study [79]. Increase saccadic distractability, an eye movement controlled by GABA projections, has also been reported in TD subjects [10].
Neurodegeneration hypothesis
According to the neurodegeneration hypothesis, TD is the consequence of neuroleptic-induced neuronal loss, particularly in the striatum [80]. Gunne and Andren [81] proposed that the degeneration occurred in the GABAergic neurones projecting from the globus pallidum to the thalamus. Two interacting mechanisms have been proposed for the pathogenesis of cell damage: production of free radicals and excitotoxicity, both of which lead to apoptotic death of neurones.
Support for the free radical hypothesis and increased oxidative stress comes from the following.
Neuroleptics increase catecholamine turnover which leads to excess production of free radicals, especially in catecholamine-rich areas such as the basal ganglia. Because of the high oxidative metabolism in these regions, neurones are particularly vulnerable to membrane lipid peroxidation and cell death.
Neuroleptics lead to the accumulation of iron in the basal ganglia which may be neurotoxic through free radical mechanisms [37].
There is a reported reduction in essential fatty acids in plasma phospholipids in patients with TD and increased cerebrospinal fluid indices of lipid peroxidation in these patients [82], although the findings are not always consistent.
The possible beneficial role of an antioxidant (vitamin E) in the treatment and prevention of TD has found some, albeit inconsistent, support from human work [83–85].
The hypothesis is consistent with the roles of age, diabetes, smoking and brain damage as risk factors for TD, the irreversibility of a proportion of TD, and the occurrence of spontaneous dyskinesias in schizophrenia.
The excitotoxicity mechanism for cell damage was proposed because dopamine has an inhibitory effect on the release of excitatory neurotransmitters, and dopamine D2 blockade leads to an increase of glutamate (Glu) and aspartate release in the striatum [86]. The persistent activation of N-methyl-D-aspartate (NMDA) receptors and non-NMDA glutamate receptors leads to oxidative damage to cellular proteins, membranes and DNA and, ultimately, cell death. There is considerable secondary interaction of glutamatergic and free radical mechanisms, leading to a vicious cycle promoting oxidative damage in the striatum [87]. A recent report presented preliminary evidence of higher concentrations of N-acetylaspartate, N-aspartylglutamate and aspartate in the cerebrospinal fluid of TD patients [87].
A synthetic view
Since there is evidence to support each of the above hypotheses, a synthesis is necessary, and an attempt at this is presented in Figure 1. In summary, free radical and excitatory mechanisms lead to neurotoxicity and cell death, particularly of the GABAergic striatal neurones, because of the high levels of catecholamine turnover and oxidative metabolism in the striatum. This leads to the disinhibition of the lateral pallidal neurones and consequently the hyperkinetic state of TD, the manifestation of which is influenced by the prevalent dopaminergic tone. Withdrawal of DA antagonism will lead to the expression of a latent hyperkinetic state, whereas DA blockade produces the reverse. The DA receptor antagonism of neuroleptics is therefore important in the pathogenesis of TD, but the mediating mechanisms are multiple and DA receptor supersensitivity may be only one aspect of the pathogenesis, and not the pre-eminent one.
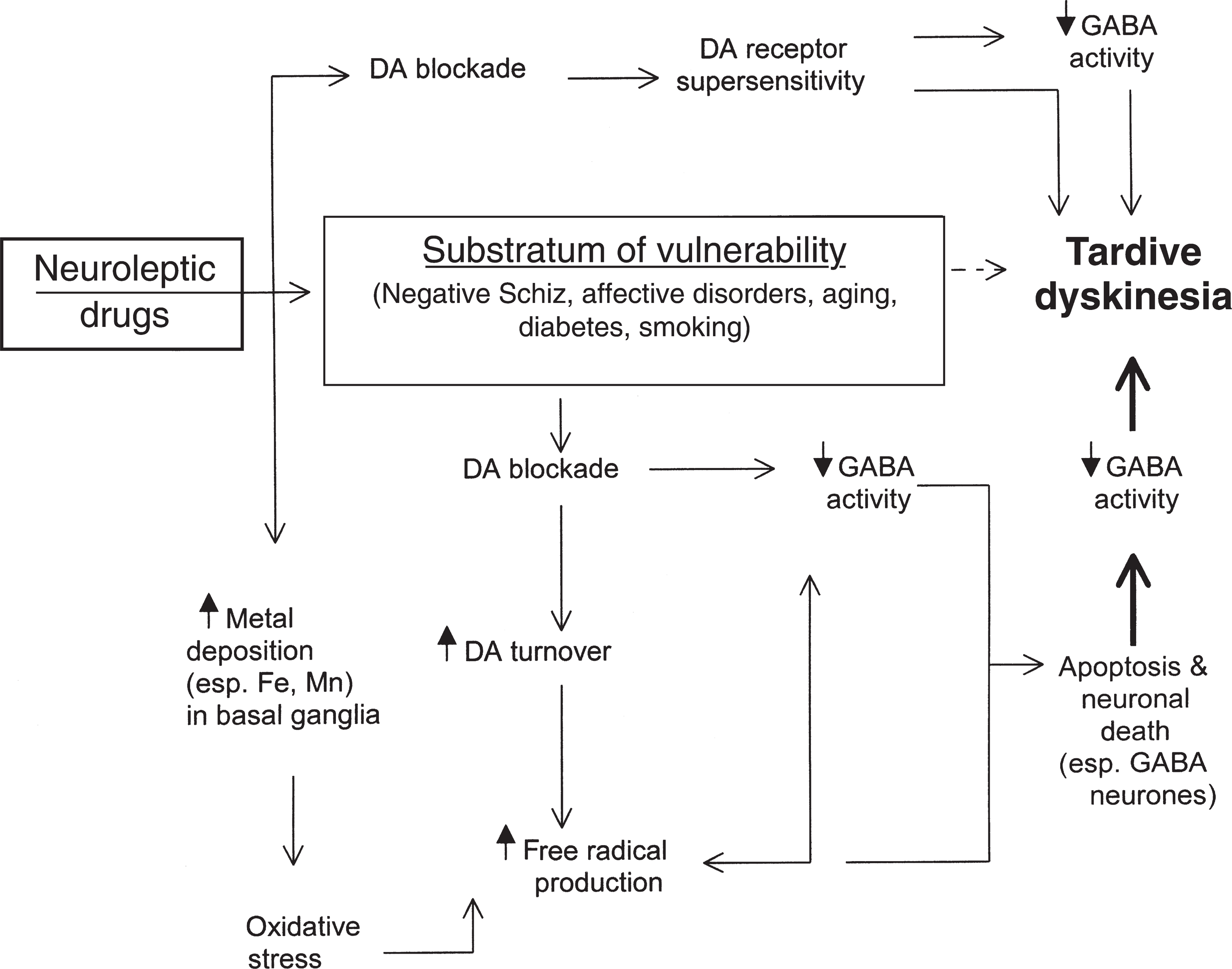
Pathogenetic mechanisms for tardive dyskinesia (see details in text). Neuroleptics may cause dyskinesia either by direct action on receptors or by promoting free radical and excitotoxic injury leading to neuronal loss, particularly of striatal GABAergic neurones. Both mechanisms probably play a role and act on a substratum of brain vulnerability neuropsychiatric disorder or other risk factors. DA, dopamine; GABA, γ-amino butyric acid; Fe, iron; Mn, manganese.
Management
No effective treatment for TD is currently available, although a number of drugs have been tried, based on the still preliminary understanding of the pathophysiology of the disease. The primary strategy in its management remains preventative. Once TD becomes established, attempts are made to minimise its symptoms and reduce ongoing risk factors for the worsening of the disorder over time.
Prevention
Neuroleptic drugs continue to remain the main treatment for a number of psychiatric disorders, in particular acute and chronic schizophrenia and other psychoses, acute mania, psychotic depression and Tourette's syndrome. Their use should be restricted to established indications, and alternatives should be considered whenever possible. Caution is advised in their use for agitation, personality disorders, anxiety or insomnia, and their prolonged use in affective disorders. Their use in children and the elderly should involve extra care. When neuroleptics are appropriate, an attempt should be made to minimise the patient's exposure to the drug by using the smallest dose for the shortest period while taking the risk of psychotic relapse into consideration. The use of atypical antipsychotics as first-line drugs cannot yet be recommended unequivocally, but this strategy is being increasingly adopted by many clinicians. When patients are maintained on long-term neuroleptic medication, they should be periodically evaluated (about every 3 months) for the continuing need for the drug and the early features of TD for which a standard assessment such as the AIMS examination is recommended. These evaluations should be adequately documented in the patient's medical record.
Throughout the period of treatment with antipsychotic drugs, informed consent of the patient and the involvement of the family are necessary. The latter is even more important if the patient is judged to be unable to understand the information or incompetent to provide informed consent. In such cases, consent should be sought as soon as the patient is considered to be competent again. The patient and the family need to be periodically educated about the continuing need for the drug and the risks and benefits. Obtaining informed consent is a continuing process in the setting of the doctor–patient relationship, and should not be taken to mean a ritualised signing of a detailed and legalistic document. Documentation of this process in the medical record is the best safeguard for the psychiatrist against a medico-legal challenge.
Treatment
If a presumptive diagnosis of TD is made in an individual, the first step is a detailed evaluation of the psychiatric status, and the assessment of the need for ongoing treatment with neuroleptics. While TD does not necessarily worsen with ongoing antipsychotic drug treatment, the best chances of remission seem to be present if the offending drug can be discontinued. Alternatives to neuroleptics should be considered. If discontinuation is inadvisable because of the clinical status, it is advisable to reduce the dose to the smallest effective level although this advice is not based on firm empirical evidence [88]. A switch to an atypical drug should be considered with the understanding that only clozapine has been demonstrated to be safe in relation to TD, but that the other atypical drugs may be relatively safer than the conventional drugs. Anticholinergic drugs should be discontinued if they are being administered. The patient's dental status should be optimised and prosthetic dental treatment prescribed if considered necessary. Education and information of the patient and family is again emphasised, as is informed consent. It is usually inadvisable to attempt to suppress TD by increasing the dose of the neuroleptic drug, as there is the distinct possibility of a breakthrough dyskinesia which may be more difficult to manage. The only exception to this is the temporary suppression of a withdrawal-emergent dyskinesia so that a more gradual reduction of the neuroleptic can then be attempted.
Most patients with TD do not need any further intervention but should be periodically reviewed to chart their progress. In moderate to severe TD, drug therapy to specifically treat the movements may be necessary. A large number of drugs have been investigated in clinical trials, but none has emerged to be clearly superior or effective in all cases or free from side-effects. It is difficult to predict an individual's response to a particular drug and it is therefore advisable to have a systematic plan of management in which the order of drugs to be tried is clearly stated. Some of the commonly used drugs will be discussed here and have been recently reviewed by Egan et al. [83] and Gardos and Cole [88].
Strategies for altering DA activity have sometimes been successful. As stated, the use of higher doses of DA antagonists is only rarely justified. Catecholamine depleters such as tetrabenazine and oxypertine do have a role. Oxypertine has been demonstrated to be superior to a placebo by one group of investigators, but its efficacy may not be sustained over long periods [89]. Tetrabenazine has, on the other hand, not been systematically investigated, but clinical experience suggests its effectiveness in a proportion of patients. The most positive results have been reported by Jankovic and colleagues who support its long-term efficacy [90]. Doses of tetrabenazine used are usually in the range of 25–150 mg per day. Common side-effects are drowsiness, parkinsonism, depression, insomnia, nervousness and akathisia. Tetrabenazine is the drug most commonly used in our clinic. Of the atypical neuroleptics, clozapine has been suggested to have a specific antidyskinetic effect although this remains controversial with the alternative that clozapine may merely permit the reversal of the pathology of TD and thereby its resolution [66]. The strategy of using DA agonists (bromocriptine, pergolide) or precursors (
GABAergic drugs have received much attention, including some controlled investigations of their efficacy. They may work in some patients and are worthy of a trial. The most commonly used drugs in this class are sodium valproate, clonazepam, diazepam, baclofen and progabide. The third class of drugs to be tried is the anti-adrenergic drugs, particularly propranolol and clonidine. The benefit of cholinergic drugs (choline, lecithin, deanol) is doubtful. Many other drugs have been tried in TD, occasionally with some success, and these are listed in Table 5.
Drugs used in the treatment of tardive dyskinesia

A recent approach has been to use anti-oxidants for both treatment and prevention, even though the clinical studies of the role of anti-oxidants so far have focused on treatment. Of 11 pilot studies published until recently, nine demonstrated a beneficial effect of vitamin E [83], but all these studies were for short periods. A recent study by Adler et al. [85] examined the effect of 1600 IU per day of vitamin E over 8 months in 28 subjects and showed a significant effect from 10 weeks that continued through the full 36 weeks. The larger, yet unpublished, Veterans Affairs Cooperative Study (CS 394), which included 158 subjects and used the same dosage of vitamin E over 1 year, showed no difference from a placebo. Examining this evidence, Lohr and Lavori [84] concluded that the effect of vitamin E was small and probably negligible, and ‘vitamin E is not proven to be effective for TD in general’ (p 862). They went on to add, however, that a trial of vitamin E carried virtually no risk. The preventative role of vitamin E has not been examined in humans, and work with other anti-oxidants is still preliminary.
In cases with severe and refractory TD, heroic measures such as deep brain stimulation and pallidotomy may be justified, with occasional reports of their usefulness [91,92].
Future directions
There are many lacunae in our understanding of TD which explains why rational therapy or effective preventative strategies are still not available. The increasing use of atypical neuroleptics has made it necessary that the epidemiology of TD be revisited, with the expectation that the newer drugs will provide new insights into its pathophysiology. Tardive dyskinesia therefore continues to present itself as a ‘natural’ experiment to study the pathophysiology of movement disorders. Its careful study may also provide a window on the underlying neurobiology of schizophrenia. Longitudinal studies of patients who receive atypical neuroleptics as their primary treatment from the first episode of psychosis onwards are necessary to assess the incidence of TD with these drugs. The current hypotheses for its pathogenesis need further empirical support and refinement so that they can be translated into treatment strategies. The subsyndromes of TD should be investigated further for their risk factors and pharmacological properties. More antipsychotic drugs need to be developed that are devoid of extrapyramidal side-effects and carry little or no risk of producing an irreversible neurological disorder.
Acknowledgements
The author is grateful to Julian Trollor, John McGrath and Jenny Bergen for comments on earlier drafts, and to Wanda Schinke for manuscript preparation.