
Abstract
Zirconolite (CaZrTi2O7) has been identified as a candidate ceramic wasteform for the immobilisation and disposal of Pu inventories, for which there is no foreseen future use. Here, we provide an overview of relevant zirconolite solid solution chemistry with respect to Ce, U and Pu incorporation, alongside a summary of the available literature on zirconolite aqueous durability. The zirconolite phase may accommodate a wide variety of tri- and tetravalent actinide and rare-earth dopants through isovalent and heterovalent solid solution, e.g. CaZr1–xPu x Ti2O7 or Ca1–xPuxZrTi2–2xFe2xO7. The progressive incorporation of actinides within the zirconolite-2M parent structure is accommodated through the formation of zirconolite polytypoids, such as zirconolite-4M or 3T, depending on the choice of substitution regime and processing route. A variety of standardised durability tests have demonstrated that the zirconolite phase exhibits exceptional chemical durability, with release rates of constituent elements typically <10−5 gm−2·d−1. Further work is required to understand the extent to which polytype formation and surrogate choice influence the dissolution behaviour of zirconolite wasteforms.
Introduction
The resurgence of nuclear power, as a driver towards cleaner energy production, will necessitate the implementation of advanced spent fuel management strategy, and development of advanced nuclear materials capable of safely conditioning highly radioactive waste [1-3]. After nuclear fuel is removed from a reactor, nation states have the option to chemically recover a significant portion of the fissile inventory, or treat the fuel as waste for disposal. These fuel cycle options are considered closed or open respectively; the unit operations associated with these are illustrated in Figure 1. For many years the U.K. has operated a closed fuel cycle, in which a PUREX (plutonium-uranium-reduction-extraction) reprocessing step is implemented, with the primary motive to recover U/Pu from spent fuel. In the PUREX process, nuclear fuel pins are stripped of cladding and dissolved in 9M HNO3; the aqueous nitric solution is then contacted with tri-butylphosphate (TBP). U6+ and Pu4+ form TBP complexes and are extracted to the organic phase; U6+ and Pu4+ are converted to oxides and calcined before storage. The remaining aqueous nitrate solution is comprised predominantly of high fission products and metalloids (Cs, I, Sr, As, Nd, Pd, Pr, Eu, La, Gd), minor actinides (Cm, Am, Np, Th), corrosion products (Mn, Ni, Cr), and entrained U/Pu. This effluent is referred to as high level liquid waste (HLLW) and is stored on the Sellafield site, before blending and calcination, prior thermal conditioning. The HLLW remains highly radioactive due to the long halflife of certain elements (e.g. t1/2237Np = 2.1 × 106 y, t1/2129I = 1.57 × 107 y). The current baseline thermal treatment for HLLW is vitrification in alkali borosilicate glass. In the vitrification process, HLLW is calcined and melted with glass forming additives, allowing complete dissolution of waste species into the vitrified network via incorporation into the glass forming structure; incorporation as network modifiers; and incorporation by encapsulation [4]. Although borosilicate glasses can incorporate a wide variety of elements, and vitrification is a well-established process that remains relatively insensitive to variations in feedstock chemistry, it is not the optimal choice for waste streams consisting of high actinide fractions, such as waste PuO2. Actinides have exhibited low solubility in borosilicate glass matrices, alongside leach rates that are considerably higher than alternate wasteforms such as crystalline ceramics. The Pu4+ solubility in the French R7T7 glass has been limited at around 1.5 wt-% [5]. Several notable publications have indicated the solubility of plutonium can be increased to 4 wt-% when reduced to the Pu(III) species [57]. A series of borosilicate glasses containing 1 wt-% PuO2 were fabricated Wellman et al. in order to elucidate the effect of self-irradiation on the elemental dissolution of the glass phase. Although it was determined that the release rate of Pu into the extraction phase was insensitive to dose rate (measured by 238Pu/239Pu ratio), temperature, and pH, was of the order 10−3 gm−2d−1 [8]. The development of SYNROC technology (synthetic-rock) in the 1980s has led to development of alternative wasteforms for nuclear waste based on ceramic systems [9]. The SYNROC formulation comprises an assemblage of chemically durable titanate crystalline phases (zirconolite, hollandite, perovskite, pyrochlore), based on natural mineral hosts that have demonstrated resistance to weathering on geological timescales; these can act as dedicated hosts for specific elements via accommodation in specific lattice sites in the host phase, providing a marked increase in solubility and chemical durability. The host phase for actinides is zirconolite – nominally CaZrTi2O7. The aim of this work is to provide an extensive literature review into the suitability of the zirconolite phase as a host for PuO2. An assessment of the current UK situation regarding Pu will first be outlined, followed by a critical assessment of zirconolite as a ceramic host phase for Pu.
Illustration of unit operations associated with open and closed nuclear fuel cycles. Orange arrows indicate closed fuel cycle operations, while blue arrows represent those for an open fuel cycle (© IOP Publishing. Reproduced with permission. All rights reserved. [10]).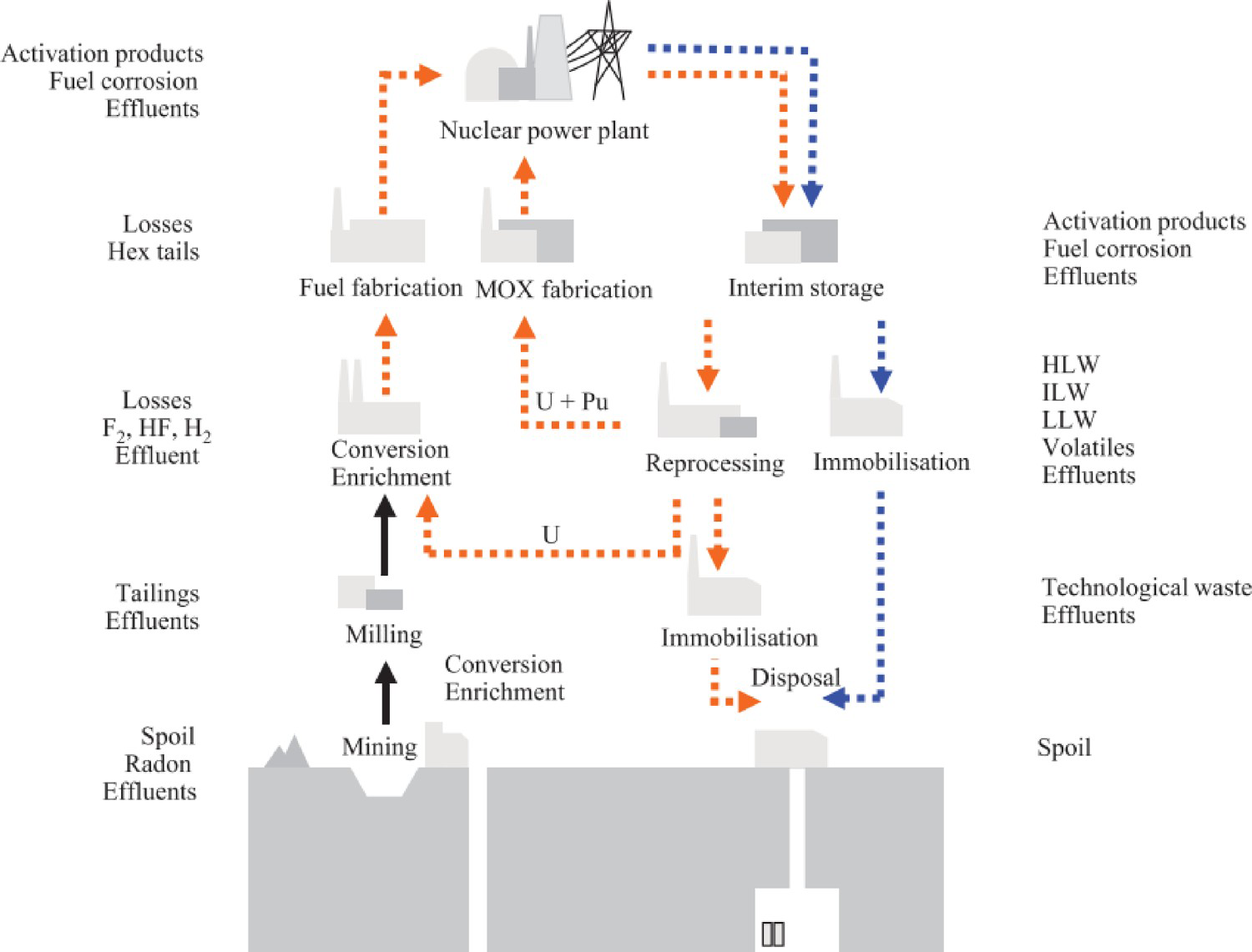
Actinide immobilisation in ceramic materials
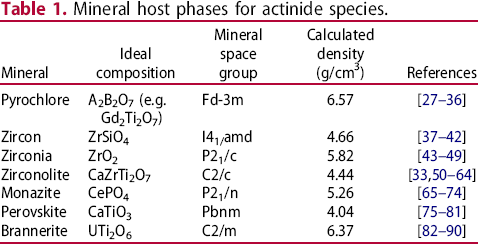
Owing to their relatively long half-lives and radiotoxicity, radionuclides must be separated from the biosphere and permanently disposed. The current intended disposal route for many countries is deep geological disposal, in an engineered repository known as a geological disposal facility (GDF). A GDF utilises the multibarrier concept, wherein a series of engineered barriers are constructed to prevent the egress of radionuclides until the radioactive output of the waste has decayed to levels comparable to the original uranium ore from which uranium fuel is derived, see Figure 2. The primary containment for radionuclides in this scenario is known as the wasteform, a passively safe material designed to prevent the release of radionuclides. Proposed wasteforms include cementitious, glass, ceramic, and glass-ceramic composite materials. Comprehensive analyses of these materials for nuclear waste applications are provided elsewhere [4,11-20]. In the context of the immobilisation of actinides, e.g. Pu, ceramic materials are considered to offer performance, including waste-loading and aqueous durability, when compared to cement-based systems, typically used for encapsulation of intermediate level wastes (ILW) and borosilicate glasses used for high level waste (HLW) immobilisation [12,16,19,21,22]. There have been many notable publications investigating potential single phase and multiphase ceramic wasteforms for the immobilisation of actinides, a selection of proposed host matrices for actinides is given in Table 1. Titanate and zirconate minerals have been particularly well-studied as a result of their excellent resistance to chemical alteration, and relatively high degree of resistance to radiation induced amorphisation [2326]. Actinide incorporation in ceramic phases is achieved by allowing the waste component to be readily accepted into solid solution in the host lattice, either by direct substitution or partial incorporation with an appropriate charge compensation mechanism. Generally, the choice of solid solution mechanism is dictated by the relative ionic radii of the radionuclide and host cation site, and accessible valence states. For example, the zirconolite structure may accept Pu4+ in solid solution via homovalent substitution for Zr4+, i.e. CaZr1–xPu
x
Ti2O7, or by a coupled substitution if Pu4+ is substituted for Ca2+, with a secondary lower valence cation included to maintain charge balance, i.e. Ca1–xPu
x
ZrTi2–xMg
x
O7.
Illustration of timescales necessary for geological disposal (© IOP Publishing. Reproduced with permission. All rights reserved [10]). Mineral host phases for actinide species.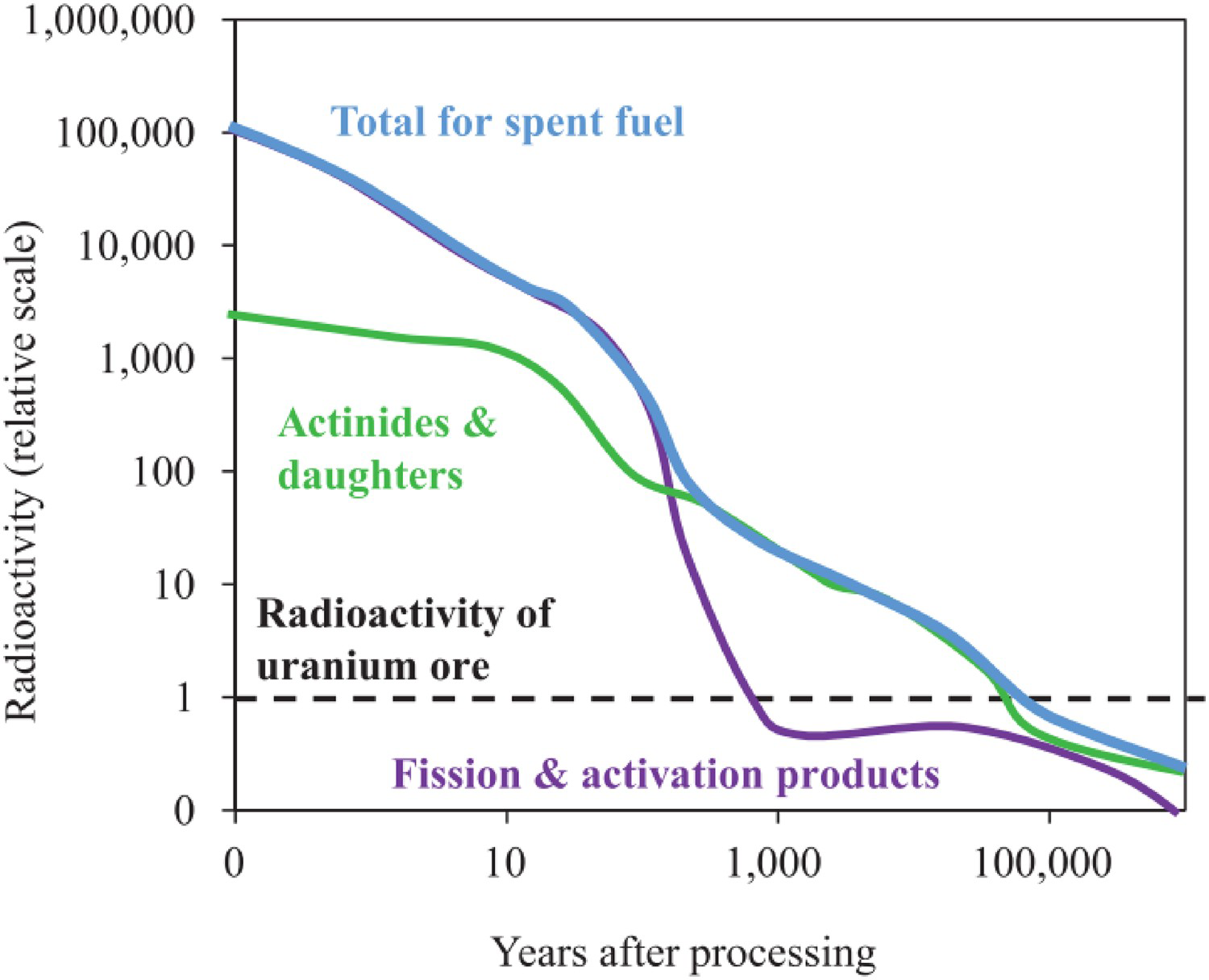
The design and implementation of ceramic actinide wasteforms is contingent on the following criteria:
Zirconolite ceramics for Pu immobilisation
Crystal structure of zirconolite
Zirconolite, ideal composition CaZrTi2O7 (ρ = 4.44 g·cm−3, Z = 8, space group C2/c) is a relatively rare accessory mineral that has been located in a variety of terrestrial geologies, with a demonstrated affinity for, but not limited to, U4+, Th4+, Ce3+4+, Al3+, Pu3+/4+, Gd3+ and Hf4+. Confidence in the zirconolite wasteform to successfully act as a host for actinides is largely underpinned through the existence of nature specimens, which have demonstrated excellent resistance to natural weathering effects over geological timescales, with several specimens found to retain significant portions of their primordial actinide inventories (∼ 20 wt-% U/Th) [33, 91-94]. The ideal zirconolite structure is an anion-deficient fluorite superstructure, and is considered to be a derivative of the pyrochlore family of minerals, with approximate formulation (Ca,Na,Ce,Th)2(Nb,Ta,Ti)2O6(OH,F), however this often generalised to A2B2O7 (Z = 8, space group Fd-3m) where A is typically some trivalent REE3+ species and B4+ = Ti, Zr [30,95,96]. During the development of the SYNROC wasteform, zirconolite was included as the host phase for actinides, due to its high aqueous durability [26,97100]. Accordingly, a significant body of work has since been undertaken in order to determine the solubility of a wide array of actinide and rare-earth cation species within the zirconolite framework, particularly Ce and U as surrogates for Pu. The ideal zirconolite unit cell is comprised of planes of corner sharing CaO8 and ZrO7 polyhedra, interleaved by hexagonal tungsten bronze (HTB) type layers along (001). Ti4+ is distributed across three distinct sites in the HTB plane, two of which are TiO6 octahedra, and one of which is a 50% statistically occupied TiO5, giving rise to trigonal biprymidal coordination [101]. In this idealised structural description (see Figure 3), cation and HTB layers are integrated 1:1 along (001), related by a 180° rotation along the c* axis. Owing to this two-layer repeat, stoichiometric CaZrTi2O7 is commonly referred to as zirconolite-2M, with reference to the two layer lamellar monoclinic motifs comprising the unit cell. The zirconolite-2M polytype has been since been demonstrated to form over the compositional range CaZr
x
Ti3–xO7 for 0.83 ≤ x ≤ 1.33, indicating considerable flexibility with regards to [Ti]/[Zr] ratio [102]. The distribution of Ti across cation sites in zirconolite has also been shown to be controlled as a function of sample preparation temperature [52]. Zirconolite also exhibits a number of crystallographically distinct polytype structures, the formation of which is observed to be controlled by the chosen substitution regime and oxygen fugacity during synthesis. Zirconolite polytyoids are characterised by variation in stacking sequence of adjacent Ca/Zr and HTB layers, for example, the zirconolite-4M structure was solved by Coelho et al. as a four layer repeating sequence, comprised of alternating zirconolite-2M and pyrochlore-type layers, resulting in a doubling of the unit cell along the c-axis, retaining monoclinic symmetry [103]. Extensive substitution of Pu within the Ca2+ site, facilitated by co-substitution of Fe3+ was reported by Gilbert et al. to produce the trigonal zirconolite-3T variation (space group P3121) [104]. Polytypes adopting three and six-layer orthorhombic symmetry have also been reported, but detailed structural solutions are lacking [64]. Ca2+ and Zr4+ sites are of particular interest as both have been shown to readily accept a range of actinide and rare-earth elements [53,55,56,64,78,105108]. Extensive solubility of Mg2+, Al3+, Ti3+, Fe3+, and Nb5+ species within the Ti4+ site has also been demonstrated, with the view to charge balance substitutions which do not exhibit isovalence across the structure, e.g. the accommodation of Pu within the Ca2+ site could be achieved by the co-substitution of Al3+ via Ca1–xPu
x
ZrTi2–2xAl2xO7, assuming all Pu is present as Pu4+ [55,98,104,109]. The simultaneous substitution of trivalent species within both Ca2+ and Zr4+, negating the need for charge balancing species, has also been demonstrated [110113]. As the manipulation of Pu in wasteform development trials is not often possible, due to the stringent handling requirements associated with radiotoxicity and the handling of fissile material, the remainder of this review will aim to provide a comprehensive discussion of Ce and U surrogate incorporation in zirconolite.
Crystal structure of zirconolite-2M (CaZrTi2O7).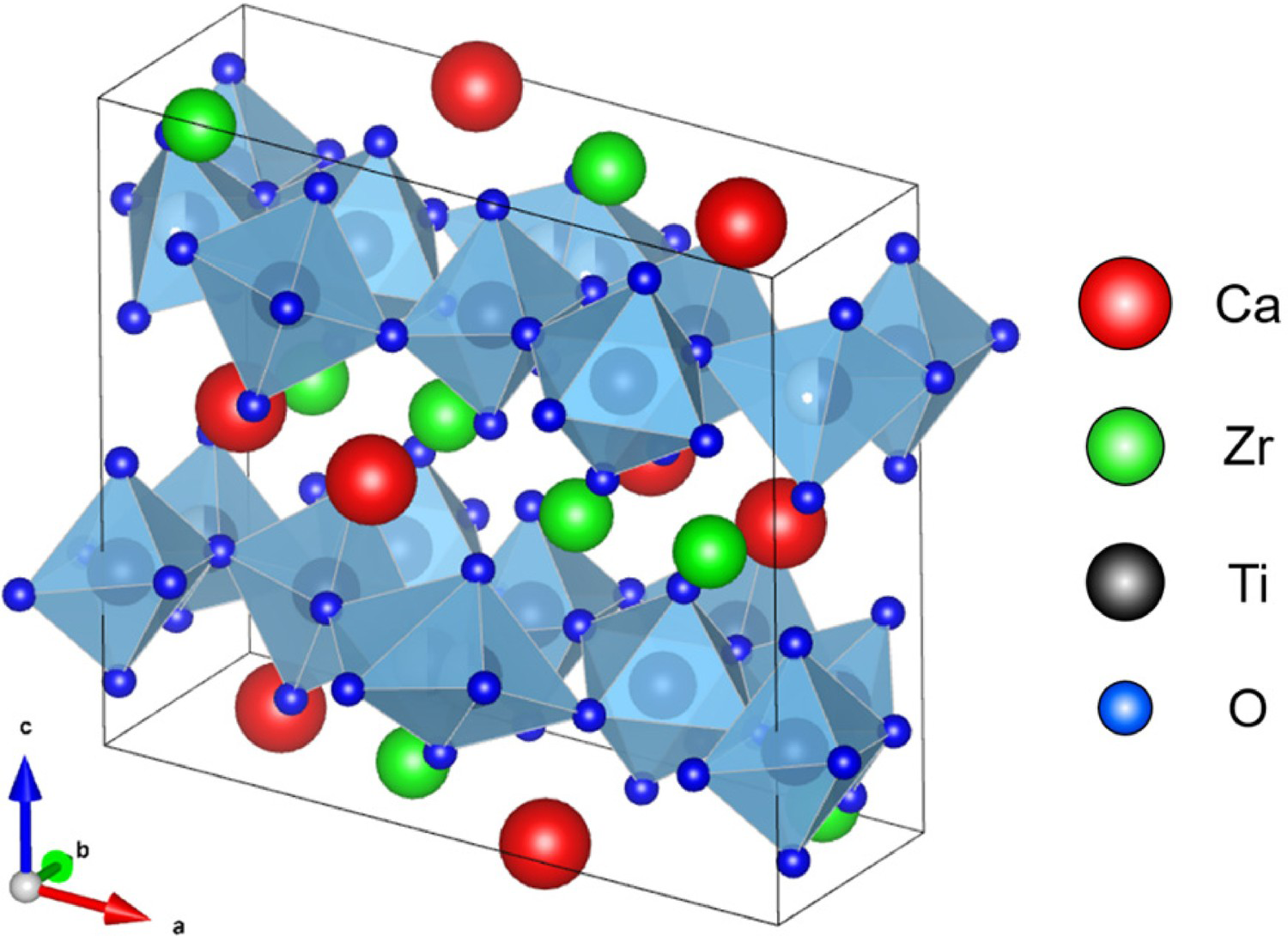
Incorporation of Ce in zirconolite
Incorporation of Ce within Zr4+ site
The formation of zirconolite with Ce targeted in the Zr4+ site (CaZr0.8Ce0.2Ti2O7) was attempted by Begg and Vance [114]. Two distinct zirconolite phases were observed to form (zirconolite-2M and zirconolite-4M) alongside a secondary perovskite phase with considerable incorporated Ce (calculated stoichiometry Ca0.72Ce0.24Zr0.02Ti1.03O3). Ce LIII edge X-ray absorption spectra (XANES) confirmed the presence of 35% Ce3+, despite extended sintering under oxidising conditions. Further annealing in air at lower temperatures resulted in total conversion to Ce4+, inferring the excess positive charge in the zirconolite-2M species may be self-compensated by cation vacancies. This work was complemented by Blackburn et al. with the CaZr1–xCe x Ti2O7 system synthesised under both oxidising and reducing conditions [115]. It was determined that sintering at 1350°C under oxidising conditions produced a transformation to zirconolite-4M above x = 0.20, although failed to stabilise the entire Ce inventory as Ce4+, with 28% Ce manifested as Ce3+. Synthesising the solid solution under a 5% H2/N2 mixture promoted the formation of a Ce-rich perovskite phase, bypassing the formation of zirconolite-4M at the result of uniform Ce3+ speciation. Blackburn et al. also fabricated a sample corresponding to nominal composition CaZr0.80Ce0.20Ti2O7 by hot isostatic pressing (1300°C dwell temperature, maintained for 4 h with isostatic pressure 100 MPa) [116]. The bulk matrix was of near theoretical density, with zirconolite-2M comprising ∼ 81 wt-% of the phase assemblage, with the remainder comprised of zirconolite-4M and Ce-perovskite. Similar phase fields were reported in the CaZr1–xCe x Ti2O7 system by Clark et al. utilising both conventional sintering and spark plasma sintering (SPS) [56]. Accommodation of Ce (x ≥ 0.20) resulted in the formation of zirconolite-4M. EDS measurements confirmed that greater Ce content was concentrated in the zirconolite-4M phase, with a secondary Ce-bearing perovskite phase produced due to partial Ce3+ speciation. At extensive targeted Ce-substitution (x = 0.5), zirconolite-4M was present at high concentration (96 wt-%). The reducing conditions imposed by the SPS process promoted Ce4+ reduction to Ce3+, destabilising zirconolite-4M in favour of Ce-rich perovskite. The CaZr1–xCe x Ti2O7 solid solution was extended by Li et al. [51]. A structural transformation from the zirconolite-2M to the zirconolite-4M polytype was observed, alongside accessory perovskite, in line with previous observations. Further attempted incorporation of Ce within the Zr site yielded a cubic pyrochlore phase (ideal composition CaCeTi2O7), and the total solubility limit of Ce was determined to be approximately x = 0.80. X-ray photoelectron spectroscopy (XPS) analysis confirmed the ratio of Ce3+/Ce4+ to decrease from 1.24 at low concentration (x = 0.20) to 0.45 at maximum Ce concentration (x = 1.00).
Incorporation of Ce in Ca2+ site with charge compensators
Ce substitutions into zirconolite were undertaken by Begg and Vance, with the successful synthesis of Ca0.8Ce0.2ZrTi1.6Al0.4O7 by sintering in air at 1400°C [114]. XANES measurements confirmed that Ce4+ was present at only 70%. Minor Al2O3 was also formed, hence Al3+ charge compensation was only therefore sufficient for 70% Ce4+. Furthermore, re-formulation targeting Ce3+ on the Ca2+ site, producing a stoichiometry of Ca0.8Ce0.2ZrTi1.8Al0.2O7 formed a single phase zirconolite, in which Ce3+ was accommodated entirely on the Ca2+ site. Further work by Begg et al. determined that zirconolite could undergo self-charge compensation via the formation of cation vacancies and trivalent Ti3+, under oxidising and reducing conditions respectively [80]. This was demonstrated by the synthesis of single phase Ca0.9Ce0.1ZrTi2O7 under both oxidising and reducing conditions (i.e. the incorporation of Ce3+ and Ce4+ on the Ca2+ site), displaying an apparent excess charge. Vance et al. confirmed that Ce3+ may be overwhelmingly incorporated into the Ca2+ site, when reacting under reducing conditions, forming a single phase up to 0.3 f.u., i.e. Ca0.70Ce0.30ZrTi1.70Al0.30O7 [117]. Similar results were obtained by Kaur et al. targeting Ca0.80Ce0.20ZrTi1.80Al0.20O7, with synthesis under air at 1400°C. Ce was observed to fully accommodate within the zirconolite-2M phase, with XPS analysis confirming the formation of 75% Ce3+, with sufficient Al3+ to charge balance [118]. Similar processing techniques were utilised by Pöml et al. targeting Ce4+ and Al3+ accommodation; specimens were sintered at 1400°C for 2 d [119]. Near single phase specimens with nominal composition Ca0.85Ce0.15ZrTi1.70Al0.30O7 and Ca0.87Ce0.13ZrTi1.74Al0.36O7 were fabricated by solid state synthesis, with EELS data confirming the formation of 80% Ce3+, without a change in phase assemblage. A complementary investigation of the efficacy of Cr3+ as a charge balancing species was reported by Blackburn et al., with the Ca1–xCe x ZrTi2–2xCr2xO7 solid solution synthesised in air at 1350°C [120]. Single phase specimens were produced in the interval 0.05 ≤ x ≤ 0.20, after which Cr2O3, CeO2 and a Ce-rich perovskite phase were observed in the microstructure, although when targeting x = 0.35 the zirconolite-2M phase remained present at ∼ 94 wt-%. Selected area electron diffraction confirmed that the 2M polytype structure was maintained throughout the phase evolution. Ce L3 XANES data confirmed partial reduction to Ce3+ varying between 15% and 27%, similar to previous studies.
Incorporation of U/Pu in zirconolite
Incorporation of U/Pu in Zr4+ site
During development of SYNROC technology, U and Pu were observed to partition overwhelmingly into the zirconolite phase, although explicit discussion of zirconolite polytype formations were not provided [99,121,122]. Although more recent attempts to fabricate titanate phase assemblages by hot isostatic pressing, targeting a high zirconolite fraction, report agreeable data [123,124], it is necessary to discuss the structural effects of U/Pu incorporation within the zirconolite phase in isolation. Kesson et al. reported the solid solution limits of U within the zirconolite structure, targeting substitution on the Zr4+ site [98]. Compositions corresponding to CaZr0.75U0.25Ti2O7, CaZr0.50U0.50Ti2O7 were fabricated by hot pressing at 1400°C; zirconolite and pyrochlore were yielded in each instance. Attempting to partition a greater amount of U within the Zr4+ site promoted the formation of the pyrochlore phase, alongside secondary (Ti,Zr,U)O2 solid solutions. Initial work by Vance et al. reported the incorporation of 0.5 f.u. of U within the Zr4+ site, targeting CaZr0.5U0.5Ti2O7, by hot pressing at 1250°C, followed by a 1400°C heat treatment under reducing conditions, with a view to stabilise U4+ [106]. Further substitution of U appeared to stabilise the pyrochlore structure, while a minor U-containing rutile was also formed in all concentrations. The crystal chemistry of the uranium pyrochlore (CaU4+Ti2O7 – betafite) is discussed elsewhere [125]. The CaZr0.80U0.20Ti2O7 composition was also produced by HIP (1300°C, 100 MPa) by Blackburn et al. yielding a significant fraction of zirconolite-2M (∼ 97 wt-%), alongside minor unincorporated UO2 and a (Zr,U)O2 solid solution [116]. A detailed investigation of U4+ accommodation in the zirconolite CaZr1–xU x Ti2O7 system was performed by Vance et al. in 2002 [55]. Synthesis of the solid solution under inert conditions produced single phase zirconolite-2M when targeting x = 0.10, with the zirconolite-4M phase preferred above x = 0.20. Further U4+ concentration increased the relative yield of the zirconolite-4M phase, with extensive incorporation (∼0.5 f.u. U4+) producing the U-pyrochlore phase, in line with previous data. Oxidation of samples corresponding to CaZr0.9U0.1Ti2O7 and CaZr0.8U0.2Ti2O7 promoted the formation of U5+, causing the destabilisation of the zirconolite-4M phase with respect to the zirconolite-2M structure. Shrivastava, Kumar and Sharma have provided an excellent structural refinement of the zirconolite-2M CaZr0.95U0.05Ti2O7 and CaZr0.90U0.10Ti2O7 compositions [126]. More recently, the CaZr1–xU x Ti2O7+x solid solution was prepared by Subramani et al., with all compositions prepared in air at 1400°C [127]. Interestingly, zirconolite-2M was observed to form as a single phase at each level of targeted U concentration, with the average oxidation state of U close to U6+ in all instances, as determined by U L3 XANES. The incorporation of Pu4+ within the Zr4+ site appears to yield broadly similar results to the corresponding U solid solution, demonstrating the efficacy of U4+ as a structural surrogate under inert conditions. Structural effects of Pu4+ substitution within the Zr4+ site in zirconolite were investigated by Begg et al. [105]. When sintering in air, CaZr0.9Pu0.1Ti2O7 was successfully synthesised as a single phase, with a secondary Pu-rich zirconolite-4M phase formed above x = 0.20. The yield of zirconolite-4M was increased with further Pu4+ substitution; a pyrochlore phase was observed to crystallise for the phase corresponding to CaZr0.60Pu0.40Ti2O7. Annealing specimens under reducing conditions (3.5% H2/N2 – 1200°C) promoted the formation of Pu3+, similar to Ce, however this reduction mechanism is not available for U, highlighting a caveat for the deployment of U as a Pu surrogate under reducing conditions. The accompanying increase in ionic radius was considered to cause the destabilisation of the zirconolite-4M phase, with respect to zirconolite-2M, stabilising a deleterious perovskite phase in agreement with cerium doped specimens, in which targeting Zr4+ substitution for Ce3+ promoted the formation of perovskite. Complementary results were obtained by Vance et al. targeting CaZr0.50Pu0.50Ti2O7; hot pressing the sample (i.e. reducing conditions) yielded approximately 50 wt-% Pu-perovskite attributed to the formation of Pu3+ [106]. Further work by Begg et al. confirmed that hot pressing the CaZr0.80Pu0.20Ti2O7 composition failed to produce a single phase product, with only 50 wt-% zirconolite yield attributed to uniform Pu3+ speciation [26,128]. Nevertheless, annealing the composition in air at 1300°C produced significant modifications to the phase assemblage, yielding ∼ 80 wt-% zirconolite, alongside a Pu-rich pyrochlore phase, eliminating the perovskite phase.
Incorporation of U/Pu in Ca2+ site with charge compensators
A selection of zirconolites targeting U4+ incorporation within the Ca1–xU x ZrTi2–2xAl2xO7 system were synthesised by Vance et al. with a view to further extend U4+ solubility without structural transformation to the closely related pyrochlore phase [117]. The solubility limit was determined to be 0.3 ≤ x ≤ 0.4, after which further accommodation of U4+ resulted in the formation of UO2–ZrO2 solid solutions, and minor Al2O3. Further work demonstrated that imposing reducing conditions by hot pressing yielded a secondary brannerite phase, at appreciable quantity [106]. A more systematic approach was later undertaken, in which both Al3+ and Mg2+ were targeted on the Ti4+ site in order to provide sufficient charge compensation for both U4+ and U5+ [55]. It was determined that single phase zirconolite-2M was formed in both instances when targeting values x = 0.1, 0.2, after which secondary formation of UTi2O6, ZrO2 and UO2 phases was observed. The use of Mg2+ to charge balance approximately 26 wt-% U4+ in the zirconolite structure was reported by Kesson et al. with U apparently distributed between Ca2+ and Zr4+ sites [98]. The targeted zirconolite stoichiometry was not reported. Pu-bearing zirconolites targeting Ca2+ substitution without charge compensation (i.e. Ca0.9Pu0.1HfTi2O7 – Zr4+ entirely replaced by Hf4+) were prepared by Begg, Vance and Conradson [78]. Pu was accommodated across both Ca2+ and Hf4+ sites, contrary to design; annealing under reducing conditions did not stabilise the formation of a secondary perovskite phase, despite 80% reduction to Pu3+. Deschanels et al. confirmed the synthesis of single phase Ca0.87Pu0.13ZrTi1.73Al0.30O7, exhibiting the zirconolite-2M structure, when targeting Pu4+ [53]. A similar composition was synthesised by Vance et al.: Ca0.80Pu0.20ZrTi1.80Al0.20O7, configured to accomodate Pu3+ [106]. While conventional sintering yielded a single phase zirconolite specimen, hot pressing at 1250°C yielded a secondary perovskite phase. Begg et al. determined the influence of processing atmosphere in the formulation of Pu3+ and Pu4+ doped zirconolites targeting Ca0.80Pu0.20HfTi1.80Al0.20O7 and Ca0.80Pu0.20HfTi1.60Al0.40O7, respectively, with Hf in place of Zr [128]. Targeting Ca0.80Pu0.20HfTi1.80Al0.20O7 while sintering under a 3.5% H2/N2 mixture promoted uniform Pu3+ speciation, yielding approximately 88% zirconolite, alongside a Pu-perovskite phase. Sintering under air was sufficient to allow uniform Pu4+ valence, with ∼ 96% zirconolite yield. Synthesis of the Ca0.80Pu0.20HfTi1.60Al0.40O7 composition, requiring Pu4+, failed to yield above 77% zirconolite when sintered under reducing conditions, whereas 94% zirconolite yield was produced in air. Fe3+ was deployed as a charge compensator in Pu-doped zirconolite, investigated by Gilbert et al. targeting Ca1–xPu x ZrTi2–2xFe2xO7 [104]. A transformation from zirconolite-2M to zirconolite-3T was reported for compositions above x = 0.20, with separated PuO2 identified above x = 0.40.
Chemical durability of zirconolite
Examples of dissolution methodologies used to ascertain the durability of candidate nuclear wasteforms.

As zirconolite comprised a significant portion of many SYNROC variations, as the primary actinide-bearing phase, a measure of zirconolite durability was obtained through evaluation of SYNROC dissolution studies. Early work by Oversby and co-workers demonstrated the comparative success of SYNROC with respect to borosilicate glasses for the immobilisation of HLW. Samples of SYNROC and PNL-76-68 waste glass (borosilicate glass with 33% simulated HLW) were studied on 0.5 g discs with distilled water at 85 and 200°C [130]. Release rates of 1.4 and 8.9 gm−2·d−1 were reported for the PNL-76-68 glass at 85 and 200°C respectively, while the upper limit for the SYNROC leach rate was determined to be several orders of magnitude lower, at <0.005 gm−2·d−1. Tests were repeated with powdered samples in the 100–200 μm size fraction to accelerate leaching; it was determined that the leach rates of U were between a factor of 5–9 lower for SYNROC at 200°C. In 1981, specimens of SYNROC (comprising ∼ 35% zirconolite) was crystallised by hot pressing with the addition of 20% HLW calcine readily accepted into solid solution with the constituent phases [122]. A more comprehensive investigation was performed in this instance, with variations in both temperature and leaching duration, allowing improved comparability between ceramic and glass phases for HLW immobilisation. With respect to leaching rates, SYNROC specimens were observed to decrease by approximately two orders of magnitude between 10–30 days, whereas borosilicate specimens were observed to dissolve at a consistent rate (see Figure 4). At 95°C the leachability of U from the SYNROC specimen was determined to be smaller than for the borosilicate glass by a factor of 100,000. A specimen of SYNROC with added 10 wt-% HLW (0.62 wt-% 239Pu) was hot pressed, and subject to MCC-1 durability testing by Smith et al. with extensive leach periods of 52 d and 2472 d at 70°C in deionised water, alongside carbonate and silicate leachates [100]. After 52 d, 239Pu release rates for carbonate and silicate leachates were an order of magnitude greater than for deionised water (10−4, 10−4, and 10−5 gm−2·d−1, respectively). However, after 2472 d, the normalised leach rate of 239Pu dropped to 10−6 gm−2·d−1 in deionised water, demonstrating exceptional durability over extensive time periods. Further work by Smith et al. on the SYNROC wasteform was performed using an MCC-2 dissolution assessment using deionised water, at 150°C [131].
Bulk leach rate of SYNROC specimen in comparison to borosilicate glass (This article was published in A. E. Ringwood et al., “Immobilization of High-Level Nuclear Reactor Wastes in Synroc: A Current Appraisal,” Nucl. Waste Manag., vol. 2, pp. 287–305, 1981, copyright Elsevier [122]).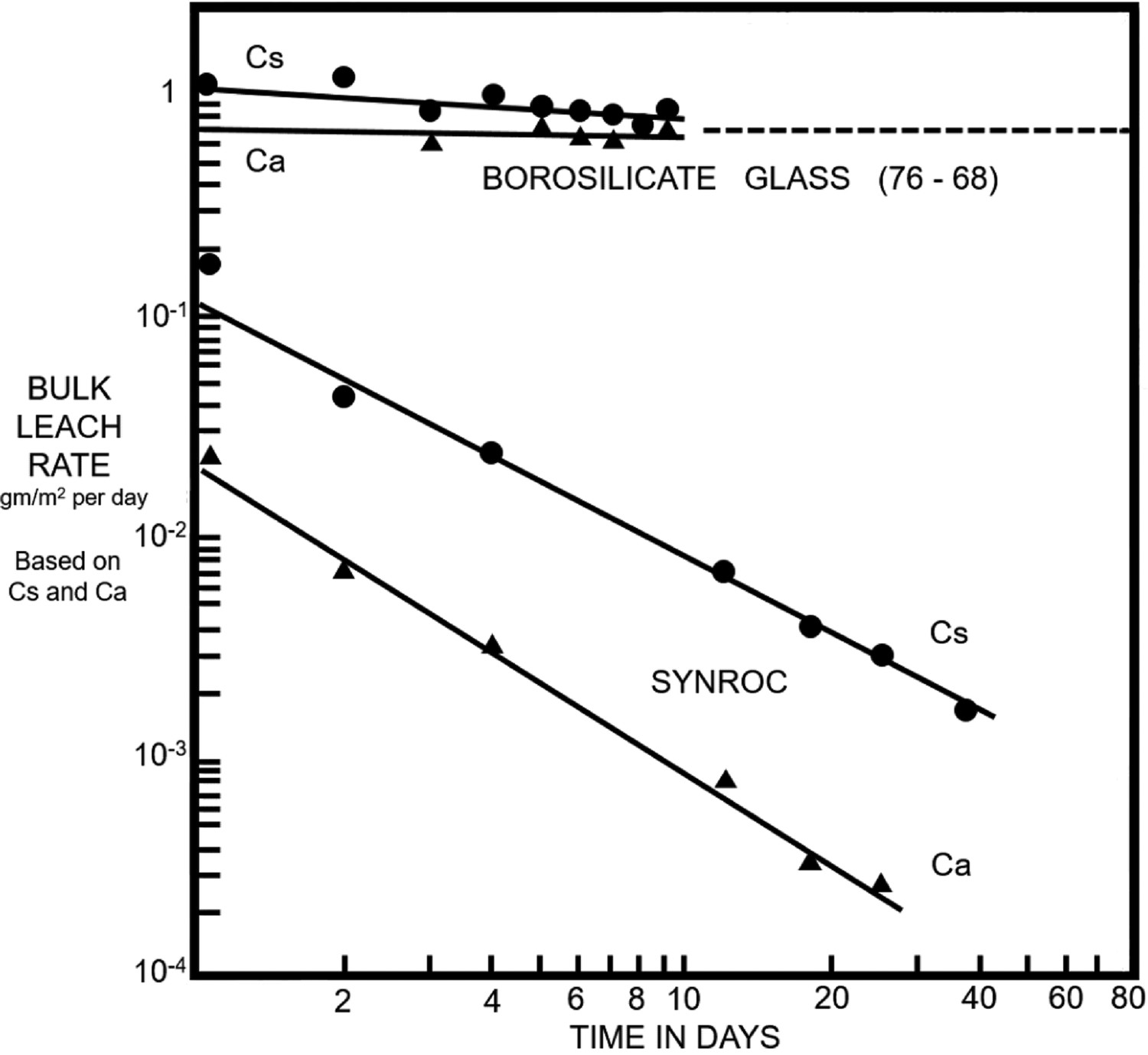
Samples were loaded with 10 wt-% simulated HLW, and after 532 d, SEM analysis determined that surface layer grains of major phases exhibited no corrosion. It was confirmed that after 337 d, 0.0045% of the original Ce inventory had been released to the leachate. These early investigations were considered sufficient to demonstrate the suitability of ceramic phases for the immobilisation of nuclear wastes. However, incongruent dissolution was a key consideration, as each phase in the SYNROC assemblage did not present comparable leach rates. A variety of zirconolite-rich and pyrochlore compositions containing approximately 12 wt-% Pu (alongside 15 wt-% Hf/Gd) were synthesised by Hart et al. with release rates of Pu were measured by MCC-1 analysis at 90°C [23]. 7-day release rates for Pu were measured to be between 10−5 and 10−6 gm−2d−1 after 300 d, with similar release rates observed for Hf. These data provide a significant contribution towards underpinning the safety case for geological disposal of Pu in the zirconolite wasteform, as the congruent release of Pu and neutron poisons is essential towards suppressing post-closure criticality. A zirconolite-rich titanate assemblage containing U/Th and Pu was synthesised by hot isostatic pressing by Zhang et al. yielding a zirconolite phase fraction of approximately 89 vol.-%. A secondary hollandite phase (nominally BaAl2Ti6O16) and UTi2O6 brannerite phase (∼ 2 vol.-%) were also formed, yet U and Th were incorporated overwhelmingly into zirconolite, with minor uptake in the brannerite phase [123]. Pu was successfully localised within the zirconolite phase, co-substituted with Gd and Hf as neutron poisons. Both samples were subject to MCC-1 durability testing in deionised H2O at 90°C, however experimental framework was modified such that the leachate was replaced after 7 and 35 days, and the test was extended to 84 days. The normalised leach rates of key elements are summarised in Figure 5. After 35 days, a normalised release rate of 10−5 gm−2·d−1 for Pu was measured, with similar accompanying release rates of included neutron poison species. Zirconolite-rich ceramics were synthesised by Zhang et al. by self-propagating synthesis, targeting a nominal CaZr1–xCe
x
Ti2O7 composition, with a view to assess the chemical durability of the composition corresponding to CaZr0.7Ce0.3Ti2O7 by a monolithic MCC-1 test in deionised H2O [132]. The normalised leach rate of Ce over the 42 d period was measured to be 2.26 × 10−6 gm−2·d−1, demonstrating exceptional resistance to alteration, despite the formation of a secondary Ce-bearing perovskite phase, accounting for approximately 35 wt-% of the phase assemblage. Meng et al. attempted the accommodation of Ce within both Ca2+ and Zr4+ sites, i.e. Ca1–xZr1–xCe2xTi2O7, anticipating the auto-reduction of Ce species may provide self-charge balance across the zirconolite structure [133]. Samples were synthesised by conventional solid state reaction and the durability was measured by PCT-B methodology. Despite a low normalised mass loss of Ce ranging between 10−6 and 10−7 gm−2·d−1, an increased order of magnitude for Ce was observed for compositions containing a greater accompanying portion of perovskite. Cerium was applied as a surrogate for Pu targeting the zirconolite phase CaZr1–xCe
x
Ti2O7 system by Wen et al. synthesised by a solid state route [134]. MCC-1 leaching was performed on the sample pertaining to composition Ca0.76Zr0.64Ce0.48Ti2.03O7; the normalised leach rate for Ce was measured to be approximately 2.3 × 10−4 gm−2·d−1 up to 10 days of exposure; after 28 days the leach rate decreased by two orders of magnitude to 2.3 × 10−6 gm−2·d−1. A Ce-bearing perovskite with composition Ca0.84Ce0.10Ti1.03O3 was also stabilised, which may have attributed to a greater release fraction of Ce, hence it is unlikely that the release rates for Ce could be attributed solely to the zirconolite phase. As the formation of perovskite is a common secondary phase in the fabrication of zirconolite wasteforms, formulations must be tailored such that the accompanying fraction is minimalised. This is an issue commonly associated with cerium surrogacy, as the tendency of Ce to undergo partial reduction, despite reaction under oxidising conditions, to form Ce3+ is commonly observed to promote the formation of a Ce-bearing perovskite [51,56,60,114]. Perovskite (nominally CaTiO3) is present as a major constituent of the SYNROC assemblage, as a host for Sr2+, despite markedly lower resistance to alteration with respect to the zirconolite phase. The relative leach rates of perovskite and zirconolite, with respect to pH dependence, was elucidated by McGlinn et al. subsequent to the demonstration of SYNROC for HLW immobilisation, as a precursor towards to implementation of single phase ceramic wasteforms. The formation of TiO2 (anatase) at 90°C on the surface of perovskite specimens was observed at low and neutral pH levels, indicating hydrothermal alteration of the perovskite phase; no evidence of dissolution was observed with XRD or SEM techniques for the zirconolite phase [135]. It has since been proposed that precipitation of TiO2 in subsequent dissolution trials was attributed to the dissociation of CaTiO3 [81,136].
Normalised release rates of key elements for zirconolite-rich titanate assemblage processed by HIP (Reprinted from Y. Zhang et al., “Zirconolite-rich titanate ceramics for immobilisation of actinides - Waste form/HIP can interactions and chemical durability,” J. Nucl. Mater., vol. 395, pp. 69–74, 2009, with permission from Elsevier [123]).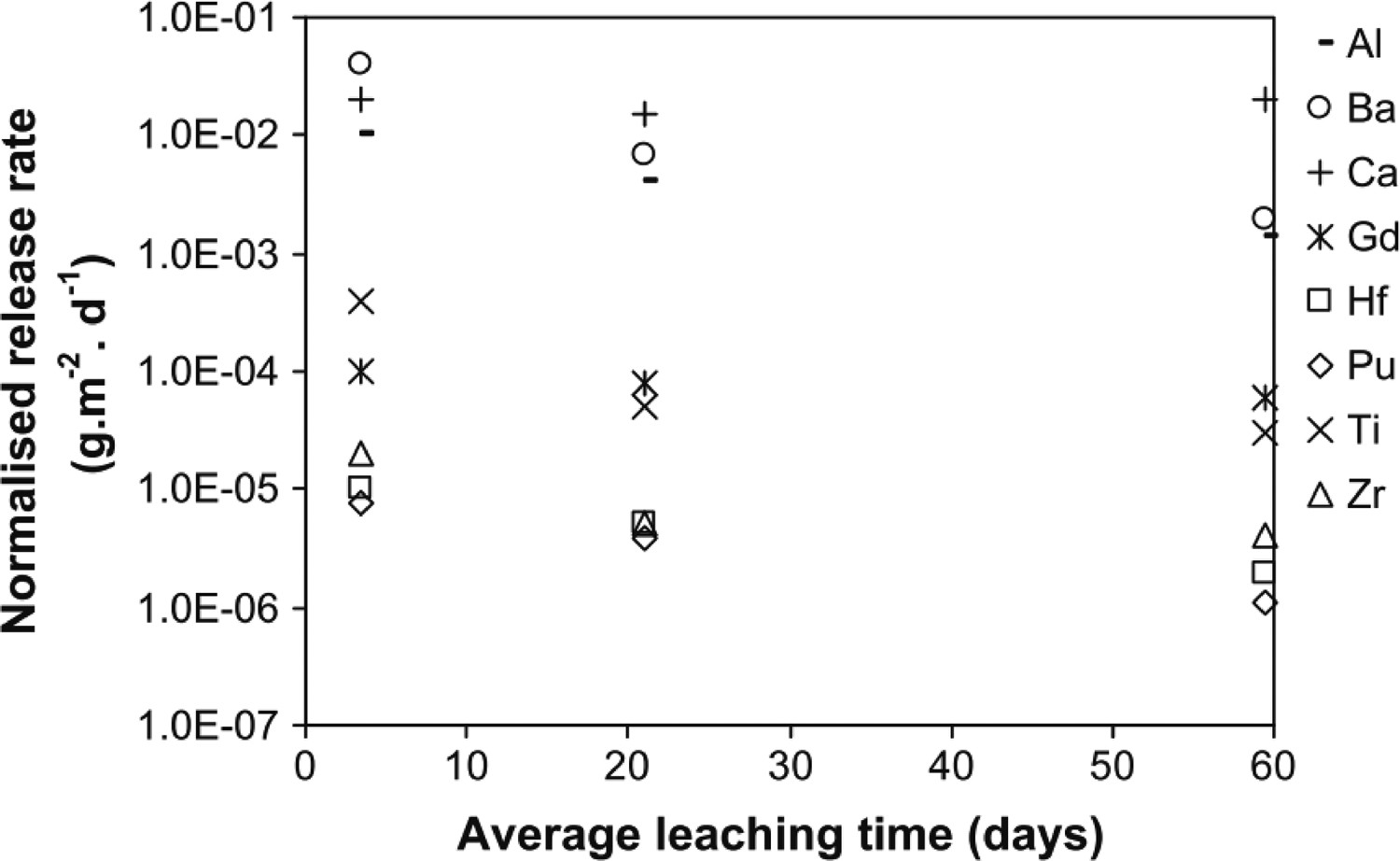
Potential zirconoliteformulations for Pu must incorporate sufficient buffer phases such that the formation of Pu-perovskite is not thermodynamically or kinetically favourable, as this may exacerbate the rate of Pu extraction from the wasteform in the geological disposal environment. Begg et al. synthesised perovskite incorporated with Pu under oxidising and reducing conditions, with a view to elucidate the substitution mechanism of Pu4+ and Pu3+, respectively, within CaTiO3 [78]. Attempting to form Ca0.9Pu0.1Ti1.9Al0.1O3 under reducing conditions yielded two perovskite phases, accounting for an 80% reduction to the trivalent species. It was determined that Pu4+ can be substituted into the perovskite structure as a single phase in excess, with Ti vacancies allowing for charge neutrality to be preserved. Moreover, the perovskite phase was observed to accommodate a considerable amount of Pu3+ and Pu4+ without the addition of charge balancing species. Further work by Begg et al. confirmed that the release of Pu from CaTiO3 under acidic conditions was indiscriminate of Pu valence [79]. More recently, the chemical durability of near single phase zirconolite has been elucidated. A sample of zirconolite with nominal composition Ca0.80Nd0.20ZrTi1.80Al0.20O7 was synthesised by hydrolysis of alkoxide nitrate precursor material, before calcination and sintering at 1400°C in air. An excess of 1.5 wt-% Ti/Zr oxides were added to discourage perovskite formation; a dense microstructure of zirconolite-2M with minor ZrTiO4 was yielded at 0.5%. MCC-2 analysis at 150°C in deionised water reported a normalised mass loss of Nd between 10−3 and 10−4 over 80 d. Single phase zirconolite doped with 0.15 f.u. of Nd (trivalent actinide surrogate) were recently fabricated by Cai et al. Subsequent to confirmation of a single product in the Ca1–xZr1–xGd2xTi2O7 system, the sample with nominal composition Ca0.925Zr0.925Gd0.15Ti2O7 was selected for PCT analysis. The durability was measured in pH 5, 7, and 9 at 90°C. The single phase specimen demonstrated a normalised Nd leach rate of 3.13 × 10−5 gm−2·d−1 after 42 d, proving insensitive to extraction under varying pH [137]. Recent work by Zhang et al. utilised Gd3+ and Hf4+ as trivalent and tetravalent actinide surrogates, with a view to eliminate the issues typically caused by Ce reduction, targeting the composition Ca1–xHf1–xGd2xTi2O7 [138]. Release rates of 4.72 × 10−7 and 1.59 × 10−8 gm−2·d−1 were measured for Gd and Hf respectively.
Critical gap analysis
Zirconolite-rich wasteforms satisfy many of the design criteria commonly applied in the design of nuclear wasteform materials, not least high chemical durability and moderate wasteloading. Nevertheless, an in-depth review of the literature has identified several gaps that have not been conclusively addressed:
Conclusions
The zirconolite wasteform is currently a candidate host phase for Pu, should U.K. Government policy adopt a strategy of immobilisation and disposal of the bulk inventory. Zirconolite chemistry permits the acceptance of a wide variety of REE3+/Ac4+ (U4+, Pu3+/4+, Th4+, Ce3+/4+, Sm3+) within solid solution, alongside a considerable selection of charge balancing species (Al3+, Mg2+, Fe3+, Nb5+) for the formation of heterovalent compositions, and neutron poison species (Gd3+, Hf4+). A review of the literature has identified that the incorporation of Pu4+ may be best achieved by homovalent substitution for Zr4+, and/or heterovalent substitution for Ca2+, with the addition of a suitable charge balancing species such as Al3+ or Mg2+. In the case of the former mechanism, the substitution would likely be facilitated by the formation of the polytypical zirconolite-4M structure above 0.15 f.u. Pu4+, a hybrid intergrowth between the nominal CaZrTi2O7 aristotype and the CaPuTi2O7 pyrochlore-structured phase. However, it is apparent that the 2M polytype may be stabilised over a wider solid solution range when favouring substitution for Ca2+, with appropriate charge compensation. The formation of deleterious secondary phases such as perovskite is shown to be dependent on the method of substitution utilised and the valence of the surrogate element, which is in turn is controlled by processing conditions rather than crystallographic design. A survey of the literature confirms zirconolite exhibits exceptional chemical durability with normalised release rates for constituent elements typically of the order 10−5 to 10−8 gm−2·d−1 under simulated disposal conditions.
Footnotes
Acknowledgements
We acknowledge financial support from the Nuclear Decommissioning Authority (NDA) and EPSRC under grant numbers EP/S01019X/1, EP/S011935/1, EP/S020659/1, EP/P013600/1 and EP/R511754/1. This research utilised the HADES/MIDAS facility at the University of Sheffield established with financial support from EPSRC and BEIS [![]() ].
].
Disclosure statement
No potential conflict of interest was reported by the author(s).