
Abstract
The effect of amyloid β-peptide (β AP), which can have both neurotrophic or neurotoxic effects on neurons and has been implicated in the pathogenesis of Alzheimer's disease (AD), was studied on astrocytes using primary cultures and astrocyte cell lines (rat C6 glioma, human 1321NI astrocytoma cells). The cultures were exposed to 0.0005–50 μg/ml) βAP fragments 1–40, 25–35, 31–35, or 40–41 (control) for 24 hr. Some of the fragments were maintained at 37°C for 48 hr to induce aggregation and some of the cell cultures were pretreated with the differentiating agent dBcAMP before the experiments. The astrocyte responses were evaluated for lysosome activity (neutral red assay) and levels of structural proteins, glial fibrillary acidic protein, vimentin, and S-100, which are altered in the dystrophic plaques with associated astrogliosis in AD. The cells frequently responded with biphasic responses, with initial (low-dose) activation-type responses (i.e., increases of indicator compared to controls), before reductions with altered morphology (increased branching of cells) at higher concentrations. However, cell death (with EC50 values) was not observed, even at the maximum concentrations of βAP fragments. The findings suggest that the astrocytes have a relatively high resistance against the β AP toxicity.
INTRODUCTION
The primary morphological signs of Alzheimer's disease (AD) are the loss of neurons associated with the presence of neuritic plaques, neurofibrillary tangles, and cerebrovascular amyloidosis. The plaques are composed of a central amyloid core surrounded by aggregates of dystrophic neurites, synaptic boutons with degenerating mitochondria, lysosomes, and activated astrocytes (Terry et al., 1964). The major component of the amyloid is a 4.2 KDa peptide termed β-peptide or A4 peptide (Glenner and Wong, 1984; Masters et al., 1985). The amyloid fibrils form a twisted, β-pleated sheet (Glenner and Wong, 1984). The amyloid β-peptide (βAP) is a derived component from at least three separate amyloid precursor fractions, termed βAPP695, βAPP770, which differ by limited numbers of amino acid sequences. The β-amyloid precursors are constitutively processed by at least two enzymatic pathways (Esch et al., 1990; Golde et al., 1992), one of which appears to be within the endosomal—lysosomal pathway, producing intact βAP (Haass et al., 1992).
Approximately half of the βAP is normally buried in the neuronal cell membranes with the other section (N-terminus) projecting extracellularly. The βAP that accumulates as insoluble aggregates (plaques) in the AD brain is a 40–42 amino acid segment of the precursor proteins (Glenner and Wong, 1984). Although the precursor and its genetic control (located on chromosome 21 in man) are highly conserved in many different species (Goldgaber et al., 1987), the reasons for the apparently aberrant accumulation in the AD patient are not yet clear, with both abnormal post-translational modifications and processing of the cell-surface moiety implicated (Robakis et al., 1987). The βAP may be liberated from cells by enzymatic cleavage at the N-terminus (Seubert et al., 1992) and/or other processing mechanisms (Golde et al., 1992). The protein is also present in the cerebrospinal fluid of normal individuals and in the media of neuronal cultures, which might suggest a normal physiological function, possibly associated with the tachykinin receptor (Yankner et al., 1990).
A major area of study in AD pathogenesis, in addition to the processing of βAP, is the involvement of the substance in neurotoxic changes. These have been investigated in a range of studies of cultures of primary neurons and clonal lines. The studies have shown that βAP can be directly neurotoxic (Yankner et al., 1990; Pike et al., 1991; Mattson et al., 1993; Borchelt et al., 1997; Weidemann et al., 1997) and can also render neurons vulnerable to excitotoxic or metabolic insults (Koh et al., 1990; Mattson et al., 1993). Altered calcium homeostasis (Mattson, 1994; Mattson et al., 1992, 1993; Weiss et al., 1994) and generation of reactive free radicals (Behl et al., 1992; Hensley et al., 1994; Muller et al., 1994) are implicated in the mechanism(s) of βAP toxicity. The neurotoxicity may be ameliorated by antioxidants (Behl et al., 1992; Goodman et al., 1994; Guo et al., 1998). However, the toxic process is complicated by the wide variations that occur with different states of aggregation, solubility, and dose of the βAP molecule. Freshly solubilized synthetic βAP (1–40) or newly solubilized βAP 1–42 are nontoxic and may in fact promote neurite outgrowth in hippocampal cultures (Pike et al., 1991). However, with increasing periods of incubation at 37°C (termed aging) the peptide adopts aggregated forms which become progressively toxic, causing rapid cell death after 24 hrs incubation (Pike et al., 1991; Burdick et al., 1992). Furthermore, certain peptide sequences within the βAP 1–42 (e.g., β25–35) exhibit a rapid aggregation and potent neurotoxicity immediately following solubilization (Yankner et al., 1990). In addition the toxicity of βAP is dose dependent, with low doses being protective and the high doses toxic (Kaltschmidt et al., 1999). Low-dose pretreatment may also protect against subsequent high dose (Kaltschmidt et al., 1999). A key mechanistic factor shown in these studies was the activation of the transcription factor NF-kB by free radicals. The activation profile of NF-kB was inverted U-shaped, with low-dose stimulation followed by inhibition at higher dose, similar to the toxicity curve of βAP (Kaltschmidt et al., 1999). The biphasic form of the βAP responses conforms to the stereotyped hormetic response, as discussed by Calabrese (2001).
In addition to the neuronal toxicity by βAP in AD, several studies have been made on astrocytes. Reactive astrocytes have a major association with the plaques (Duffy et al., 1980; Schecter et al., 1981). The reactive cells, in contrast to normal (nonreactive) astrocytes, may contain significant levels of βAP and its mRNA (Bahmanyar et al., 1987; Card et al., 1988; Siman et al., 1989; Tanzi, 1989). Cultured rat astrocytes undergo reactive morphological changes, with increased stellation, increased GFAP and S-100 immunostaining (Meske et al., 1998), enhanced cytokine and NO production (Hu et al., 1998; Hu and VanEldik, 1999), K+ and Cl− channel activities (Jalonen et al., 1997), and glutamate transporter capacity (Abe and Misawa, 2003) when exposed to βAP fragments. Cultured rat astrocytes can also respond to cytotoxic changes and cell death (Brera et al., 2000). However, although both activation and toxicity changes have been reported it is not clear whether they are parts of a graded, biphasic continuum, similar to the responses that occur to a wide range of other toxic compounds (see Pentreath and Slamon, 2000).
The present study was undertaken to further assess the reactive and toxic responses of primary astrocyte cultures and related cell lines to βAP. The aims were to evaluate and compare the responses of βAP fragments 1–40, 25–35, and 31–35 in primary, C6 glioma, and 1321NI human astrocytoma cells using quantitative assays for lysosomes and the cytoskeletal proteins GFAP, vimentin, and S-100, which are sensitive markers of the reactive and toxic responses to a range of toxic substances in vitro (see Pentreath, 1999).
MATERIALS AND METHODS
Cell Culture
Primary astrocytes were prepared from 1–3-day-old Sprague-Dawley rats. Cortical tissues were dissected into trypsin-EDTA solution (0.5 g/l) trypsin in BME-HEPES with 0.2 g/l tetrasodium EDTA; Gibco, UK). The cells were triturated with sterile pipettes, centrifuged (100g for 10 min), the supernatant removed, and 2.5 ml trypsin inhibitor solution (Sigma; 0.1% w/v in BME-HEPES) plus DNAase (100 μl of a 1 mg/ml solution; Sigma) was added. Trituration was repeated followed by centrifugation at 100g for 5 min. The supernatant was poured off and 2 ml growth medium was added; this consisted of DMEM (Dulbecco's modified Eagle's medium 4,500 mg/l glucose; Gibco). This was supplemented with 10% (v/v) heat-activated fetal bovine serum (FBS; Seralab, UK) and 0.5% gentamycin sulphate and 1% nystatin (with respect to total volume; culture media from Gibco, antibiotics and an-timycotics from Sigma). The FBS employed was certified to contain less than 6 units/ml bacterial endotoxin. The cultures were used without further purification procedures. Astrocytes prepared by this procedure are more than 90% pure as established by immunostaining for GFAP (see Cookson and Pentreath, 1994).
The C6 rat glial tumour cell line and human 1321NI astrocytoma cell lines were obtained from the European Collection of Animal Cell Cultures, Porton Down, United Kingdom. Both cell lines were subcultured as follows. The cells were initially maintained in 250 cm3 flasks (Nunclon), then washed three times in calcium and magnesium free Hank's Balanced Salt Solution (HBSS; Gibco). Cells were then trypsinized with 1 ml per flask of trypsin-EDTA (1X) solution in HBSS. Following detachment, 9 ml of growth medium (DMEM, 10% FBS, 0.5% gentamycin sulphate) was added to prevent further action of the trypsin-EDTA. A split ratio of 1:3–1:10 was employed and the media was made up to 20 ml in each flask. Subculturing was repeated every 7 days.
Further details of the cell culturing procedures for astrocytes in toxicity testing have been extensively reported (see, e.g., Cookson et al., 1994, 1995; Mead and Pentreath, 1998a; Cookson, 1999).
Toxicity Testing in Multiwell Plates
Two types of test system were employed. The cells were grown either in 96-well microtitre plates (Greiner) or on coverslips in 24-well multidishes from Nunclon. For direct plating (96 well) 100 μl of a 0.025 mg/ml solution of poly-L-lysine (Sigma) was added to each well, left for 10 min, washed off with sterile (Millipore) water and allowed to dry. Coverslips were dipped in ethanol, flamed, then placed in wells (24-well plate), to which was added 100 μl of a 0.1 mg/ml solution of poly-L-lysine (Sigma), then washed and dried.
Cells were seeded in each system at a density of 5 × 105/ml and counted by haemocytometer before addition of the cell suspensions.
Addition of Dibutyrylcyclic AMP (dBcAMP)
Treatment of cultured astrocytes (primary or cell lines) with CAMP is a widely employed procedure that induces differentiation of the cells. Levels of glial fibrillary acidic protein (GFAP) are increased and the cells develop a more branched form (Fedoroff et al., 1987). Other aspects of cell phenotype are also changed (often upregulated), with several characteristics of reactive astrocytes (Eddleston and Mucke, 1993). In the context of toxicity evaluation the changes provide a valuable adjunct to the untreated cells, because the cells may become more responsive, levels of toxicity markers (e.g., GFAP) are increased, and the increased protective capacities can shed light on mechanisms of toxicity. This is especially relevant for the cell lines (Mead and Pentreath, 1998b).
When the cells reached confluency (2–3 days), some of the cell lines were treated with 0.5 mM N6, 2′-0-dibutyryladenosine 3′:5′ cyclic monophosphate (dBcAMP; Sigma) for 48 hrs before toxicity measurements.
Indicators of Toxicity
Neutral Red (NR) Assay
NR selectively targets lysosomes in living cells by binding to anionic and/or phosphate groups of the lysosomal matrix (Borenfreund and Puerner, 1984). It can be extracted from the cells and gives a linear relationship of cell viability (in terms of lysosome activity and numbers). For this a 0.1% w/v solution of NR dye (Sigma) in phosphate buffered saline (PBS) was filtered (0.2 μm pore size) and 10 μl aliquots added to the media in the wells. The plates were incubated at 37°C for 3 hr. The media was replaced with 100 μl of acid alcohol (1% v/v acetic acid in 50% ethanol), which elutes NR from the viable cells. Absorbance was read at 550 nm on a plate reader. The mean absorbance for each concentration of compound was expressed as a percentage of the value obtained for the solvent blank and was plotted against the log of compound concentration.
Measurement of GFAP, Vimentin, and S-100 by ELISA
The cytoskeletal proteins were measured quantitavely by ELISA. The methods have been previously described (O'Callaghan, 1991; Cookson and Pentreath, 1994). Briefly, following incubation in the wells, cells were washed with PBS:ethanol. The cells were fixed in 100 μl ethanol at −20°C (30 min) and washed (three times) in PBS-CMG. To each well 100 μl of primary antibody (anti-GFAP, anti-S-100, or monoclonal antivimentin; all Sigma), diluted 1:500 with a solution of PBS-CMG, 1% bovine serum albumin (BSA; Sigma), and 0.1% Triton X-100 (Sigma) were added and the plates incubated for 1–2 days at 4°C. The cells were washed three times with a solution of PBS-CMG/0.1% Triton X-100. To each was added 100 μl rabbit antimouse IgG conjugated to alkaline phosphatase (Sigma) diluted 1:500 with PBS-CMG, 1% BSA, and 0.1% Triton X-100. For S-100 measurement the secondary antibody was goat antirabbit IgG conjugated to horseradish peroxidase diluted 1:5000 in PBS-CMG with 1% BSA and 0.1% Triton X-100. Cells were incubated with gentle shaking (2 hr, room temperature) and then washed five times with PBS-CMG/0.1% Triton X-100 and blotted dry. The plates were developed with 100 μl per well of paranitrophenylphos-phate solution (Sigma; 1 mg/ml in 10% diethanolamine buffer with 0.5 mM MgCl2, pH 9.8). The reaction was stopped after 10 min by the addition of 100 μl per well of 0.4 M NaOH. The amount of antigen per well was expressed as a percentage of the absorbance at 410 nm compared to the control (untreated) wells. Appropriate control wells (i.e., without either primary or secondary antibodies, substrate, or cells) were included in duplicate.
Treatment with AP
The cells were treated with βAP fragments 1–40, 25–35, 31–35, or 40–41 (as control), all purchased from Sigma. The treatments were started immediately after the cells had reached confluency or been treated with dBcAMP and were maintained for 24 hr.
The βAP fragment 1–40 was made up either fresh on the day of testing as a 1 mg/ml stock solution in sterile distilled water, or three days before testing in a 37°C incubator to age. Serial dilutions of both (from 50–0.0005 μg/ml) were made in DMEM containing 10% FBS and 0.5% gentamycin. The βAP fragments 25–35 and 31–35 were not aged and were made up fresh on the day of treatment. Serial dilutions were prepared as described earlier and all fragments were added to the astrocyte cultures at confluency.
Monolayer Morphology
The monolayer cultures were observed at intervals during the exposures with βAP by phase contrast microscopy.
Statistical Analysis
The values for a minimum of three sets of measurements for each βAP fragment and concentration were assessed by a paired student t-test.
RESULTS
The primary, C6, and 1321NI cells frequently responded with activation-type responses, with the aged βAP fragment 1–40 and fragment 25–35, recorded as increased in NR uptake, GFAP, vimentin, or S-100 at 24 hr (Table 1). The responses were also frequently nonlinear, with reduction in indicators at higher concentrations; however, cell death with EC50 values was rarely observed even at the maximum concentrations employed (50 μg/ml).
Activation in Primary, C6, and 1321N1 Astrocytes (% of Control) When Treated with βAP Fragment 1–40)
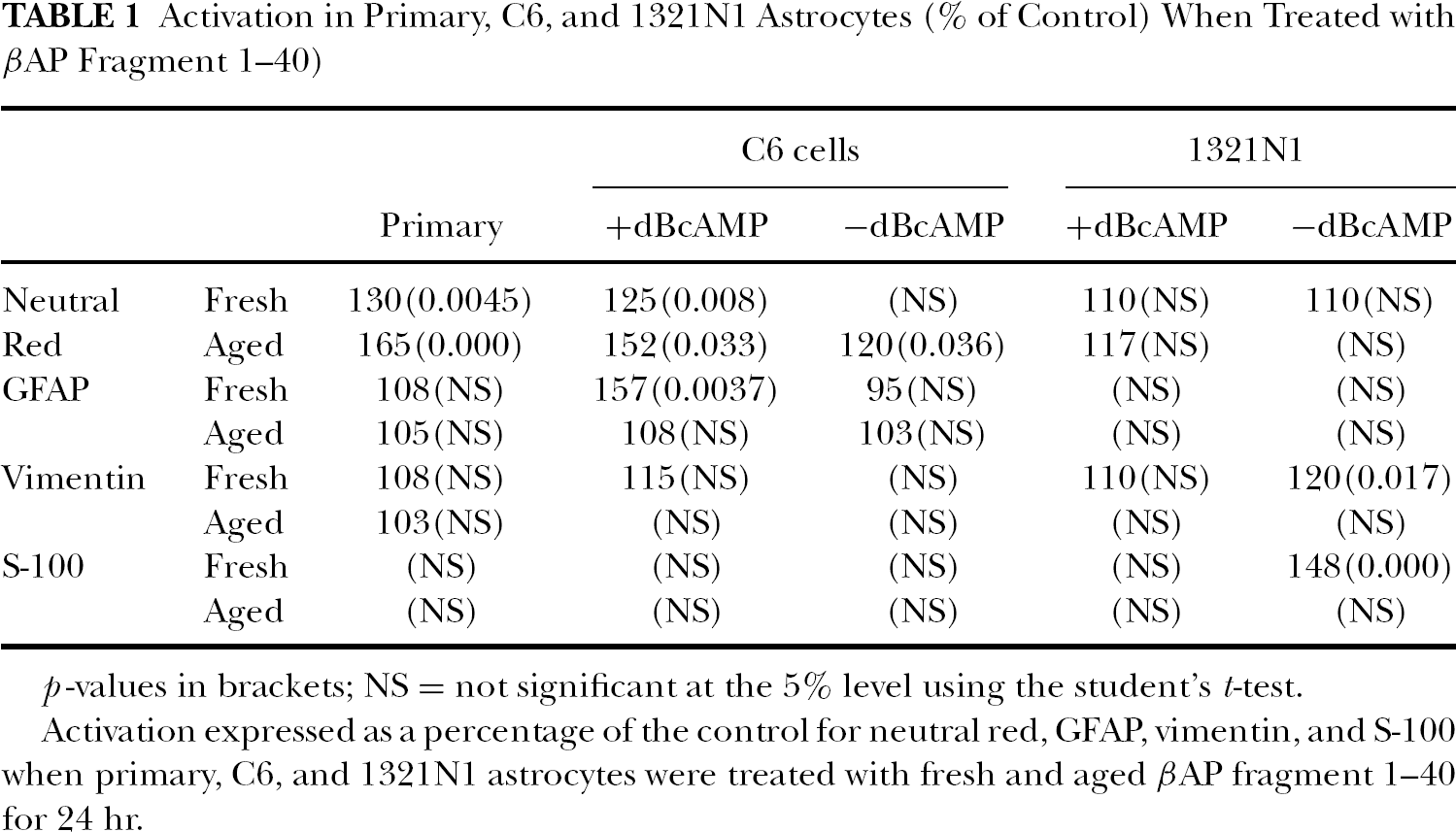
p-values in brackets; NS = not significant at the 5% level using the student's t-test.
Activation expressed as a percentage of the control for neutral red, GFAP, vimentin, and S-100 when primary, C6, and 1321N1 astrocytes were treated with fresh and aged βAP fragment 1–40 for 24 hr.
βAP Fragment 1–40
The primary astrocytes were activated by low concentrations of aged βAP (i.e., 0.0005–0.005 μg/ml), whereas fresh βAP caused activation at 50 μg/ml when assessed by NR uptake (Table 1). C6 cells also showed significant increases in NR uptake after dBcAMP treatment and the aged βAP fragment, whereas smaller responses occurred with fresh βAP and/or no dBcAMP treatment (Figure 1). The activation maxima were followed by reductions at higher concentrations (Figure 1). The 1321NI cells, on the other hand, showed only modest (not significant) biphasic changes with aged or fresh βAP 1–40, either with or without dBcAMP pretreatment (Table 1).

Effects of βAP fragments 1–40 on C6 glioma cells, either with or without dBcAMP treatment. Cells were exposed to the fresh or aged fragments for 24 hr and responses measured with neutral red. Each point is the mean of three replicates.
When evaluated with measurement of GFAP levels, small but nonsignificant nonlinear responses were recorded for the primary cells (Figure 2), but some relatively large increments, without inhibition at the highest concentrations employed, were recorded with the C6 cells and the fresh βAP (Figure 3). The 1321NI cells were generally unresponsive (Table 1). In relation to this several properties of GFAP in the 1321N1 cell line (for example, responses to dBcAMP) can differ significantly from the primary and C6 cells (see Cookson, 1999).

GFAP levels in primary astrocyte cultures following 24 hr exposure to fresh or aged βAP 1–40. Each point is the mean of seven replicates.

GFAP levels in C6 glioma cells following 24 hr treatment with fresh or aged βAP 1–40. Some of the cultures were treated with dBcAMP. Each point is the mean of three replicates.
Vimentin and S-100 levels also underwent modest, generally nonsignificant, changes in the different cell types with or without dBcAMP treatments (Table 1). For example, in the 1321NI cells the increases (maximum 20%) were not followed by cell death even with 50 μg/ml aged βAP (Figure 4).
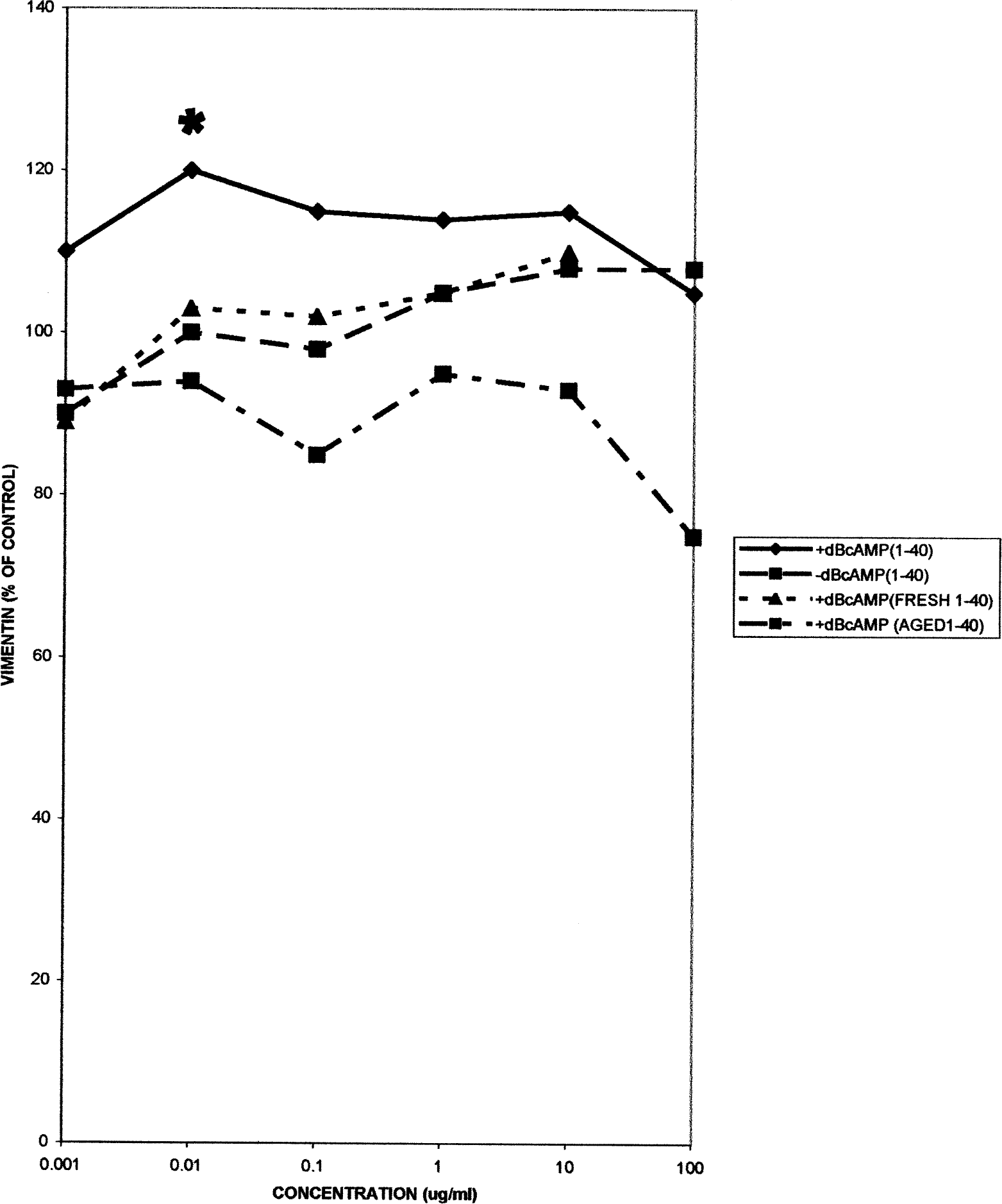
Vimentin levels in 1321N1 astrocytoma cells, either with or without dBcAMP treatment, following 24 hr exposure to fresh or aged βAP 1–40. Each point is the mean of seven replicates.
Examination of the monolayers by phase contrast showed progressive morphological changes in the primary cells and cell lines with increasing concentrations of βAP, which appeared to correspond with the quantitative measurements. These were altered fibrillar arrangements with changes in cell shape to more branched morphology, with increased gaps between the cells. However, despite the alterations of morphology there was no evidence of cell detachments or cell death, even at the highest concentrations of βAP employed.
βAP Fragments 25–35 and 31–35
Similar patterns of responses, with some modest biphasic alterations in indicator levels in the different cell types, were caused by the different fragments. The activation data are summarized in Table 2. However, many of the changes were not significant, and cell death did not take place even at the highest concentrations (50 μg/ml) of βAP fragments employed. Examples of the responses of the levels of vimentin and S-100 in the C6 cells are shown in Figures 5 and 6.
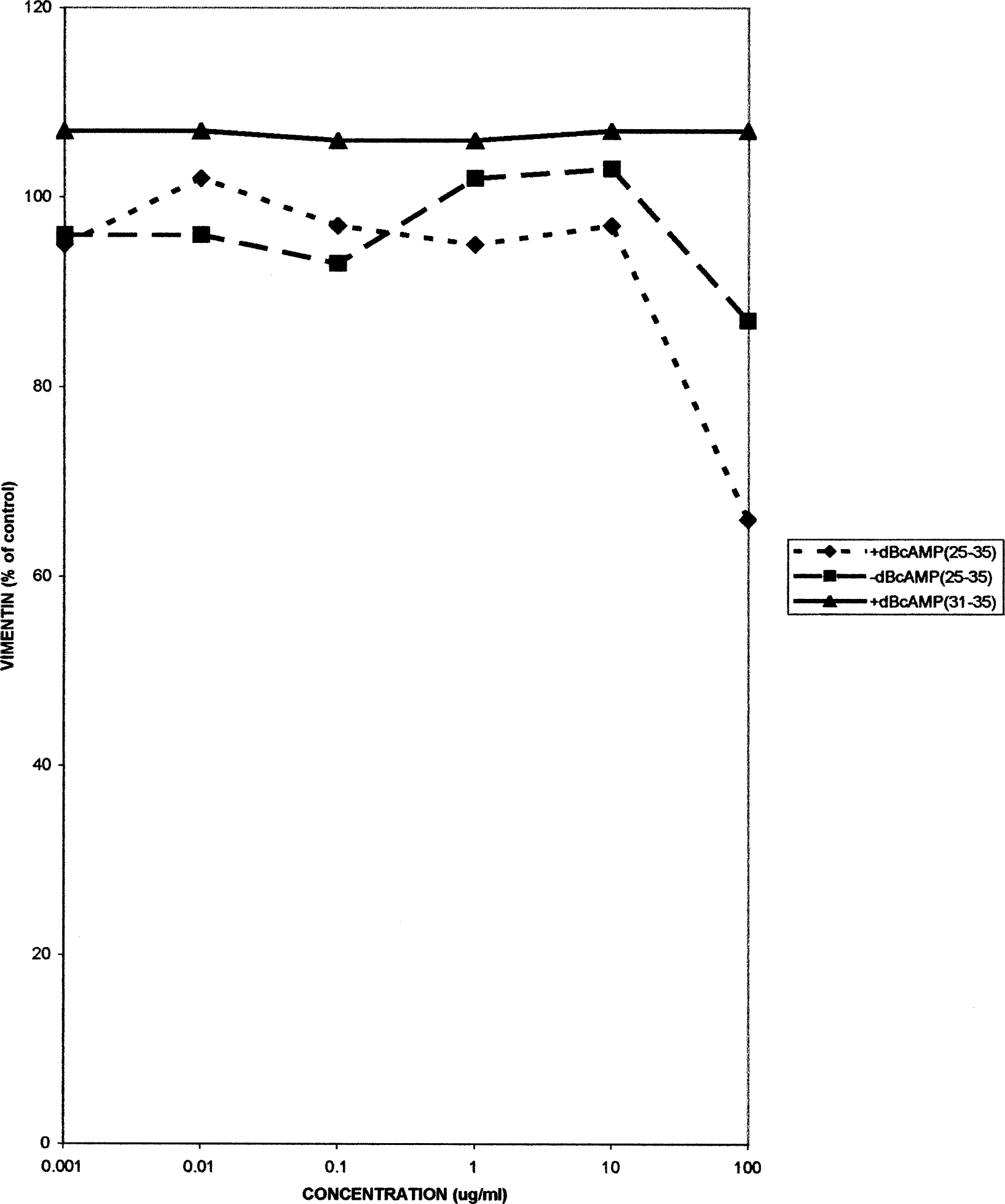
Vimentin levels in rat C6 glioma cells, either with or without dBcAMP treatment, following 24 hr treatment with βAP fragments 25–35 or 31–35. Each point is the mean of seven replicates.

S-100 levels in C6 glioma cells, with or without dBcAMP treatment, following 24 hr treatment with βAP fragments 25–35 or 31–35. Each point is the mean of seven replicates.
Activation in Primary, C6, and 1321N1 Astrocytes (% of Control) When Treated with βAP Fragment 25–35 and 31–35

p-values in brackets; NS = not significant at the 5% level using the student's t-test.
Activation expressed as a percentage of the control for neutral red, GFAP, vimentin, and S-100 when primary, C6, and 1321N1 astrocytes were treated with fresh and aged βAP fragment 1–40 for 24 hr.
Phase contrast microscopy showed altered morphology with increased branching of the cell processes after exposure to 50 μg/ml βAP 25–35. The changes were apparent after 2 hr exposure in each cell line, and progressed until 24 hrs when there appeared to be significant disruption of the cells, with gaps forming in the previously confluent cell layer. However, there did not appear to be any cell death (i.e., detachment). With βAP fragment 31–35 the changes were less marked, with no alterations in the 1321NI cells at 24 hr at a concentration of 50 μg/ml.
DISCUSSION
A range of toxicity studies of astrocytes have been made using cultures of primary, subcultured, or permanent cell lines. These have been extremely valuable in toxicity evaluation and understanding the mechanisms of action of a number of neurotoxic substances (see Pentreath and Slamon, 2000, for review). The common experimental procedure in the studies has been to expose the cultures to a range of concentrations of the toxic substances and assess the responses by different cytological and biochemical markers (e.g., Tiffany-Castiglioni et al., 1989; Cookson and Pentreath, 1994, 1996; Cookson et al., 1994, 1995; Cookson, 1999; see also Pentreath, 1999). The data concerning several features of toxicity, for example EC50 values and associated rankings of toxicants, are generally closely comparable to the in vivo studies, but additional novel mechanistic information on gliotoxicity has been obtained (see Pentreath, 1999). In particular it has been established that many of the toxic substances produce biphasic responses with the different endpoints, with the suprathreshold concentrations causing increases (i.e., activation) in endpoint values followed by reductions at higher concentrations. The suprathreshold concentration increases were frequently in the range 110–160% control values (see Figure 7). Mounting evidence shows that these responses are associated with the phenomenon of astrocyte activation (astrogliosis) and involve, in part, increased protective capacities against toxicity caused by oxidative damage (Pentreath and Slamon, 2000). The reductions at higher concentrations are associated with increasing toxic damage, which for many neurotoxicants cannot be compensated for and leads to cell death. The response patterns in most of these situations corresponds to the nonlinear, hormetic responses which appear common to multiple biological systems (see Calabrese and Baldwin, 2001).

Responses of cultured primary astrocytes and astrocyte cell lines to toxic substances. The curve depicts the generalized response derived from the study of a large number of different toxicants, assessed by different endpoints (including structural proteins, lysomal activity, and energy metabolism) over a range of concentrations (see Pentreath and Slamon, 2000). The shape, dose range, time course, and amplitude of the activation phase and the subsequent reductions vary with test substance, indicator/endpoint, and cell type. The different amyloid beta fragments studied in the present work frequently produced responses within the activation phase but did not cause cell death, even at high concentrations (i.e., reach 0% on the downward phase). There is growing evidence that the responses to the lower doses of toxicants, measured by different endpoints, reflect different types of insult/stress, which in turn initiate activation (i.e., upregulation) of different components of the cells' protective systems (Pentreath and Slamon, 2000).
The present findings are consistent with a fairly frequent biphasic toxicity dose-response for βAP and its 11-amino acid toxic fragment βAP 25–35 in cultures of primary astrocytes, C6 glioma, and 1321NI astrocytoma cells. Most of the treatments induced activation-type responses, with the aged βAP fragment 1–40 causing greater activation (increase in NR uptake) than fresh βAP fragment 1–40. However, none of the βAP fragments caused the marked toxic changes with EC50 values followed by cell death comparable to those described for a number of other chemical toxicants (see, e.g., Cookson and Pentreath, 1994; Cookson et al., 1994). A similar lack of overt toxicity has been reported by Brera et al. (2000), using the MTT assay as an indicator of mitochondrial activity, who found that neither βAP 25–35 nor βAP 1–40 caused more than 50% cell death in the astrocyte population after 24 hr. Evidence was presented that the limited toxicity was due to peroxide generation by βAP, but that the cells' high antioxidant capacities were sufficient to counteract the damage.
This contrasts with previous studies on neuronal cultures; for example, Pike et al. (1991) found that βAP 1–42 in the aggregated or “aged” form caused marked cell death of cultures of hippocampal neurons after 24 hr at concentrations of 10–50 μg/ml. Similarly, Yankner et al. (1990) have shown that βAP fragment 25–35 has a direct toxic effect on neurons in rat hippocampal cultures. However, the findings are consistent with the well-established high levels of protective systems, especially against oxidative/radical mediated damage, in astrocytes compared to neurons (see review by Pentreath and Slamon, 2000). Further experiments using extended culture periods may shed light on this.
Astrocyte activation with enhanced staining for GFAP and invasion of the neuritic plaques is a hallmark of AD (Duffy et al., 1980; Schechter et al., 1981). The calcium-binding protein S-100, which is normally concentrated in glial cells (reviewed by Kligman and Hilt, 1988), is also elevated in the AD patient. It is concentrated in the neuritic plaques of dystrophic dendrites (Kosik et al., 1986) and may be released from astrocytes to promote neurite extension (Kligman and Hilt, 1988).
Increased GFAP and S-100 immunostaining in astrocytes exposed to toxic βAP fractions, but without significant cell death, has been observed by Meske et al. (1998). Although the staining showed altered morphology with condensation (i.e., apparent increases in fluorescence) of the proteins, it did not provide quantitative data on the changes.
Vimentin is another cytoskeletal component, chiefly of immature, undifferentiated glia, which is coexpressed with GFAP in reactive astrocytes following damage to nervous tissue (Calvo et al., 1991). Treatment with dBcAMP induces activation-associated increases in the cytoskeletal components along with many other structural and metabolic constituents of astrocytes (Eddleston and Mucke, 1993) and, in accord with this, greater activation by the βAP fragments when measured by NR uptake, GFAP, vimentin, and S-100 expression. Several studies have demonstrated that βAP fragments induce other components of the reactive response in astrocytes, including upregulation of 1L-1β, NO synthase (Hu et al., 1998, 1999), K+ and Cl− channel activities (Jalonen et al., 1997), neprilysin (a putative βAP degrading enzyme; Apelt et al., 2003), and glutamate transport (Abe and Misawa, 2003). Thus, taken together, the present work and those of others demonstrate that the significant responses of astrocytes to the toxic βAP fractions are the activation-type changes, with relatively limited toxicities. They also strongly imply that βAP may be an etiologic agent in the glial changes that accompany AD.
The significance of the astrocyte activation in response to the βAP is not yet clear. Although the activation repertoire embraces a change in cell phenotype with alterations (normally upregulation) of a large number of enzymes (see Eddleston and Mucke, 1993), substantial evidence is available for a number of toxic compounds showing that part of the changes confer increased protection against oxidative/radical mediated cell damage (reviewed by Pentreath and Slamon, 2000). In relation to this the toxic forms of βAP can initiate oxidative damage in neurons, which in turn causes activation of NF-κB, which is responsible for initiating the cellular antioxidant defense program (Kaltschmidt et al., 1997, 1999). High, chronic exposure to increased levels of βAP may exceed the capacities for repair and lead to neuronal apoptosis and death (Kaltschmidt et al., 1997, 1999; see also Calabrese, 2001). The likely protective roles of astrocytes in βAP toxicity are important areas for further study.