
Abstract
Copper-coated steel containers are part of the engineered barrier system to permanently store Canadian nuclear fuel waste in a deep geological repository. This work models the dose rates (D Rs) at the container surfaces as a function of fuel age. It also utilises a humid-air radiolysis model to study the effects of D R and humidity on radiolytic oxidant production for conditions where unexpected early water intrusion reaches clay seal materials. Radiolysis of humid air produces HNO3. The HNO3 production rate in a condensed water droplet formed on a container surface was conservatively estimated by assuming that every •OH produced by primary radiolytic processes was immediately converted to HNO3 in the gas phase and that all of the HNO3 was absorbed in the water droplet. Also assuming that all of the nitric acid absorbed in the water droplet is consumed in corroding copper and using a hemispherical water droplet geometry, the corrosion depth of the copper coating induced by humid-air radiolysis is conservatively estimated to be 9.4 μm over the permanent storage time.
This paper is part of a supplement on the 6th International Workshop on Long-Term Prediction of Corrosion Damage in Nuclear Waste Systems.
Keywords
Introduction
Canada's long-term plan for used nuclear fuel includes storage in a deep geological repository (DGR) using a multiple-barrier system [1]. A key barrier is the used fuel container (UFC), which was recently redesigned. The current UFC design consists of an inner vessel made of carbon steel (CS) for structural strength and an outer Cu coating as an external corrosion barrier [2]. Compared to the previous design, the CS vessel thickness is reduced considerably (from 10 to 4.6 cm) and the 25-mm-thick outer copper shell is replaced with a 3-4-mm thick integrally applied copper coating [1,2]. The use of a pressure-grade CS vessel and copper coating improves the mechanical integrity by eliminating many fabrication issues associated with the previous design. However, the possibility of localised CS corrosion near the weld region and the general corrosion of the copper coating of the new design must be assessed carefully to ensure the integrity of the inner vessel and the adequacy of the copper coating for corrosion protection.
Concerns arise about whether moisture trapped inside a UFC could condense on the internal surface, particularly near the CS weld regions. Corrosion within the gap between the hemispherical head and the body of the container assembly or in the stressed regions near the welds could lead to localised corrosion and potential failure of the container. The local environment (liquid water, water vapour, and humid air) and the UFC metal components will be exposed to the ionising radiation (particularly γ-radiation) emitted by decay of radionuclides in the used fuel. The radiation energy absorbed by metal is dissipated mainly as heat but it can induce ionisation and decomposition of water and gas-phase molecules to yield redox active species [3,4]. For example, γ-irradiation of water droplets would produce oxidants such as H2O2, while humid-air radiolysis would produce NOx and HNO3 that can dissolve in the water droplets. Since the thinner container design will provide less shielding for γ-radiation, the effect of γ-radiation on copper corrosion must be evaluated to ensure that the coating provides corrosion protection.
In this paper, the effects of radiation on container corrosion during long-term disposal in the anticipated DGR environments are evaluated. Model calculations are used to determine the concentrations of radiolytically produced oxidants that may potentially contact the container. The evolution of γ-radiation dose rates (D Rs) with time for both internal and external surfaces of the current container design is calculated in the different aqueous environments (aerated and deaerated water, waters with impurity levels of solutes, saline water, water vapour) a container may experience under DGR conditions. This paper presents model calculations with a focus on the radiolytic production of HNO3.
Dose rates at the internal and external surfaces of a UFC
In these calculations, only the γ-radiation from used Canada Deuterium Uranium fuel inside a container was considered since the emitted α- and β-particles will be adsorbed within the fuel and the fuel cladding and, hence, will not contribute to either internal or external surface D Rs.
Internal and external UFC surface γ-radiation D Rs as a function of storage time were estimated for a base case involving used fuel with a burnup of 220 MWh kgU−1 using Microshield v9.05 and appropriate fluence to D R conversion factors [5]. Because the software is not designed to model non-homogeneities in the shielding materials, the source was represented as a single cylinder of used fuel (UO2) encapsulated in a layer of Zircaloy (modelled as zirconium) and the UFC was modelled as a layer of steel [6–8]. Moreover, MicroShield does not allow backscatter calculations (i.e. all dose points must be on the exterior of the modelled geometry). Therefore, the D Rs on the internal surface of the container were modelled without the steel vessel and copper coating. As a conservative measure, these internal surface D Rs were then multiplied by 1.3 to account for secondary electrons backscattering [9,10]. The full details of the radiation dose model, including the UFC and source geometries, with further details on the calculation assumptions and accuracy, and the results of three case that assess the sensitivity of the results to key assumptions, are provided in the supplementary information (SI-1). In general, the calculated D Rs are very sensitive to the source parameterisation and should therefore be interpreted with an appropriate level of uncertainty.
The D Rs calculated at the internal and external UFC surfaces, expressed as absorbed D Rs in air, are presented in Table 1 and Figure 1. For the first 500 y, the γ-D R is dominated by the decay of the fission products in the fuel and, hence, it decreases nearly exponentially with time. The D R is subsequently dominated by the decay of the actinides present and decreases nearly linearly from 500 to 1,000,000 y. The accumulated dose on the container surfaces is also provided for reference, although there is no simple relationship between accumulated dose and the extent of corrosion damage in a changing radiation field as claimed elsewhere [11], see further discussion later. To approximate the accumulated dose on each surface, the D R curves were fitted to mathematical functions (see supplementary information) and then integrated. The accumulated doses are shown in Figure 1.
Calculated D Rs and accumulated doses at the internal and external surfaces of the Canadian UFC, starting with 10-year-old fuel. Summary of internal and external UFC surface γ-radiation D Rs. aIncludes assumed contribution from back-scattered β-radiation near the internal surface. Fuel age refers to the time since the fuel was removed from the reactor.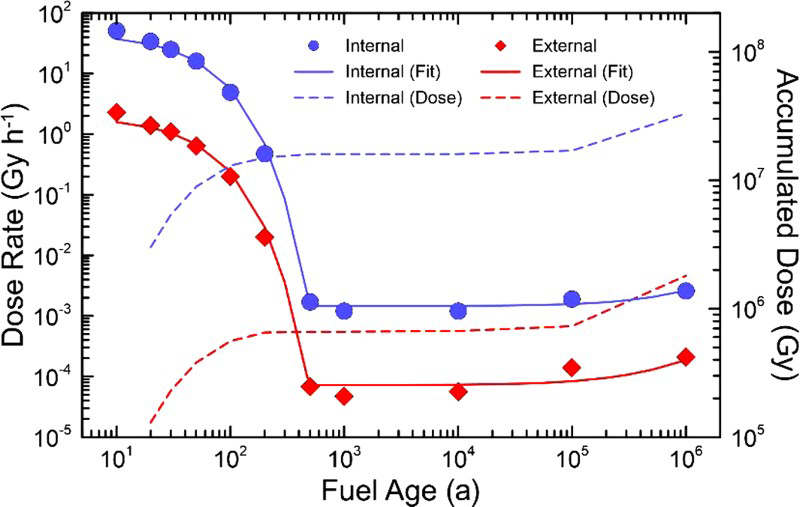

The D Rs anticipated at the internal surface would be 20-25 times greater than those on the external surface of the container in the first 10,000 years and then only about 10 times greater thereafter, owing to the time-dependent change in the γ-radiation spectrum, as described above. The D R to the external surface of a thin-walled container would decay from ∼2.3 to ∼0.02 Gy h−1 after 200 years. This is a significant (>20-fold) increase in the external fields compared to those anticipated in the previous thick-walled container [2].
Modelling approach for γ-radiolytic production of oxidants
The main objective of γ-radiolysis modelling is to determine the radiolytic oxidant concentrations that may affect the localised corrosion of the weld inside a UFC or the general/localised corrosion of the external copper coating. The initial environment inside a sealed UFC could be humid due to the combination of the ambient humidity of the air at the time of container sealing and that due to trapped water in the fuel bundles before encapsulation. For the localised corrosion of the CS weld region, γ-radiolysis of humid air and water droplets is the main concern. For the external copper coating, the DGR environment will evolve through a sequence of possible stages: (1) an initial aerated period with no condensed H2O on the Cu surface, (2) a period of aerated vapour in equilibrium with a condensed H2O layer on the surface, (3) a transition period to fully water-saturated air and potentially oxidising aqueous conditions, and (4) a final aqueous anoxic period after container corrosion and reactions with minerals and organic matter in the surrounding clay has consumed all of the available O2 [2].
To address the different corrosion exposure environments, calculations using three different radiolysis kinetic models have been performed: (1) water radiolysis, (2) humid-air radiolysis, and (3) radiolysis of highly saline water. Each calculation determines the changes in radiolysis product concentrations ([i]t) as a function of time by solving the rate equations of all the strongly coupled reactions that describe the chemistry of the environment. The processes that control the concentration of primary radiolysis product i under a continuous flux of radiation are as follows:
the primary radiolysis process that produces i; and the chemical reactions of i with itself and other chemical species, j, including other radiolysis products, and dissolved chemical or reactive surface species present in the corresponding water phase.
The primary radiolysis processes that ionise and dissociate molecules act nearly instantly upon absorption of radiation energy [12–15]. This leads to radiolysis decomposition products uniformly distributed within an irradiated volume within ∼100 ns following a radiation pulse input. The radiolysis products formed on this timescale are commonly referred to as primary radiolysis products whose chemical yields depend primarily on the total absorbed radiation energy and hence the primary radiolysis product yields are expressed per unit of absorbed radiation energy and are commonly known as g-values in units of μmol·J−1. Because the rate of radiation energy absorption by an interacting medium (D R) depends primarily on the density of the medium, the D R is typically expressed per unit mass in units of Gy (J kg−1)). For example, the g-values for liquid water radiolysis at room temperature (T) are given in brackets in reaction (R.1) [12,16]
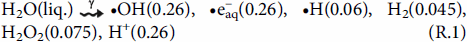
For the analysis of the radiolysis product concentrations on the timescale of corrosion reactions (>1 ms), modelling of the detailed kinetics of the radiolysis processes at very short timescales (<1 μs) is not necessary. Thus, the production rates of primary radiolysis products can be simplified to a rate proportional to its g-value (gi), D R, and the density of the medium, ρW
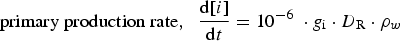
However, the primary radiolysis products are reactive and rapidly undergo chemical reactions with each other, with water and its acid and base ions, and with solute species, producing secondary radiolysis products (such as O2,  and
and 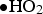 ). Owing to the long penetration depth of a typical γ-ray (∼20 cm for a half reduction in intensity in liquid water), the radiolysis products are created uniformly within a reasonably large volume of water. A consequence is that the concentration of a reactive species in the bulk phase is determined by a combination of the rates of primary radiolysis production and the chemical reactions of the species in solution
). Owing to the long penetration depth of a typical γ-ray (∼20 cm for a half reduction in intensity in liquid water), the radiolysis products are created uniformly within a reasonably large volume of water. A consequence is that the concentration of a reactive species in the bulk phase is determined by a combination of the rates of primary radiolysis production and the chemical reactions of the species in solution
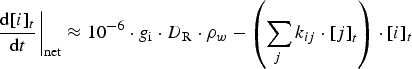
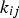 represents the rate constant of the chemical reaction between species i and j. Note that for a primary radiolysis product, the rate of any secondary production of this species via chemical reactions is typically negligible compared to the rate of the primary radiolysis process. The complex chemical kinetics involving many chemical species with strongly coupled reactions are solved numerically using commercially available software.
represents the rate constant of the chemical reaction between species i and j. Note that for a primary radiolysis product, the rate of any secondary production of this species via chemical reactions is typically negligible compared to the rate of the primary radiolysis process. The complex chemical kinetics involving many chemical species with strongly coupled reactions are solved numerically using commercially available software.
Specific models and the model calculation results that address the γ-radiolysis kinetics of liquid water containing dissolved oxygen, nitrate/nitrate, organic compounds (e.g. methyl ethyl ketone) and dissolved ferrous ions at a concentration level below <0.01 M have been reported elsewhere [17–19]. This paper focuses on the HNO3 production by humid-air radiolysis.
Nitric acid production by humid-air radiolysis
Homogeneous radiolytic production of HNO3 in the gas phase
Humid-air radiolysis produces nitric acid [20–22]. The HNO3 formed in the gas phase will be continually absorbed in the condensed water droplets in contact with the humid air. This will lower the pH of the water in the droplet and increase the concentration of nitrate, a potential oxidant for CS and copper. The homogeneous radiolytic production of HNO3 in the gas phase (HNO3(g)) is described first. The section ‘Aqueous concentrations of HNO3 in water droplets’ describes how the production rate in the gas phase may be used to estimate the production rate of HNO3 in a water droplet (HNO3(aq)). The section ‘Bounding estimates for corrosion extents by radiolytically produced HNO3’ describes how it may be used in obtaining the upper bounding estimate for the extent of corrosion that can occur due to the radiolytic production of HNO3 over the disposal period, and how the radiolytic production rate of HNO3 could be incorporated in a corrosion model are discussed.
To calculate the production rate of HNO3(g) by humid-air radiolysis and its rate of accumulation in condensed water droplets, a humid-air radiolysis model (HARM) has been constructed. The model is primarily based on the reaction set initially reported by Matzing and later modified by others [23–25]. For humid-air radiolysis, the absorption of radiation energy by the three main components of air (N2, O2, and H2O) results in the formation of a range of primary products that include electronically excited and ionised molecules, ions and free radicals [26].
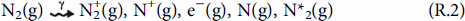
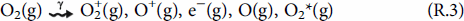
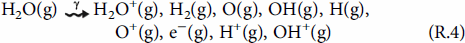
The current version of HARM consists of the primary radiolytic production processes and about 730 chemical reactions involving 25 primary radiolysis products and 95 secondary species.
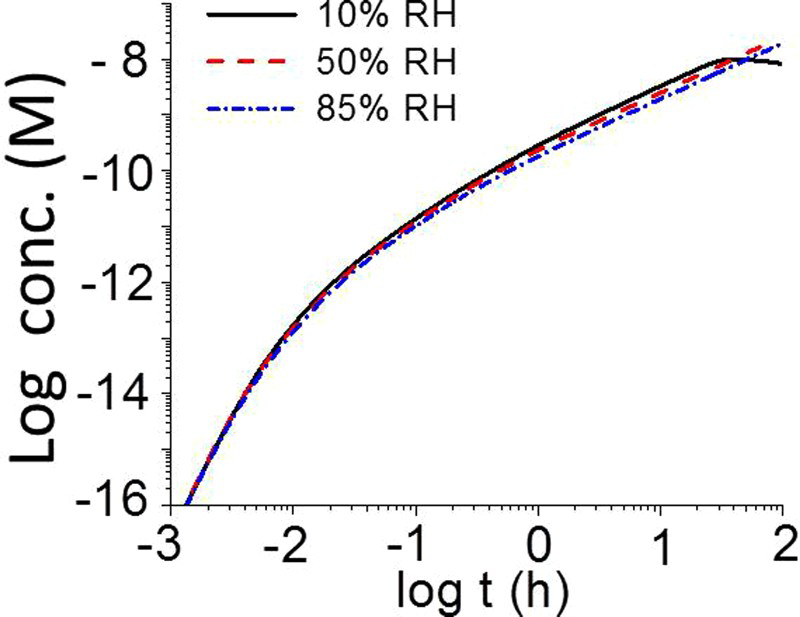
The HARM model was used to calculate the radiolytic production rate of HNO3 in the gas phase as a function of D R, relative humidity (RH), and T. The calculation results obtained as a function of RH at 75°C and at D R 1 Gy h−1 are presented in Figure 2. At a given T and D R, the main radiolysis product of dry air (0% RH) is O3(g) at very short irradiation times, but this changes to NO2(g) at longer times. When water vapour is present (as little as 10% RH), the main products are •OH(g), •HO2(g) at very short times, and HNO3(g) at longer times.
Time evolution of the concentrations of key radiolysis products formed during γ-radiolysis of 0, 10%, and 85% RH air at 75°C and a D R of 1 Gy h−1.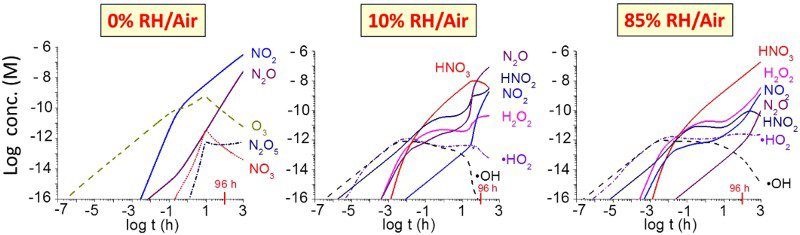
Detailed kinetic analysis of the modelling results suggests that the precursors for HNO3(g) production are •OH(g) and NO2(g), where NO2(g) is a secondary product of the γ-radiolysis of air.
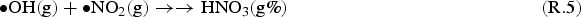
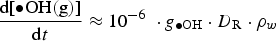
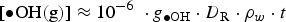
The full model calculation results (Figure 2) show that the overall production rate for HNO3(g) at times longer than a minute is mainly determined by the primary radiolytic production of •OH(g) and the competition rates for •OH(g) between •NO2(g) and H2O2(g). That is, the overall radiolytic production of HNO3(g) at times longer than a minute can be approximated as
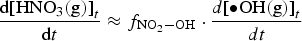
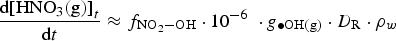
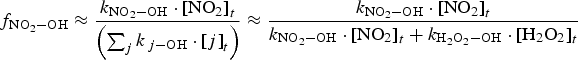
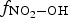 is the fraction of •OH(g) reactions leading to the formation of HNO3(g), and
is the fraction of •OH(g) reactions leading to the formation of HNO3(g), and 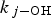 represents the rate constant of the chemical reaction between species j and •OH(g).
represents the rate constant of the chemical reaction between species j and •OH(g).
The fraction, 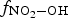 , changes rapidly with time at early times, but the change becomes much slower. Thus, for a given D R, RH, and T, Equation (7) shows that the overall production rate of [HNO3(g)]t can be approximated as
, changes rapidly with time at early times, but the change becomes much slower. Thus, for a given D R, RH, and T, Equation (7) shows that the overall production rate of [HNO3(g)]t can be approximated as
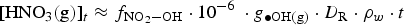
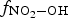 at times longer than 1 min is nearly one at 10% RH. As the humidity level ([H2O(g)]) increases while the concentrations of air molecules ([N2(g)] and [O2(g)]) remain constant, the primary radiolytic production of •OH(g) increases due to an increase in
at times longer than 1 min is nearly one at 10% RH. As the humidity level ([H2O(g)]) increases while the concentrations of air molecules ([N2(g)] and [O2(g)]) remain constant, the primary radiolytic production of •OH(g) increases due to an increase in 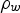 . However, the production of H2O2(g) also increases due to higher production of •OH(g) and •HO2(g) (via •H + O2). On the other hand, [H2O(g)] has a negligible effect on •NO2(g) production.
. However, the production of H2O2(g) also increases due to higher production of •OH(g) and •HO2(g) (via •H + O2). On the other hand, [H2O(g)] has a negligible effect on •NO2(g) production.
The overall effect of RH on Time evolutions of [HNO3(g)] produced by γ-radiolysis of air for different RHs at 75°C and at D R of 1 Gy h−1.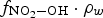 at times >1 min is thus relatively small. The plot of log [HNO3(g)]t vs. log t (Figure 3) is shifted by log (½ RH), showing that the overall production rate of HNO3(g) decreases by about a factor of ½ RH.
at times >1 min is thus relatively small. The plot of log [HNO3(g)]t vs. log t (Figure 3) is shifted by log (½ RH), showing that the overall production rate of HNO3(g) decreases by about a factor of ½ RH.
Equation (8) shows that the overall production rate of [HNO3(g)]t is proportional to D R for a given RH and T which is consistent with computational modelling results. The time evolutions of [HNO3(g)]t calculated for γ-radiolysis of 85% RH air at 75°C and different D Rs (0.01 to 10 Gy h−1) are compared in Figure 4. The computational modelling results show that the linear plot of log [HNO3(g)]t vs. log t also shifts to a higher [HNO3(g)]t proportionally to log (D R), confirming the linear dependence of [HNO3(g)]t on D R as defined in Equation (8). That is, the rate of production of HNO3(g), and [HNO3(g)]t at a given t, increase proportionally with D R.
Time evolutions of [HNO3(g)] produced by γ-radiolysis of 85% RH air at 75°C at different D Rs.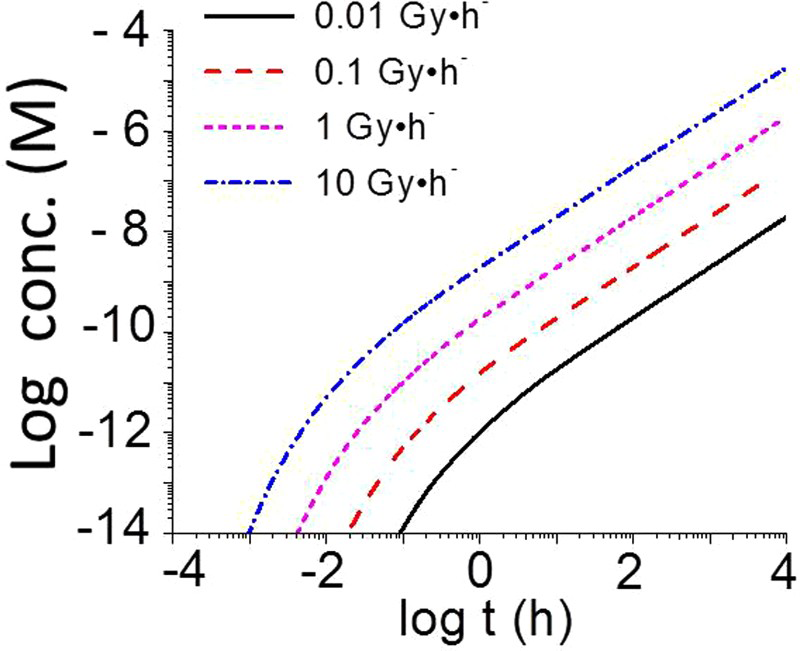
Aqueous concentrations of HNO3 in water droplets
As noted earlier, the calculations presented in Figures 2 –4 do not take into account any adsorption or surface reactions involving the radiolysis products. Furthermore, the calculated concentrations are those in the gas phase. For the degradation of metal, aqueous corrosion is the main concern. Thus, for the corrosion of the CS inner vessel and the copper coating of the container, the main concern is the HNO3 that condenses in water droplets on the container surfaces which can then participate in the electrochemical reactions associated with aqueous corrosion. The water in the droplets will also be subject to radiolysis, but γ-radiolysis of liquid water does not produce HNO3. Thus, the main route for the production of HNO3(aq) in a water droplet in contact with humid air in the presence of continuous flux of γ-radiation is the deposition of HNO3(g) formed in the gas phase by the humid-air radiolysis.
The accurate modelling of [HNO3(aq)]t in water droplets will require solving the kinetics of liquid water radiolysis coupled with humid-air radiolysis via gas–liquid interfacial transfer of radiolysis products and its consumption due to corrosion. The accurate modelling of [HNO3(aq)]t in the water droplets that may form on the Cu-coated container surface under the DGR conditions is futile and impractical because of an infinite combinations of water droplet and the headspace geometries. However, we can obtain the bounding estimates for [HNO3(aq)]t in a water droplet to assess the corrosion behaviour of a waste container. This section explores these bounding estimates.
The production rate of [HNO3(aq)]t in a water droplet is determined by the deposition rate of HNO3(g) formed by humid-air radiolysis onto the water droplet surface. This deposition rate increases with [HNO3(g)]t. The maximum [HNO3(g)]t can be achieved when the removal rate of HNO3(g) by the gas-phase chemical reactions of HNO3(g) is negligible, i.e.
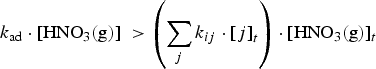
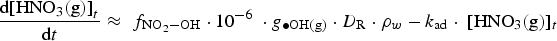
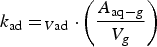
The corresponding rate equation for [HNO3(aq)]t, when the aqueous-phase and surface reactions of HNO3(aq) is negligible, is
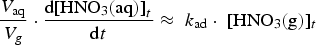
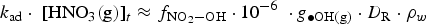
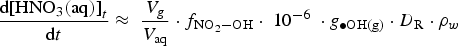
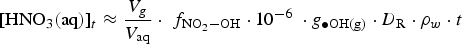
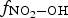 depends on
depends on 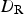 .
.
The current version of HARM calculates that the production rate of HNO3(g) in the gas phase at 85% RH and 75°C would be about 2.0 × 10−10 M h−1 at a D R of 1 Gy h−1. To determine the corresponding production rate of HNO3(aq) in a water droplet, we need to determine the effective ratio of V g/V aq. This ratio is estimated to be 100 based on the different diffusion coefficients of a compound in the gas and solution phases. The diffusion coefficient of a compound in the gas phase (D g) is about 10,000 times greater than that in the solution phase (D aq) [27]. Because HNO3 can adsorb on any surface equally easily, the diffusion of HNO3 from the gas phase to the container surface would be one dimensional. The ratio of one-dimensional diffusion length (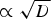 ) in the gas phase to that in the water droplet is 100.
) in the gas phase to that in the water droplet is 100.
For the volume ratio of air to a droplet of 100, this results in a [HNO3(aq)]t production rate in the water droplet of about 2.0 × 10−8 M h−1. If [HNO3(aq)]t continues to accumulate at this rate (without being consumed), the [HNO3(aq)]t in the water droplet could reach up to about 0.5 μM in a day, 0.18 mM in a year or 18 mM in 100 years at a constant D R of 1 Gy h−1. The D R at the external surface of the container is calculated to decrease from 2.3 to 0.2 Gy h−1 in 100 years (Figure 1). After 100 years, the D R decreases more rapidly and decreases to a value below 0.1 mGy h−1 in 400 years, and the radiolytic production of HNO3(aq) after 100 years would be significantly less. The simple analysis suggests that the [HNO3(aq)]∞ that would accumulate over the full waste disposal period would be less than 100 mM, assuming that all of the HNO3(g) produced radiolytically within a gas volume 100 times larger than the volume of a water droplet is absorbed in the water droplet.
Bounding estimates for corrosion extents by radiolytically produced HNO3
The rate of metal corrosion depends on many environmental parameters including the solution pH, T, and redox conditions. The rate of corrosion also tends to evolve with time, even under constant exposure conditions, because the metal surface changes as corrosion progresses. Efforts are under way to develop detailed models of the corrosion of the container materials, although the present use of these models to predict an exact corrosion allowance would be premature. However, the upper limit for the extent of copper corrosion due to radiolytically produced HNO3 can be estimated. Corrosion of copper produces two possible copper cations
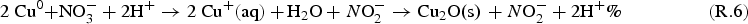
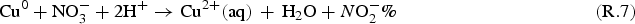
Assuming the volume ratio of air to droplet is about 100, the maximum cumulative nitric acid concentration over the permanent disposal period, [HNO3(aq)]∞, would be less than 100 mM. Using this concentration and a hemispherical droplet geometry illustrated in Figure 5, the upper limit to the depth of copper corrosion, d Cu, can be calculated as follows:
Schematics of (a) radiolytic and overall corrosion reactions, and (b) geometric parameters used in calculating corrosion depth.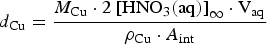
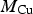 is the atomic mass of Cu (63.55 g mol−1),
is the atomic mass of Cu (63.55 g mol−1), 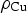 is the density of copper metal (8.96 g cm−3), and
is the density of copper metal (8.96 g cm−3), and 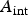 is the interfacial surface area covered by a droplet.
is the interfacial surface area covered by a droplet.
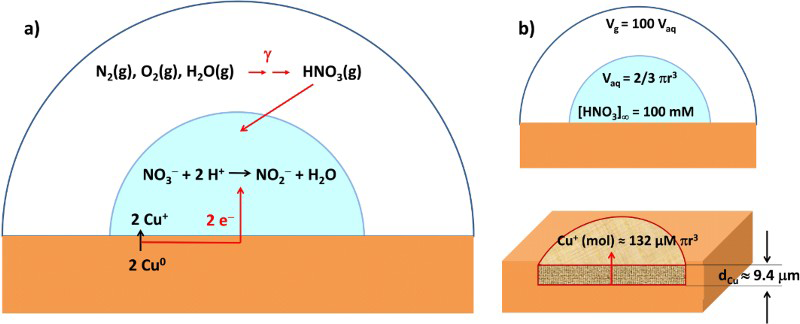
For a droplet with a radius of 1 cm and the [HNO3(aq)]∞ of 100 mM, the depth of copper corrosion by radiolytically produced HNO3 over the permanent disposal period would be
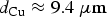
This value was obtained assuming that all of the HNO3(g) produced radiolytically within a gas volume 100 times larger than the volume of a water droplet is absorbed in the droplet. For a given volume ratio, the corrosion depth would increase with water droplet radius. If the air volume is fixed, the [HNO3(aq)]∞ would increase with decreasing water droplet radius. However, decreasing the water droplet radius also decreases the water droplet volume. The net effect of water droplet radius, r, on corrosion depth is that d Cu is inversely proportional to r 2.
It is often suggested that the extent of corrosion damage depends on accumulated dose and not on D R [11]. There are cases in which the overall corrosion may be determined by accumulated dose rather than D R but that is more of a coincidence than fundamentally determined. Any chemical or electrochemical reaction rate depends on the concentration of a reactant and not on the total amount of the reactant introduced into the reaction system over the entire reaction time. For chemical or electrochemical reactions (such as corrosion) induced by radiation, accumulated dose is not a fundamental parameter [16]. The concept that total dose rather than D R determines the effect of radiation on any chemical processes is only applicable for very short irradiation times when bulk phase chemical reactions of radiolysis products are not occurring at any substantial rates.
It has been also suggested that it is •OH and not HNO3 that is the dominant species that causes copper corrosion in the presence of radiation [28]. The mechanism by which humid-air radiolysis affects copper corrosion requires in-depth discussion that is beyond the scope of this paper. Irrespective of whether •OH or HNO3 is the dominant species driving corrosion, the estimate that we obtained for corrosion depth should still be bounding because it assumes that every •OH produced by the primary radiolysis process would eventually produce HNO3 and that all of the HNO3 produced that way would be consumed in corroding copper. However, it should be emphasised that the corrosion estimate does not take into account any radiolysis of water droplets. Radiolysis of liquid water produces H2O2 and O2 that will more effectively contribute to copper corrosion than •OH.
Conclusions
The γ-radiation D Rs at the internal and external surfaces of a Canadian copper-coated UFC were calculated using MicroShieldv9.05. As the used fuel ages from 10 to 106 a, the internal and external surface D Rs decay from 51 to 2.6 mGy h–1 and from 2.3 to 0.21 mGy h–1, respectively. Radiolysis of dry air initially produces O3(g) and later NO x (g) at longer irradiation times. In humid air, radiolysis produces initially •OH which can then react with air molecules and their decomposition products to produce nitric acid. The RH (10-80%) has no significant effect on the rate of HNO3 production. The HNO3 production rate in a condensed water droplet formed on a container surface was conservatively estimated by assuming that every •OH produced by primary radiolytic processes was immediately converted to HNO3 in the gas phase and that all of the HNO3 was absorbed in the water droplet. Also assuming that all of the HNO3 absorbed in the water droplet is consumed in corroding copper and using a hemispherical water droplet geometry, the corrosion depth of the copper coating induced by humid-air radiolysis is conservatively estimated to be 9.4 μm over the permanent storage time.