
Abstract
The linear nonthreshold (LNT) model plays a central role in low-dose radiation risk assessment for humans. With the LNT model, any radiation exposure is assumed to increase one's risk of cancer. Based on the LNT model, others have predicted tens of thousands of deaths related to environmental exposure to radioactive material from nuclear accidents (e.g., Chernobyl) and fallout from nuclear weapons testing. Here, we introduce a mechanism-based model for low-dose, radiation-induced, stochastic effects (genomic instability, apoptosis, mutations, neoplastic transformation) that leads to a LNT relationship between the risk for neoplastic transformation and dose only in special cases. It is shown that nonlinear dose-response relationships for risk of stochastic effects (problematic nonlethal mutations, neoplastic transformation) should be expected based on known biological mechanisms. Further, for low-dose, low-dose rate, low-LET radiation, large thresholds may exist for cancer induction. We summarize previously published data demonstrating large thresholds for cancer induction. We also provide evidence for low-dose-radiation-induced, protection (assumed via apoptosis) from neoplastic transformation. We speculate based on work of others (Chung 2002) that such protection may also be induced to operate on existing cancer cells and may be amplified by apoptosis-inducing agents such as dietary isothiocyanates.
INTRODUCTION
The shape of the dose-response curve for stochastic effects (mutations, neoplastic transformation, cancer) after exposure to low doses of ionizing radiation has for years been the topic of enormous debate, yet this debate continues (Crawford-Brown and Hofmann 1990, 1993; Chen and Wei 1991; Bond et al. 1995; Rossi and Zaider 1997; Becker 1998, 2002; Bogen 1998; Calabrese and Baldwin 1998, 1999; Calabrese et al. 1999; Kondo 1999; Pollycove and Feinendegen 1999; Brenner et al. 2001; Feinendegen and Pollycove 2001; NCRP 2001; Schöllnberger et al. 2001a, b, 2002). The key discussion relates to whether the linear nonthreshold (LNT) model for low-dose extrapolation of cancer risk is valid. This model is widely used by regulatory agencies and in radiation and chemical protection. With the LNT hypothesis, risk progressively increases as dose increases. Any amount of carcinogen exposure increases one's risk of cancer. Thus, for radiation, any exposure is assumed to increase one's risk of cancer. Tens of thousands of cancer deaths in the U.S. have been calculated to arise from fallout from nuclear weapons testing (CDC/NCI 2001). Similar numbers could possibly emerge if one chose to use the LNT model to calculate cancer deaths from inhaling smoke from forest fires that have occurred over the years (which contains naturally occurring and manmade radionuclides).
Other possible dose-response curves (linear-threshold, sigmoid, u-shaped, etc.) are considered to be more in line with known mechanisms of carcinogenesis (Feinendegen et al. 1999, 2000; Pollycove and Feinendegen 1999; Feinendegen and Pollycove 2001; Schöllnberger et al. 2002). The principal worker protection and public health implication is that if a threshold response were assumed, then exposures below the threshold value would be considered safe (Calabrese and Baldwin 1999).
It is highly unlikely that use of the LNT model will be abandoned by regulatory agencies and in radiation/chemical protection unless substantial evidence of thresholds can be demonstrated from epidemiological studies and from mechanisms-based experimental and theoretical investigations. Now there is growing evidence from epidemiological, experimental, and mathematical modeling studies that does not support use of the LNT model for central estimation of cancer risks at low doses. Instead, the results support the existence of thresholds (quite large in some cases) for radiation-induced excess cancers, possibly in association with complex dose-response relationships (e.g. u-shaped). The u-shaped dose-response relationship is well known among researchers of hormesis (Calaberse and Baldwin 1998, 1999; Calabrese et al. 1999; Ducoff 2002).
The focus of this paper is to use a mechanisms-based model (called NEOTRANS2) for radiation-induced neoplastic transformation (considered an early stage in cancer development), to show how thresholds for specific radiation-induced excess stochastic effects (problematic nonlethal mutations and neoplastic transformation) could arise in some cases as a result of natural protection (resilience) from radiation-induced damage. Results for radiation may also apply to genotoxic chemicals. The NEOTRANS2 model was mainly developed over the last 4 years largely in the absence of supporting data initially for key modeling assumptions. However, over these years, supporting data have slowly emerged via publications and presentations by others in a variety of fields. Here, we introduce and explain the supporting data and present the NEOTRANS2 model as though it were developed based on existing data to support key modeling assumptions. This form of presentation was selected to help the reader feel more familiar with these assumptions (as some relate to novel mechanisms) when presented.
MOLECULAR MECHANISMS ASSOCIATED WITH CARCINOGENESIS
Macromolecular Changes
Ionizing radiation induces a range of DNA damage similar to that which arises endogenously from reactive oxygen species generated as byproducts of metabolism (Jeggo 2002). Daniel Billen (1990), in discussing the concept of negligible dose in the context of naturally occurring DNA damage and repair, has reported that thousands of spontaneous DNA-damaging events occur in each cell each day. Robert Stewart (1999) reported an estimate (best estimate) numerically equivalent to 105 spontaneous “Locally Multiple Damage Sites” (in particular double strand breaks) occurring in DNA, per million cells, per day. These lesions are quickly repaired, essentially error free in most cases. It is highly plausible that adding a few tens or hundreds more of such lesions through low-dose radiation (especially low-LET radiation) or low-dose chemical exposure is unlikely to overwhelm the cell's highly efficient damage repair machinery. Therefore, it is reasonable that error-free repair could operate after very low doses of low-LET radiation or genotoxic chemical.
Numerous repair processes are now known and include nucleotide excision repair, base excision repair, transcription-coupled repair, mismatch repair, and nonhomologous end joining (Friedberg et al. 1995; Scicchitano and Mellon 1997; Hanawalt 2001). The indicated repair processes operate at the individual cell level and provide for individual cell resilience to vulnerable states. A complex cell signaling network regulates the individual-resilience system. The failure of this system can lead to repair errors, which in turn can lead to problematic lethal and nonlethal mutations (forms of genomic instability).
Operationally, two types of mutations (heightened vulnerability states) are used to classify genes: (1) those where a mutation causes a gain in function (protooncogene to oncogene change); and (2) those where mutations cause function loss (tumor suppressor genes). In the development of leukemia and lymphoma, the first step is considered to be activation of a protooncogene into an oncogene, which arises via a translocation of a promoter beside the active site of a normally repressed growth-promoting gene site (Young 1994).
In the case of thyroid cancer, specific genes are rearranged that involve activation of the ret protooncogene (Jacob et al. 1996; Rabes and Klugbauer 1998; Smida et al. 1999). Whereas oncogene activations are quite specific, for tumor suppressor gene mutations, random deletions of large amounts of DNA, large parts of a gene, an entire gene, or several genes could occur. For many solid tumors, the inactivation of a tumor suppressor gene is considered to be the first step in the cancer induction process and is commonly assumed to affect a tissue-specific “gatekeeper” (Sidransky 1996; Trott and Roseman 2000). After loss of the gatekeeper function, clonal expansion of tissue-specific stem cells is allowed (Sidransky 1996).
Radiation mutagenesis may principally proceed via DNA deletions through misrepair and misrecombination at DNA double-strand breaks (ICRP 1999; Trott and Roseman 2000). In our modeling of radiation-induced neoplastic transformation, mutations are assumed to arise from misrepair of DNA damage, and nonlethal mutations are assumed responsible for the initial persistent genomic instability. Here, we have not distinguished between misrecombination of DNA double strand breaks, misrepair, or incomplete repair. Presently, we only distinguish between lethal and nonlethal mutations.
Genomic Instability and Mutations
The concept of genomic instability was introduced by W. F. Morgan and colleagues (1996) and is now widely accepted. Genomic instability can propagate over successive cell generations (Morgan et al. 1996; Wright 1998). We consider all mutations to represent genomic instability. Problematic nonlethal mutations among dividing cells we consider to possess persistent problematic instability (PPI) transferable to progeny. Most radiation-induced mutations directly involve the loss of large parts of the tested gene, leading to loss of heterozygosity (Trott and Roseman 2000). However, most radiation-induced mutations associated with genomic instability are point mutations and small deletions (Little 1999). In modeling radiation-induced genomic instability, we do not assume PPI to be associated with a specific type of mutation. We only distinguish between lethal and nonlethal mutations, and we assume that neoplastic transformation arises as a stochastic process among cells (including progeny) with PPI.
Some useful findings related to genomic instability have been reported in a study of 20 liver tumors, which were diagnosed in a cohort of people treated with Thorotrast (Iwamoto et al. 1999). It was found that 95% of the cases showed p53 point mutations. Iwamoto et al. (1999) concluded that the relevant genetic alterations leading to liver cancer result from an induced genetic instability (indirect effect), rather than from radiation exposure directly. In our modeling of neoplastic transformation, we have characterized PPI as an indirect effect (arising via misrepair) of irradiation (or chemical exposure) that can be passed to cell progeny. We have also introduced a new class of genomic instability (transient; Scott 1997), which is now modeled as a direct effect (hit hypersensitive cells) and indirect effect (including deleterious bystander effects) of irradiation.
Apoptosis: Protector of the Cell Community from Stochastic Effects
In contrast to the necrotic mode of cell death, apoptosis protects from problematic cells in the body via their elimination without causing inflammation (Mendonca et al. 1999). Strasser et al. (2000) summarized key points associated with apoptosis signaling as follows:
Apoptosis, a physiological process for killing cells, is critical for the normal development and function of multicellular organisms. Abnormalities in cell death control can contribute to a variety of diseases, including cancer, autoimmunity, and degenerative disorders. Signaling for apoptosis occurs through multiple independent pathways that are initiated either from triggering events within the cell or from outside the cell, for instance, by ligation of death receptors.
New research results indicated that problematic cells in the body may be detected via molecular biological mechanisms and selectively eliminated via apoptosis to protect the cell community (Yang et al. 2000). A key assumption of the NEOTRANS2 model to be introduced is that existing problematic cells (e.g., problematic mutants, neoplastically transformed cells) in the cell community can be signaled to undergo apoptosis and selectively eliminated via low-dose-induced protective bystander mechanisms. These mechanisms of reduction in cell community vulnerability status we presume to explain, at least in part, reported low-dose hypersensitivity to cell killing among cancer cell lines (Joiner et al. 1999) as well as virally transfected cells (Seymour and Mothersill 2000).
Thus, the NEOTRANS2 model to be presented includes both deleterious and protective bystander effects.
Possible Mechanisms for Recognizing and Selectively Eliminating Problematic Cells
As already indicated, we have hypothesized the existence of a protective apoptotic bystander effect for neoplastic transformation. Such an effect is necessary to adequately explain existing data whereby risks for neoplastic transformation (Azzam et al. 1996; Redpath et al. 2001) and lung cancer (Rossi and Zaider 1997) decrease rather than increase at very low doses.
A crucial missing link related to our modeling is identification of mechanisms whereby problematic cells already present in a population can be recognized and signaled to undergo apoptosis, while nearby normal cells are essentially unaffected. Some progress is being made by researchers to identify and characterize such a protective process for the cell community.
Cucinotta et al. (2002) point out that ionizing radiation produces DNA damage that causes protein fluctuations through binding damage recognition proteins to DNA breaks and subsequent downstream events. The type of fluctuations may depend on the type of DNA break such as simple or complex single-strand breaks and double-strand breaks or base damage (Cunniffe and O'Neil 1999).
Barcellos-Hoffand Brooks (2001) point out that bystander effects after low doses of radiation are extracellular signaling pathways that modulate both cellular repair and death programs. The authors also indicate that transforming growth factor β (TGFB1) is known to be an extracellular sensor of damage. They further indicate that extracellular signaling relevant to carcinogenesis in normal tissue can eliminate abnormal cells or suppress neoplastic behavior.
Dr. C.-R. Yang and colleagues (2000) at Case Western University have reported clusterin [CLU, a.k.a. TRPM-2, SGP-2, or radiation-induced protein-8 (XIP8)] tobe implicated in apoptosis. In a recent study (Yang et al. 2000), they reisolated CLU/XIP8 by yeast two-hybrid analyses, using as bait the DNA double-strand break repair protein Ku70. They showed that low-dose, radiation-induced nuclear CLU/XIP8 protein coimmunoprecipitated and colocalized in vivo with Ku70/Ku80, a known DNA damage sensor and key double-strand break repair protein, in human MCF-7:WS8 breast cancer cells. Their key finding was that enhanced expression and accumulation of nuclear CLU/XIP8-Ku70/Ku80 complexes appear to be an important cell death signal after irradiation. Further, their data suggest that CLU/XIP8 may play an important role in monitoring cells with genomic instability and/or infidelity, created by translesion DNA synthesis, by facilitating removal of genetically unstable cells as well as severely damaged cells. Yang et al. (2000) strongly suggest that the CLU/XIP8 protein is a general cell death signal, monitoring overall cell health.
Yang et al. point out that the recent findings that Ku70, but not Ku80, knockout mice are cancer prone appear consistent with the notion that formation of nuclear CLU/XIP8 with Ku70 may play an important role in eliminating carcinogenic initiated (problematic) cells.
It is now known from in vitro studies of viral-induced neoplastic transformation (Bauer 1996) that:
Increasing plating density reduces transformation frequency.
Transformed cells are selectively killed via apoptosis.
Cytokines and reactive oxygen produced by nontransformed neighboring cells trigger apoptosis.
TGFB1 enables nontransformed cells to trigger apoptosis among transformed cells.
Given the above information, we consider our key modeling assumption of the existence of an inducible protective bystander apoptosis effect whereby problematic cells are recognized (after signaling from other cells) and selectively eliminated from the cell community to be highly plausible.
Another assumption we make is that neoplastic transformation is a necessary early step for cancer induction (a widely held view). Thus, demonstrating low-dose-induced protection from neoplastic transformation in vitro is consistent with the possibility of low-dose-induced protection from cancer in vivo.
Cellular Differentiation
The current view is that some problematic cells may undergo differentiation (group resilience), and this also protects the cell community from propagating stochastic adverse effects. Presently, the NEOTRANS2 model does not include this feature. We consider differentiation to be more important in vivo than in vitro. Our modeling applications presented in this paper relate to in vitro studies.
Deleterious Bystander Effects
Deleterious bystander effects (Ballarini et al. 2002) whereby unirradiated cells are damaged have been examined in two general types of cellular systems. In the first, monolayer cultures have been exposed to very low fluences of alpha particles either from an external source (Nagasawa and Little 1992; Azzam et al. 1998; Little et al. 2002) or focused microbeam (Hei et al. 1997; Prise et al. 1998). The second technique involves harvesting medium from irradiated cells and incubating it with nonirradiated cells (Mothersill and Seymour 1997; Lyng et al. 2000). Both techniques have demonstrated that cells not being irradiated can still be damaged. Further, the bystander effect does not arise from simply irradiating media. Cell damage and intercellular signaling are essential.
We also allow for the possibility of deleterious bystander effects via model parameters that account for both direct and indirect deleterious radiation effects.
THE NEOTRANS2 MODEL AND ITS PREDECESSOR NEOTRANS1
Our modeling research focuses on characterizing excess stochastic effects (mutations, neoplastic transformations) after very low doses of radiation by using mechanisms-based models. While many in vitro experimental studies have been conducted on radiation-induced neoplastic transformation, only limited experimental data are available for doses < 100 mGy (Azzam et al. 1994, 1996; Redpath and Antoniono 1998; Redpath et al. 2001).
In previous work, we introduced a class of models (that included NEOTRANS1) for characterizing neoplastic transformation of cells that relates the probability of neoplastic transformation to the state of genomic instability (Scott 1997; Schöllnberger et al. 2001a; Scott et al. 2001). With NEOTRANS1, the target cell population was modeled as heterogeneous with both hypersensitive- and resistant-cell subpopulations (considered the simplest case of heterogeneity). NEOTRANS1 has now been refined, leading to a model called NEOTRANS2 (Figures 1 and 2) that includes apoptotic and necrotic death pathways. In this paper, NEOTRANS2 is applied to in vitro data for low-radiation-dose-induced neoplastic transformation. We have focused only on data with several dose groups ≤ 100 mGy.
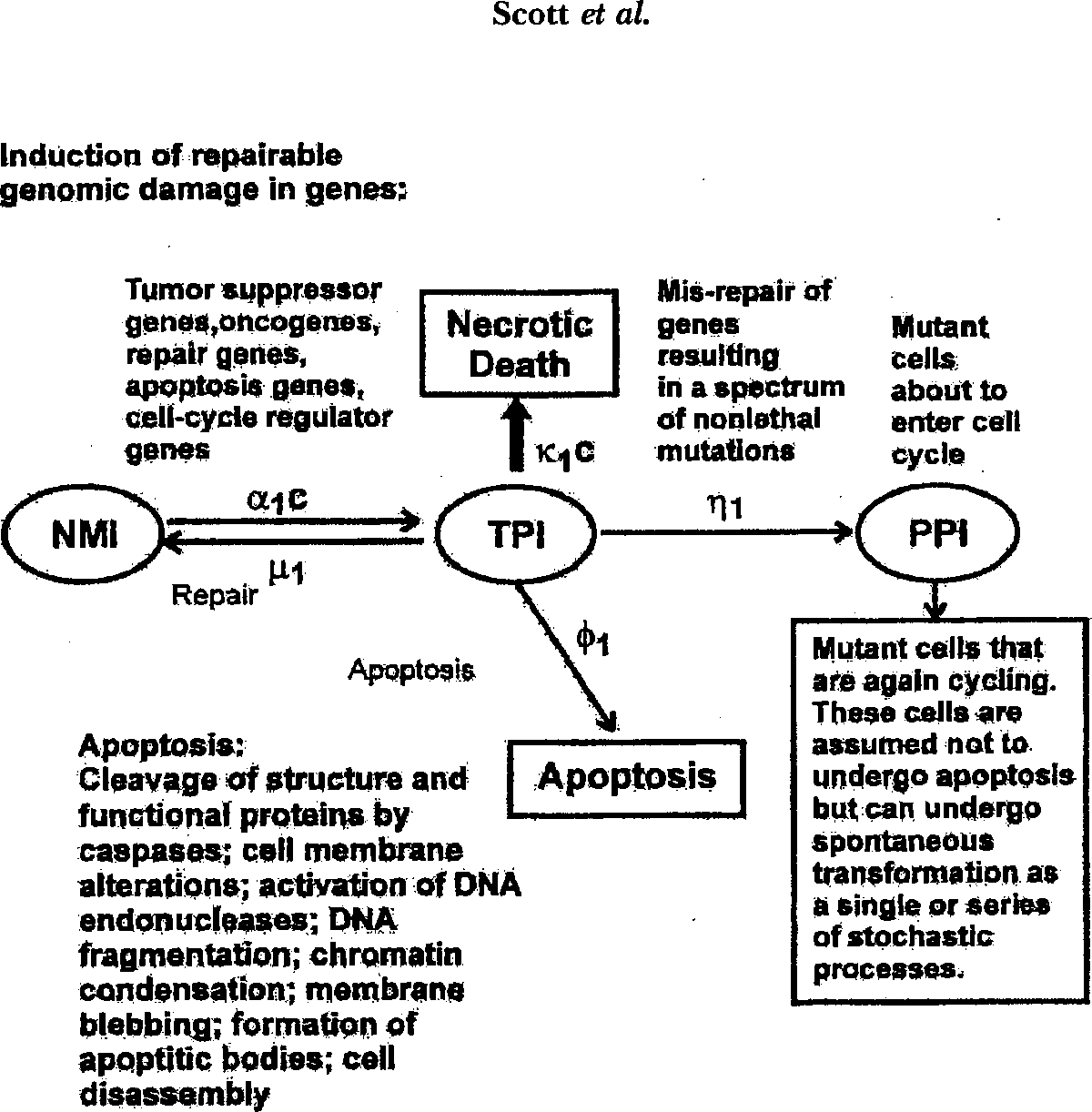

Genomic Instability States Used in NEOTRANS2
Our use of terminology related to genomic instability is the same as used in earlier publications (Scott 1997; Schöllnberger et al. 2001a). The expression “genomic instability state” refers to any spontaneous or toxicant-induced instability in the genome, including any initial transient instability, as well as any persistent instability that can be passed to cell progeny. In addition to a stable (ST) genome, the NEOTRANS2 model (as well as NEOTRANS1) involves four types of genomic instability (Figures 1 and 2): (1) Normal-minor instability (NMI), associated with normal cell function and normal genome status; (2) Transient-minor instability (TMI), associated with toxicant-induced genomic damage that is fully repairable (without any significant errors); (3) Transient-problematic instability (TPI), associated with genomic damage mat may sometimes be fully repaired but can be misrepaired; and (4) PPI, which arises from misrepair that yields nonlethal mutations. Thus, PPI can be passed to progeny, increasing their potential for stochastic effects such as neoplastic transformation. We use the term misrepair in a broad sense as already indicated. We consider TPI and PPI to be vulnerability states (for additional deleterious stochastic effects).
Other Model Features
With the NEOTRANS2 model, a very small fraction, T0 ≪ 1, of the cell population is presumed to have already undergone neoplastic transformation over their life history. The discussion that immediately follows relates to the remaining vast majority (1 − T0 ≈ 1) of the cells. With both NEOTRANS2 (Figures 1 and 2) and NEOTRANS1, only cells in the high vulnerability state PPI (viable mutants) can produce neoplastically transformed progeny. Only genomically ST cells, those with NMI, and those with PPI progress through the cell cycle and divide. Other cells are assumed arrested at cell cycle checkpoints (resilience facilitation) where genomic damage is repaired or misrepaired. Irradiation times were assumed quite short relative to cell cycle transit times, so that no equations were used to account for progression through the cell cycle during irradiation. Neoplastic transformations are assumed to occur as a stochastic process, and the transformed cells may have an altered cell cycle transit time distribution.
With NEOTRANS2, target genes are specified (Figures 1 and 2) and include tumor suppressor genes, oncogenes, repair genes, apoptosis genes, and cell-cycle regulator genes. Unlike NEOTRANS1, with NEOTRANS2 cell killing is explicitly addressed and not treated as independent of neoplastic transformation. Two modes of cell death are considered: apoptosis (assumed to predominate at very low doses) and necrotic death (assumed important only at moderate and high doses). Again, nonlethal mutations are assumed to arise via misrepair. Lethal mutations are assigned to the apoptosis pathway (including delayed lethal mutations). The analytical solutions in the present paper apply only to very low-radiation doses where necrotic death can be assumed negligible.
Model parameters α1, α2, and α3, common to both NEOTRANS] and NEOTRANS2 reflect genomic sensitivity to initial and higher levels of damage production and should be multiplied by the dose rate c. The parameters μ1 and μ2 are also common to both models and govern the commitment rate of damaged cells to an error-free repair pathway. In addition, the parameters η1 and η2 are common to both models and govern the commitment rate of damaged cells to a misrepair pathway that leads to nonlethal mutant cells (PPI cells).
In light of new evidence that protracted exposure to low-LET radiation can lead to large dose thresholds for cancer induction, we allow η1 and η2 to be step functions of dose rate. Below a critical dose-rate value c* (presently undetermined), the parameters take on a value of zero. This dose-rate threshold is presumed to depend on the type of radiation and type of cancer. For dose rates above c*, the parameters then take on fixed values > zero. The parameters φ1 and φ2 appear only in NEOTRANS2 and govern the rate of commitment of damaged cells (including lethal mutations) to the apoptotic pathway. The parameters κ1 and κ2 (which are important only for moderate and high doses) appear only in NEOTRANS2 and when multiplied by dose rate, govern the rate at which already damaged cells enter the necrotic death pathway. Typical units for αi and κi are mGy−1. Typical units for μi, ηi, and φi are min−1.
Parameters α1, α2, and α3 should be viewed as being comprised of two parts: (1) one part relates to direct damage to DNA; (2) the other part relates to indirect damage to DNA and includes deleterious bystander effects.
For very low-radiation doses, only hypersensitive cells are assumed to be induced to transform (new transformations) and cells are modeled as being killed only via the apoptotic pathway. Thus, only Figure 1 applies for very low doses and to the hypersensitive subfraction, f1, of cells at risk.
Further, with our current version of the NEOTRANS2 model, a fraction T0 (stochastic quantity) of cells at risk is assumed to have already undergone spontaneous neoplastic transformation, based on genomic alterations over their life history, but prior to dosing with radiation (or chemical). Because the life history of cells spans a long time compared to the short time period over which cells are irradiated during in vitro studies, this is considered a highly plausible assumption when applying NEOTRANS2 to data from in vitro irradiation studies. For in vivo exposure, additional protective mechanisms could be important (Stecca and Gerber 1998; Barcellos-Hoff and Brooks 2001).
Model Solutions for Very Low Doses
Evidence is now strong that death via apoptosis at low radiation doses can occur via a bystander mechanism (Mothersill and Seymour 1998a, b; Lyng et al. 2000; Belyakov et al. 2001a, b, 2002a, b; Prise et al. 2002). We consider the highly plausible possibility that a fraction f0 of the T0 cells already neoplastically transformed is killed via a bystander effect for apoptosis (a key modeling assumption). In such cases, the dose response at very low-radiation doses could decrease rather than increase. Indeed, this type of dose response has now been demonstrated experimentally with 60Co-gamma irradiation of C3H 10T1/2 cells (Azzam et al. 1996) and with 137Cs-gamma irradiation of HeLa × skin fibroblast human hybrid cells (Redpath and Antoniono 1998; Redpath et al. 2001).
The analytical solutions that follow apply to very small dose increments. As indicated, a small fraction T0 of cells in the population is modeled as already having the problem of interest (e.g., neoplastic transformation in this case, but a similar equation would apply to nonlethal problematic mutations). At such doses, newly induced neoplastic transformations are modeled as arising from a small (in number), hypersensitive subfraction of remaining (1 − T0) cells at risk. This hypersensitive subfraction is given by f1(1 − T0) ≈ f1. From Figure 1 (which shows only hypersensitive cells), it can be seen that a very small dose increment ΔD (where ΔD ≈ c Δt, for a small time increment Δt) to the fraction f1(1 − T0) of hypersensitive cells will lead to an expected fraction f1(1 − T0)α1ΔD of cells in the state TPI (assuming all hypersensitive cells are initially in the state NMI); for this fraction entering the transient state TPI, the conditional probability of subsequently undergoing misrepair (leading to PPI) is just η1/(μ1 + η1 + φ1).
The dose-response function for radiation-induced, neoplastic transformations per surviving cell, TFSC(ΔD), at very low doses ΔD is thus given by the following:
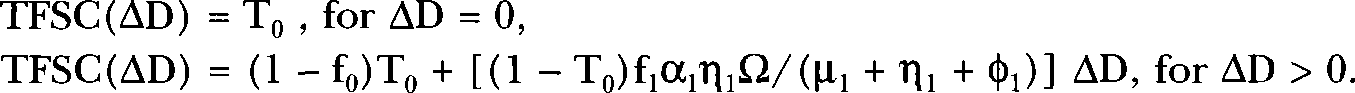
For ΔD > 0, Equation 1 has a fixed slope of (1 − T0)f1α1η1Ω/(μ1 + η1 + φ1). The parameter Ω is the proportion of the newly induced parental PPI cells that produce neoplastically transformed progeny. The parameter Ω, therefore, depends on follow-up time. It is also likely influenced by the signaling characteristic of the cellular Community (Barcellos-Hoff and Brooks 2001). Equation 1 leads to the LNT model only when f0 = 0 (i.e., when the protective apoptosis effect is absent) and η1 > 0 (misrepair occurs).
Equation 1 is based on the assumption that intercellular signaling that leads to the protective bystander apoptosis effect occurs without a radiation dose threshold. Data to be presented later (Azzam et al. 1996; Redpath et al. 2001) support this hypothesis for ionizing radiation. However, this may not be the case for genotoxic chemicals (Walker et al. 2002).
With Equation 1, the dose-response relationship is discontinuous at zero dose [steps down from T0 to (1 − f0)T0]. The dose-response associated with Equation 1 is linear but with a zero-dose intercept of (1 − f0)T0 rather than T0 when fitted to low-dose data with the zero-dose group excluded (see hypothetical dose-response curve in Figure 3). As indicated in Figure 3, T0 is stochastic.

The dose-response curve for TFSC will exceed T0 (a random variable) only for AD in excess of a stochastic threshold (StoThresh) dose DTh (Figure 3) given by:
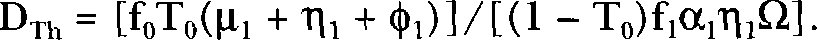
Here, we assumed that cell survival is very near 100% at the very low doses considered and that η1, f1, α1, f1, and Ω are all > 0. This is consistent with observations of Azzam et al. (1996). A StoThresh (as apposed to a deterministic threshold) is considered to occur because T0 as well as all other model parameters are treated as stochastic.
Since T0 is on the order of 10−4 to 10−5 for most in vitro studies of neoplastic transformation, selectively killing all T0 cells (i.e., f0 = 1) would still lead to a cell survival fraction > 0.999. Unfortunately, currently available data at low doses for which equations apply are inadequate to derive distributions for individual model parameters μ1, η1, φ1, α1, f0, and Ω. However, more general forms of Equations 1 and 2 are derived and used in obtaining estimates of f0, T0, and DTh. Since demonstrating that DTh > 0 has important implications for radiation protection and radiation risk assessment, these more general solutions are quite useful.
Equation 1 can be rewritten in the more general form:
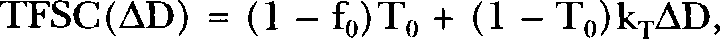
where

Equation 3 can be considered a generalized, three-parameter (stochastic parameters f0, T0, and kT) form of the NEOTRANS2 model for application to very low-radiation doses. A corresponding equation may also apply to highly genotoxic chemicals, with ΔD then representing a very small dose of the agent of interest. For a constant exposure time (for a chemical), AD could be replaced by the concentration with the parameter kT redefined to include the exposure time in the numerator.
Equation 2 can also be rewritten in the more general form:

Figure 3 shows a hypothetical mean dose-response curve based on Equations 3 and 4. In the figure, hypothetical distributions F(T0) (shown vertically) and G(DTh) (shown horizontally) are presented for T0 and DTh, respectively.
For very low doses and in the framework of the NEOTRANS2 model, it is possible that the protective bystander effect may predominate (f0 ≫ kTΔD) when the spontaneous frequency T0 of transformation is relatively high and when f0 > 0 and ΔD is very small (e.g., less than about 100 mGy low-LET radiation). Implied here is a relative small value for the slope parameter kT in combination with a small dose. In such cases, the data for radiation-associated neoplastic transformation (and for specific problematic nonlethal mutations) should be adequately represented by the relationships:

Further, TFSC (ΔD) should be uncorrelated with dose over the dose range for which the above applies. This requirement only applies to doses in excess of background. We later apply Equation 5 to two data sets for gamma-ray-induced neoplastic cell transformation for doses up to about 100 mGy.
We describe later how distributions for T0 (stochastic), DTh (stochastic), and the slope parameter kT have been obtained for induced neoplastic transformation.
FITTING THE NEOTRANS2 MODEL TO DATA
Data Used to Estimate Model Parameters
We fitted the protective bystander effects version of the model to available data for radiation-induced neoplastic transformation (two data sets) and low-dose apoptosis (one data set):
Data Set 1 — Gamma-ray-induced neoplastic transformation data of Redpath et al. (2001) (delayed plating):
HeLa × skin fibroblast human hybrid cells
137Cs gamma rays
Dose rate: 3.3 mGy/min for dose < 100 mGy; 41.3 mGy/min otherwise
Doses: 0, 1, 5, 10, 50, 100, 300, and 500 mGy
Data Set 2 — Gamma-ray-induced neoplastic transformation data of Azzam et al. (1996) (delayed plating):
C3H 10T1/2 mouse embryo fibroblast clone 8 cells
60Co gamma rays
Dose rate: 2.4 mGy/min
Doses: 0, 1, 10, and 100 mGy
Data Set 3 — Gamma-ray-induced cell killing (via apoptosis) data of Seymour and Mothersill (2000):
Human keratinocytes (immortalized via viral transfection but not transformed)
60Co gamma rays
Dose rate: 750 mGy/min
Doses: 0, 10, 30, 50, and 100 mGy
Estimating Model Parameters
For the narrow dose range (0 to 100 mGy), all data (for ΔD > 0) for transformation and cell survival were uncorrelated with dose. This is in line with characteristics of the NEOTRANS2 model that predicts that the largest effect at very low doses is the protective bystander apoptosis effect, which is modeled as being independent of dose.
For data in the dose interval 0 to 100 mGy (excluding the zero dose group), the parameter f0 was evaluated for both the data of Redpath et al. (2001) and Azzam et al. (1996) as follows based on Equation 5. For 0 < ΔD ≤ 100 mGy, f0 for transformation was calculated as a function of the mean observed transformation frequency, TFSC, and reported mean for T0 using the relationship

Equation 6 was used for each dose in the dose range indicated leading to different estimates of f0 and corresponding values (1-f0)T0. Mean values for (1-f0)T0 and the associated standard deviation were obtained. Dose-response relationships (horizontal line) were based on these means and the associated 95% confidence intervals assuming a normal distribution.
Bayesian methods (Siva 1998) were used only for the neoplastic transformation data of Redpath et al. (2001) and only when doses over the wider range of 0 to 500 mGy were evaluated. For this dose range, Equations 5 and 6 do not apply. Equation 3 applies and was therefore used. WinBUGS software (Spiegelhalter et al. 1999) was used to carry out the Bayesian inference via use of Markov Chain Monte Carlo (MCMC) analyses. Transformants were modeled as having Poisson distributions. For the Bayesian analysis, the prior distribution for kT was uniform over the interval (4 × 10−8 − 7.5 × 10−8); for f0, a uniform prior distribution over the interval 0 to 1 was used; for T0, a normal prior distribution was used with a mean of 2.24 × 10−5 and standard deviation of 2.8 × 10−6 [same values as reported by Redpath et al. (2001)]. Five thousand MCMC iterations were first run. Auto correlations were then examined to judge how many additional iterations were needed for convergence. Fewer than 30,000 iterations (total) were found needed to ensure convergence. Iterations were then increased so that the total was 60,000. These iterations were more than were needed, but they essentially guaranteed convergence of the Markov chains. The first 40,000 iterations were then discarded as burn-in. Analysis of posterior distributions was then based on the final 20,000 MCMC realizations.
MODELING RESULTS
Figure 4 shows results obtained for our analysis of the Azzam et al.'s (1996) data for gamma-ray-induced neoplastic transformation of C3H 10T1/2 cells in vitro. Only data in the very low dose range (0 to 100 mGy), where Equation 5 applies, were used. For this dose range (with the zero dose group excluded), there was no significant correlation between transformation frequency and dose (R2 = 0.18, p > 0.5).

The corresponding results for application of the NEOTRANS2 model to the Redpath et al. (2001) data for gamma-ray-induced neoplastic transformation of HeLa × skin fibroblast cells are presented in Figure 5. Solid points in these figures represent the experimental data, and smooth and dashed curves represent model-associated results with means (central curve) and 95% confidence regions. For these data and for doses above zero, there was no significant correlation of transformation frequency with dose (R2 = 0.4, p = 0.2).

In both Figures 4 and 5 the risk of neoplastic transformation clearly drops immediately below the spontaneous frequency to a fixed value independent of radiation dose, as predicted by the NEOTRANS2 model (Equation 5).
The mean and standard deviations for f0 were 0.32 + 0.04 and 0.71 ± 0.04 for the data of Redpath et al. (2001) and Azzam et al. (1996), respectively. The parameter f0 mean was therefore 2.2 times larger for the C3H 10T1/2 cells than for the HeLa × skin fibroblast human hybrid cells. Similarly, the spontaneous frequency mean was about 76 times larger for the C3H 10T1/2 cells than for the HeLa × skin fibroblast cells. These results suggest that f0 may be correlated with genetic sensitivity, being larger (more protective) for the more sensitive target cells. However, what implication this has for sensitive individuals is unclear.
Figure 6 shows results of applying the NEOTRANS2 model to a wider range of doses (0 to 500 mGy) based on the Redpath et al. (2001) data for gamma-ray-induced neoplastic transformation of HeLa × skin fibroblast human hybrid cells. Equation 3 was used in this analysis in conjunction with Bayesian methods. Transformants were modeled as having dose-dependent Poisson distributions with the expected frequency given by Equation 3. Solid points in Figure 6 represent experimental data. Upper and lower lines drawn represent the upper and lower 95% credibility bands (from Bayesian posterior distribution), and the central line represents the posterior mean. Model parameter estimates are presented in Table 1 along with standard deviations, and 5% (percentile), 50%, and 95% values.
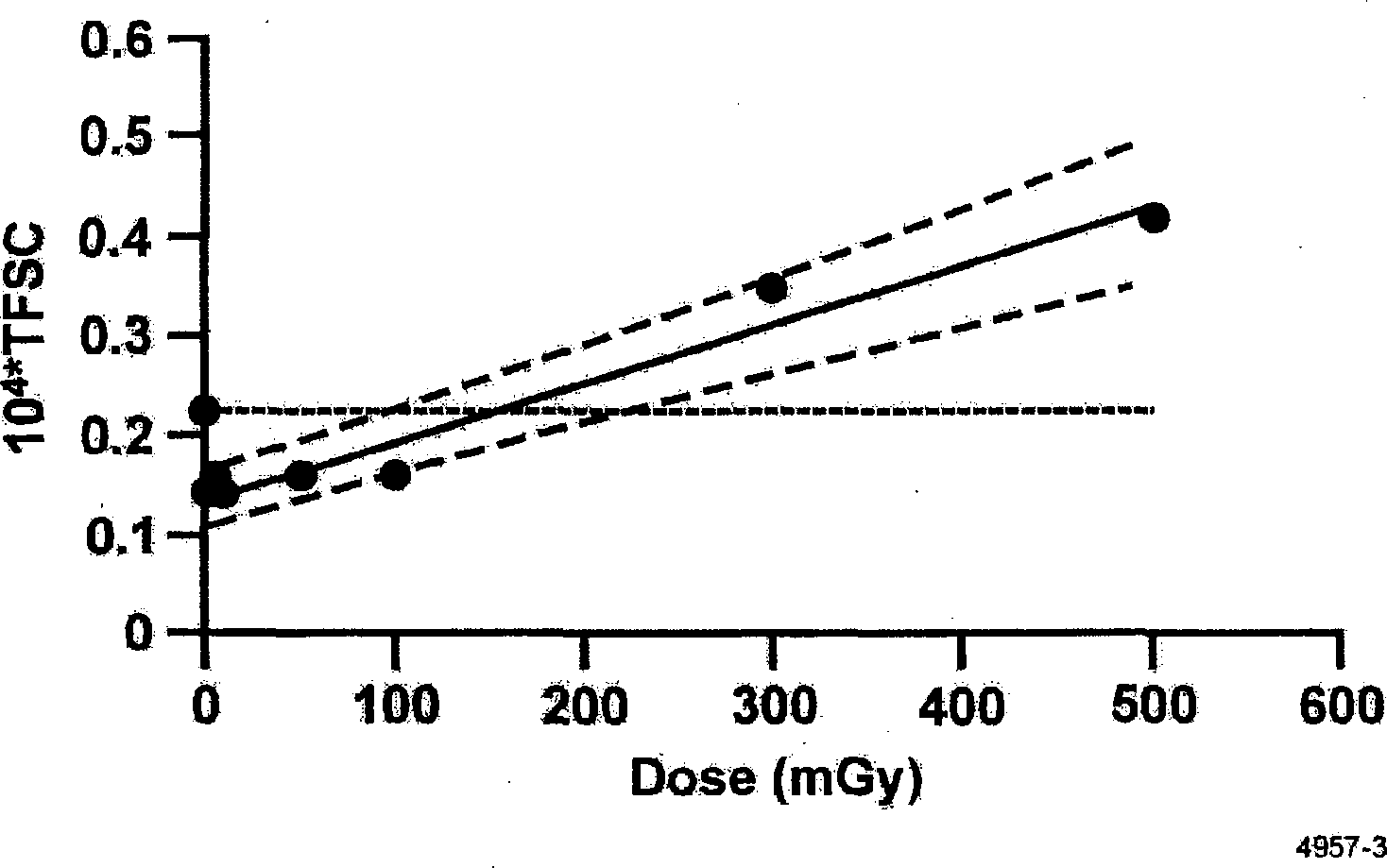
NEOTRANS2 model parameter and threshold estimates for data of Redpath et al. (2001) for gamma-ray-induced transformation of HeLa × skin fibroblast human hybrid cells.

The central line in Figure 6 has been used to demonstrate a protective effect of low-dose radiation against neoplastic transformation. Figure 7 shows the benefit (expected number of spontaneous transformants eliminated) to harm (expected number of newly induced transformants) ratio. A benefit/harm ratio ≫ 1 demonstrates potential for possibly eliminating early stage cancer cells from the body via low-dose irradiation (e.g., from radon in the home, living at a high altitude where cosmic ray doses are higher, etc.). Similar potential protection likely also exists for other inducers of apoptosis signaling (e.g., apoptosis-inducing chemicals in food such as isothiocyanates [Yang et al. 2002]). Note that the benefit/harm ratio increases steeply as the dose decreases below about 50 mGy. The lowest dose featured on the curve is 10 mGy. For this dose, the benefit/harm ratio exceeds 600,000. This means that on average, for each newly induced transformant, more than 600,000 assumed already present spontaneous transformants are eliminated via the presumed protective bystander apoptosis effect. This is a pronounced protective effect since relatively little harm to a human would be expected to be associated with a 10 mGy radiation dose, especially if protracted. Further, the benefit/harm ratio may increase as the period over which the dose is delivered increases because extending exposure also would be expected to prolong the period over which the protective bystander effect was operating.

Benefit (spontaneous transformants eliminated) to harm (newly induced transformants) ratio based on the central line in Figure 6 for the neoplastic transformation data of Redpath et al. (2001).
Similar protection has also been demonstrated in cancer chemoprevention studies where apoptosis-inducing isothiocyanates in the diet have prevented the occurrence of benzo(a)pyrene-induced lung tumors in mice (Yang et al. 2002).
Radiation may also induce the elimination of virally transfected cells via the protective bystander apoptosis effect. Figure 8 shows results obtained in modeling the cell survival data of Seymour and Mothersill (2000) for gamma ray-induced apoptosis in human papillomavirus type 16 transfected (Pirisi et al. 1988) human keratinocytes. The cell killing for the dose range 0 to 100 mGy was modeled as arising from a protective bystander effect that was independent of dose for ΔD > 0. As seen in Figure 8, the data are in excellent agreement with the modeling assumptions. For the indicated data and for doses > 0, there was no correlation between survival and dose (R2 = 0.04, p > 0.5). The parameter f0 (for removal of problematic cells) was found to have a mean and standard deviation of 0.37 ± 0.08 (i.e., 37% of problematic virally transfected cells are expected to be removed via a protective bystander apoptosis effect).

DISCUSSION
With the NEOTRANS2 model, neoplastic transformation arises as a stochastic process but only for cells in the high vulnerability state PPI. The PPI (spontaneous or induced) is assumed to arise via nonlethal mutational events. These mutations are the type described by Mothersill and Seymour (1998b) as nonlethal but possibly cancer-facilitating mutations.
Results in Figures 4 and 5 are consistent with predictions of the NEOTRANS2 model that for very low doses of low-LET radiation, TFSC(ΔD) should be below the spontaneous frequency and independent of dose (e.g., in the range 0 to 100 mGy). Results in Figure 6 for gamma ray-induced neoplastic transformation of HeLa × skin fibroblast human hybrid cells have 5% (percentile), 50%, and 95% values for the Bayesian posterior distribution for the StoThresh of approximately 50, 150, and 250 mGy, respectively. We can state that the data used are consistent with a StoThresh and that the Bayesian posterior distribution for the threshold assigns essentially a zero probability for the threshold being zero. A value of zero corresponds to the LNT model. The dose-response curve for excess transformations is clearly of the nonlinear, threshold type.
A similar analysis was attempted for fission neutron (Hill et al. 1984; Balcer-Kubiczek and Harrison 1991) and for alpha particle data of others (Bettega et al. 1992) for induced neoplastic transformation. However, a close examination of posterior distributions for the StoThresh revealed modes (highest values in the distribution function) very close to zero with the distributions having a tail to the right but not to the left of the modes. Such results indicate that a zero-dose threshold for the high-LET radiation-induced neoplastic transformation could not be excluded based on the very limited data used. Central (mean) estimates of the thresholds were < 10 mGy, which would be very difficult to demonstrate experimentally.
We now introduce the term group (G) adaptation to describe any cell community adaptation initiated by a group of cells that protects the cell community (including the elimination of problematic cells as well as reducing cell vulnerability by cell differentiation). G-adaptation therefore also applies to low-dose radiation therapy for cancer (used in Europe but currently forbidden in the U.S.) assuming problematic cancer cells to be signaled via irradiated normal cells to undergo apoptosis.
Where an individual cell adapts to an environmental stress by changing its resilience or vulnerability status, we called this individual (7) adaptation. Thus, ≥I-adaptation can be beneficial or problematic, depending on the endpoint considered. An example of beneficial I-adaptation would be the induction of an error-free repair process, thereby increasing the cells' resilience to the induction of stochastic effects. An example of problematic I-adaptation would be a stress-related induction of an error-prone repair process producing viable mutations that facilitate the occurrence of neoplastic transformations.
Another example of problematic I-adaptation is when an individual cell through adapting to a different environment has a net reduction in its capacity to undergo apoptosis. A reduced capacity to undergo apoptosis increases the risk for stochastic effects such as neoplastic transformation and cancer. Studies at our Institute suggest that background radiation can influence cells through problematic I-adaptation. Dr. Saxena and colleagues (2002) have conducted studies whereby cells irradiated after maintenance at background radiation levels were compared to corresponding cells irradiated after being maintained at lower than background (via special shielding). The cells maintained at lower than normal background had reduced competence for undergoing apoptosis. These finding are in line with the view that background radiation may serve as a natural nutrient. Moving from a region of moderate background radiation to a region of lower background may over time increase one's susceptibility to the induction of stochastic effects.
Note that the view that problematic I-adaptation can lead to an increase in cancer risk is consistent with reported cancer risks from radon in homes (Cohen 1995). Dr. Cohen (1995) reported lung cancer mortality related to residential radon exposure in the U.S. After adjusting for smoking, lung cancer mortality was found to decrease with increasing mean residential radon levels.
Some early modeling publications attempted to explain high-LET radiation inverse dose rate effect in vivo based on cell culture studies where cells were irradiated over several hours (Brenner and Hall 1990; Elkind 1994). These models were essentially based on the hypothesis that cells would be repeatedly hit at sensitive stages of the cell cycle during protracted irradiation. Such a hypothesis is inconsistent with the observation that damaged cells are arrested at cell-cycle checkpoints rather than progress through the cell cycle. Further, biological kinetics over a few hours cannot explain inverse dose rate effects from radiation doses delivered over months to years. We speculate that inverse dose rate effects, such as those seen by Dr. Cohen (1995) that involve dose delivery over months to years, are more likely related to problematic I-adaptation, where cells in lower radiation environments (relative to an optimum) lose ability to undergo apoptosis. Additional problematic I-adaptation may be associated with the known decrease in DNA repair efficiency with increasing age.
With the current version of NEOTRANS2, a fraction f0 of the spontaneous neoplastic transformants is eliminated via a radiation- (or chemical-) induced protective bystander apoptosis effect. The parameter f0 can be viewed as representing the number of present spontaneous neoplastic transformants (of the type of interest) eliminated via the protective bystander apoptosis effect divided by the total number of spontaneous neoplastic transformants present. We give the parameter f0 the special name protection factor (PROFAC). Thus, the PROFAC represents the removal efficiency via intercellular signaling mechanisms of existing problematic cells among the problematic cells present. The PROFAC is considered to relate to -adaptation but its magnitude may be influenced by I-adaptation.
Using our NEOTRANS2 model, which includes a protective bystander effect via apoptosis, we have found a rather large StoThresh for gamma-ray-induced neoplastic transformation of HeLa × skin fibroblast human hybrid cells. Others (Kondo 1999; Feinendegen et al. 2000; Tanooka 2000; Yamamoto and Seyama 2000; Feinendegen and Pollycove 2001) also put forth the notion that a threshold for radiation-induced excess stochastic effects could arise from such a protective mechanisms.
In the current version of NEOTRANS2, the misrepair pathway is assumed to apply only for a dose rate in excess of a critical value c*. This has been changed in the model to account for others' observation that chronically administered (low-dose rate) low-LET irradiation appears to induce cancer (at certain sites in the body) only after very large radiation doses, if at all (Ootsuyama and Tanooka 1991, 1993; Rossi and Zaider 1997; Yamamato et al. 1998; Kondo 1999; Yamamato and Seyama 2000; Tokarskaya et al. 2002).
Harold Rossi and Marco Zaider (1997) critically reviewed the literature on radiogenic lung cancer and concluded that “at radiation doses generally of concern in radiation protection (< 2 Gy), protracted exposure to low-LET radiation (x- or -rays) does not appear to cause lung cancer. There is, in fact, indication of a reduction of the natural incidence.”
Hoel and Li (1998) have demonstrated that use of a threshold-type, dose-response model leads to better characterization of both the leukemia incidence and mortality data for atomic bomb survivors than use of the LNT model. In addition, Zoya Tokarskaya and colleagues (1995, 1997) reported a threshold close to 1 Gy for lung cancer induction by alpha radiation, based on Mayak workers who inhaled plutonium-239. R. E. Rowland (1994) has reported a large threshold for bone cancer induction by alpha radiation based on data for radium dial painters.
The recent Hanford Thyroid Disease Study did not find evidence for any excess risk of thyroid cancer induction for persons who were exposed to radioactive iodine released from the Hanford facility (USDHHS 2002). For doses in the range of 0 to 100 mGy, risk was not correlated with dose and was less than for the control group based on persons outside what was considered the irradiation zone. Further, for several health effects, the mean slope of the risk vs. dose relationship was negative.
It is important to point out implications of our findings related to low-dose risk assessment for chronic exposure. Firstly, G-adaptation can lead to protection of the cellular community from low-dose-induced stochastic effects. However, there is a possibility that some risk gains and risk losses can occur. Persons who would have developed spontaneous cancers may be protected by low-dose exposure, while others exposed and who would not have developed spontaneous cancer may have added risk (likely very small if any) for cancer induction. The net population effect could be a reduction in the number of cancer cases after low-dose exposure, but the subgroup that would not have developed spontaneous cancers might have a higher cancer incidence. This poses some “food for thought” for the future discussion of protecting the public from low doses of genotoxic agents.
The NEOTRANS2 model has also been adapted for ethylene oxide (EO)-induced mutations in T cells of mice after inhalation exposure to a genotoxic chemical (Walker et al. 2002). The International Agency for Research on Cancer has classified EO (an immediate oxidative metabolite of ethylene, a normal body constituent) as a Group 1 human carcinogen based on sufficient evidence in animals with strong evidence in humans of a relevant carcinogenic mechanisms, rather than on support from epidemiological studies of EO-exposed workers (IARC 1994). For application to chemicals such as EO, the variable c then becomes the time-dependent exposure concentration or dose rate (e.g., for a critical metabolite) to target tissue. The indicated adapted model, called NEOTRANS2-EO, includes a postulated threshold concentration for the induction of intercellular signaling that leads to the protective bystander apoptosis effect as well a second threshold for excess mutations (Walker et al. 2002).
We have explained the low-dose protective effects against radiation-induced neoplastic transformation as being due to a bystander apoptosis effect. Another possible explanation would be the induction of a highly efficient repair process that is not available at background levels of irradiation. However, we do not consider the notion of induced efficient repair adequate to explain a decrease in risk for neoplastic transformation. For one reason, the induced repair process may not be highly efficient (for protecting against problematic cells) as recently demonstrated (Oudalova et al. 2002). The cited researchers demonstrated that moderate doses (> 400 mGy) induce a system of SOS response leading to elevated survival and elevated yield of chromosomal aberrations among surviving cells (meristem cells of spring barley). Secondly, with induced efficient repair, plating efficiency for low-dose-irradiated cells should then be significantly higher than for controls, which appears not to be the case.
It is important to mention the issue of genetic sensitivity. Some humans are highly sensitive to low-dose radiation-induced stochastic effects, due to inherited abnormalities in DNA repair. How the protective bystander apoptosis effect presented here relates to such individuals has not been resolved. One could speculate that such individuals would have high frequency of spontaneous neoplastic transformations. If so, how these individuals will be affected by low-dose radiation or low-dose chemicals would be expected to depend on their ability to mount a protective response via apoptosis. Since both DNA repair and apoptosis are regulated through signaling pathways that involve p53, it is possible that persons with inherited deficiencies in DNA repair will also have deficiencies in apoptosis. If so, such individuals may not benefit from the protective bystander apoptosis effect discussed.
CONCLUSIONS
Nonlinearity in the dose-response relationship for the risk of radiation- (or chemical-) induced problematic, nonlethal mutations and neoplastic transformation could arise via several mechanisms:
The induction of a protective bystander apoptosis effect whereby existing problematic cells are selectively eliminated.
Only error-free repair below a specific dose rate with misrepair occurring above that dose rate.
An induction of an error-prone repair process above a dose threshold whereby cells with significant genomic damage are more likely to survive. This would be expected to lead to an increase in mutations and neoplastic transformations and possibly for cancer cases (rather than a decrease).
Both 1 and 2 above would be expected to lead to a threshold for excess (relative to the spontaneous level) problematic mutations, excess neoplastic transformation, and possibly for excess cancers.
Footnotes
ACKNOWLEDGMENTS
Research supported by the U. S. DOE Office of Science and Office of Environmental Management. We are grateful to Drs. E. I. Azzam and J. L. Redpath for allowing use of their data. We are also grateful to Ms. Paula Bradley and her staff for editorial and graphics support provided and to the journal reviewers for their helpful comments.