
Abstract
The effect of chloride on copper corrosion in anaerobic aqueous sulphide solutions has been investigated using corrosion potential measurements, electrochemical impedance spectroscopy, scanning electron microscopy, focused ion beam and inductively coupled plasma atomic emission spectroscopy. Long-term exposure experiments were performed in 10−3 mol L−1 Na2S solutions containing chloride concentrations in a range from 0.1 to 5 mol L−1. Chloride was found to (i) inhibit Cu2S film growth by displacing adsorbed sulphide from the Cu surface; (ii) induce film porosity; (iii) facilitate transport of Cu+ to the solution (at very high chloride concentrations).
This paper is part of a supplement on the 6th International Workshop on Long-Term Prediction of Corrosion Damage in Nuclear Waste Systems.
Introduction
Since copper is highly corrosion resistant and thermodynamically stable in the anoxic saline groundwater anticipated in deep geologic nuclear waste repositories, it is the candidate material for the manufacture of high level nuclear waste containers in Sweden, Finland and Canada [1–3]. Once oxygen, trapped in the repository on sealing, is consumed by microbial activity and mineral reactions in the bentonite clay compacted around the container, and by minor container corrosion, the major threat to the long-term durability of the container is corrosion by sulphide (SH−) produced in the groundwater by mineral dissolution and/or microbial activity involving sulphate-reducing bacteria [4,5]. For Swedish repository conditions, [SH−] is expected to be in the range 10−7 to 10−4 mol L−1, while [Cl−] can be up to 0.5 mol L−1.
Our previous work [6–8] showed that the corrosion product deposit formed on a Cu surface in anaerobic sulphide solutions in a range from 5 × 10−5 to 10−3 mol L−1 is a single layer Cu2S (chalcocite) film. Film growth occurs at the Cu2S/solution interface and is controlled by a combination of Cu+ diffusion in the film and SH− diffusion in solution. In stagnant solutions with a low [Cl−]/[SH−] ratio (≤500), the film appears compact, and its growth obeys a parabolic law. Under these circumstances, film growth is controlled mainly by Cu+ diffusion in the Cu2S film indicating the film is protective. However, when the [Cl−]/[SH−] ratio is ≥1000, the film has a cellular structure and its growth kinetics are linear. The film growth process is controlled primarily by SH− diffusion in solution, implying that the Cu2S film is not protective under these conditions [9]. These results suggest that the properties of the Cu2S film formed in anaerobic SH− solutions not only depend on [SH−] and [Cl−] but also on their ratio. However, when convection is involved, the properties of the Cu2S film are determined by the relative rates of diffusive sulphide flux and the interfacial reaction leading to the deposition of Cu2S [10].
The aim of the present study is to clarify the role of Cl− in the formation of the Cu2S films in stagnant anaerobic SH− solutions containing Cl−. A longer term goal is to determine whether the structure and properties of the sulphide films are determined by the SH− flux at the Cu2S/solution interface [10] or by the [Cl−]/[SH−] ratio.
Experimental
Sample preparation
All experiments were performed with phosphorous-doped (30-100 ppm), oxygen-free Cu (Cu-OF) provided by the Swedish Nuclear Fuel and Waste Management Co. (SKB), Stockholm, Sweden. Working electrodes were Cu discs, with a diameter of 1 cm, threaded onto a stainless steel shaft painted with a non-conductive lacquer to prevent contact of the Cu/steel junction with the aqueous solution. After painting, electrodes were heated at 60°C for 12 h to promote adhesion between the paint and the sample. Copper surfaces were ground successively with 240, 600, 800, 1000, 1200 grade SiC paper and then polished to a mirror finish using 1, 0.3, and 0.05 μm Al2O3 suspensions. Before experiments, electrodes were washed with Type I water (ASTM D1193-6
Electrochemical experiments
All experiments were performed in an Ar-purged anaerobic chamber (Canadian Vacuum Systems Ltd), maintained at a positive pressure (2-4 mbar) by an MBraun glove box control system, to ensure anoxic conditions were maintained (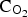 < 1 ppm). The [O2] in the chamber was analysed with an MBraun O2 probe.
< 1 ppm). The [O2] in the chamber was analysed with an MBraun O2 probe.
Electrodes were exposed to a 10−3 mol L−1 Na2S solution containing [Cl−] in the range 0.1-5.0 molL−1. The solutions used were prepared with Type I water, reagent-grade sodium sulphide (Na2S·9H2O, 98.0% assay), and reagent-grade sodium chloride (NaCl, 99.0% assay). Electrodes were immersed in the [SH−] solutions (1L) for various times under natural corrosion conditions. A standard three-electrode system was employed with a Pt plate as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. All potentials, unless otherwise specified, are quoted on this scale. Before each experiment, electrodes were cathodically cleaned at −1.6 V/SCE for 2 min, and then at −1.15 V/SCE for 2 min.
Because of the long duration of experiments, it was necessary to periodically monitor the [SH−] over the course of an experiment. As demonstrated previously, the pH of a SH− solution can be used to monitor the [SH−]. Consequently, the pH was recorded weekly, and SH− added, when required, to readjust its concentration to the original value [7].
Experiments were performed at ambient temperature, 25 ± 2°C. The corrosion potential (E CORR) was monitored, and electrochemical impedance spectroscopy (EIS) measurements were performed at E CORR using a Solarton 1287 electrochemical interface and a Solarton 1255B frequency response analyser. A sinusoidal potential perturbation with an amplitude (peak-to-zero) of 10 mV was applied over the frequency range from 105 to 10−3 Hz. Data were obtained at 10 frequencies per decade. The validity of the impedance spectra was checked using the Kramers–Kronig transform.
Surface and solution analyses
Electrodes removed from solution for surface analyses were rinsed with Type I water for 10 min and dried with cold Ar gas. Analyses were then performed immediately with the minimum period of interim storage (<30 min). The surface and cross-sectional morphologies of corroded specimens were observed using a Leo 1540 scanning electron microscope (SEM) equipped with a focused ion beam (FIB) (Zeiss Nano Technology Systems Division, Germany). The composition of sulphide films was qualitatively analysed by energy dispersive X-ray spectroscopy (EDS) using a Leo 1540 FIB/SEM microscope. The Cu content of solutions was analysed using plasma atomic emission spectroscopy (ICP-AES). Analyses were performed within two weeks to avoid conversion of the copper sulphide to insoluble copper oxide in the acidified sample used for the analysis.
Results and discussion
E CORR and EIS measurements
Cu electrodes were immersed in 10−3 mol L−1 Na2S solutions containing different Cl− contents in the range from 0.1 to 5.0 mol L−1 for an exposure time up to 1691 h (∼71 days). Over this exposure period, E CORR was monitored continuously with the rapidly attained steady-state E CORR being independent of [Cl−] within the range ∼ −960 ± 20 mV/SCE, Figure 1. This value is close to the equilibrium potential for Cu/Cu2S at this [SH−] (−1.01 V/SCE), suggesting that the corrosion film deposited on the Cu surface was Cu2S, as demonstrated previously by XRD [7].
Evolution of E CORR of copper with immersion time in 10−3 mol L−1 sulphide containing chloride in the concentration range of 0.1-5.0 molL−1.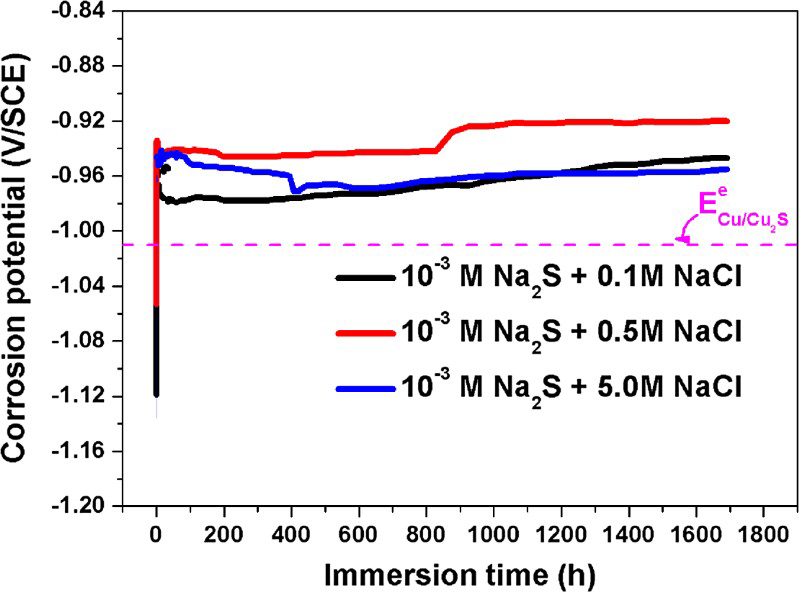
When [Cl−] was low (i.e. 0.1 mol L−1), EIS spectra exhibited two time constants attributable to the formation of Cu+ species at the Cu/Cu2S interface and the impedance properties of the Cu2S film (Figure 2(a)). With time, the resistance at the low frequency limit increased, consistent with the thickening of an, at least partially, protective film. When the [Cl−] was increased to 0.5 molL−1 the EIS spectra exhibited three time constants indicating a clear influence of Cl− on the properties of the film. The detection of a high frequency response, Figure 2(b), implied the charge transfer process at the Cu/Cu2S interface was accelerated suggesting porosity in the film. A further increase in [Cl−] (to 5.0 mol L−1) also yielded EIS spectra with three time constants with the high frequency response shifting to even higher frequencies, consistent with the presence of open porosity in a non-protective Cu2S film. Despite this apparent increase in porosity the impedance at the low frequency limit remained almost unchanged. A more detailed analysis of this impedance study will be published elsewhere.
Nyquist (a) and Bode (b) plots on copper after 1691 h immersion in: (▪) anoxic 10−3 mol L−1 Na2S + 0.1 mol L−1 NaCl solution; (●) anoxic 10−3 mol L−1 Na2S + 0.5 mol L−1 NaCl solution; and (▴) anoxic 10−3 mol L−1 Na2S + 5.0 mol L−1 NaCl solution.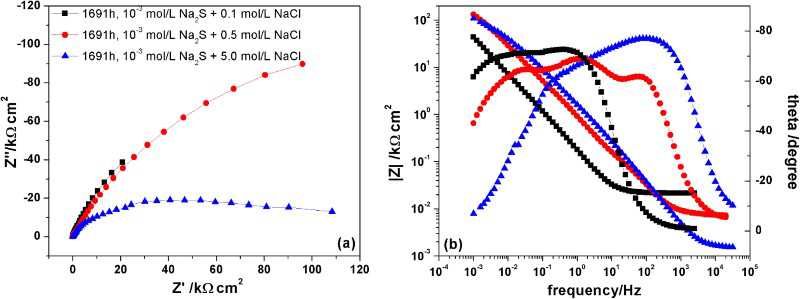
Surface and cross-sectional morphologies of Cu2S film
The surface morphologies of the Cu2S films formed after 1691 h immersion in 10−3 mol L−1 Na2S solution containing different [Cl−] are shown in Figure 3. In SH− solutions containing 0.1 mol L−1 Cl−, well-defined Cu2S crystals were formed and grew rapidly as corrosion continued, some up to ∼20 µm in dimension after 1691 h, Figure 3(a). For a [Cl−] of 0.5 mol L−1, the surface also became covered with a crystalline film but with crystals with much smaller dimensions than that formed at the lower [Cl−] over the same immersion period (Figure 3(b)).
Morphology of the Cu2S film formed on Cu after 1691 h immersion in: (a) 1 × 10−3 mol L−1 Na2S + 0.1 mol L−1 NaCl solution; (b) 1 × 10−3 mol L−1 Na2S + 0.5 mol L−1 NaCl solution; and (c) 1 × 10−3 mol L−1 Na2S + 5.0 mol L−1 NaCl solution.
When the [Cl−] was increased to 5.0 mol L−1, Figure 3(c), the Cu2S film grew rapidly but two-dimensionally and with a high porosity. Subsequently, for interim exposure periods (not shown here), the pores became blocked with agglomerates of nanoparticulates. However, over the extended exposure period (1691 h) these particulates were either lost or re-dissolved. The formation and subsequent disappearance of these particulates suggests Cu was mobile in extremely saline solutions, a process which appears to be attributable to the variation in chemical conditions within the rapidly grown porous film. We have speculated that this could be attributed to the formation of CuCl2 − soluble complexes which allowed transport of Cu+ out of the pores before precipitation of Cu-sulphide clusters. That Cu transport occurred in this concentrated Cl− solution was confirmed by analyses of the solution which showed concentrations of 520 ppb. Analyses after the experiments at lower [Cl−] detected no soluble Cu (detection limit 4 ppb). The mode and form of Cu transport has been addressed elsewhere [11].
Figure 4 shows cross-sections cut using FIB after 1691 h of immersion in the three different Cl− solutions. It is clear that the Cu2S deposit was comprised of a single layer in all cases. At the lowest [Cl−] the film is compact and protective, Figure 4(a) as indicated by the EIS measurements. As the [Cl−] was increased the film became increasingly more porous, Figure 4(b,c), again consistent with the EIS measurements.
FIB cut cross-sections of the Cu2S film formed on Cu after 1691 h immersion in: (a) 1 × 10−3 mol L−1 Na2S + 0.1 mol L−1 NaCl solution; (b) 1 × 10−3 mol L−1 Na2S + 0.5 mol L−1 NaCl solution; and (c) 1 × 10−3 mol L−1 Na2S + 5.0 mol L−1 NaCl solution.
Film growth kinetics
Besides influencing the morphology and porosity, the [Cl−] influenced the film growth rate, the film thickness decreasing as [Cl−] increased, suggesting Cl− inhibited the film growth process. Because the deposits are non-uniform, the thickness varies with location. Consequently, film thicknesses were measured at various locations and the variability represented in the plots by the standard deviations. This procedure yielded a reasonable measure of film growth kinetics at the two lowest [Cl−] but not at the highest concentration for which the apparent film thickness was not representative of film growth kinetics due to the high porosity and the loss of Cu to the solution.
In general, the film growth kinetics can be empirically described by the power law [7,8]
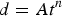
Figure 5 shows the evolution of the corrosion film thickness as a function of immersion time for the two lower [Cl−]s. For [Cl−] = 0.1 molL−1, the film growth kinetics obey a parabolic law, a linear least squares fit to the data yielding values for A and n of 58 and 0.5, respectively, with a fitting dependency of >0.98. This parabolic growth law suggests the diffusive SH− flux was larger than the interfacial film formation rate consistent with control of the film growth process by Cu+ diffusion in the film [7,9], as suggested by the EIS results.
Sulphide film growth laws on Cu in anaerobic sulphide solutions containing different chloride concentrations.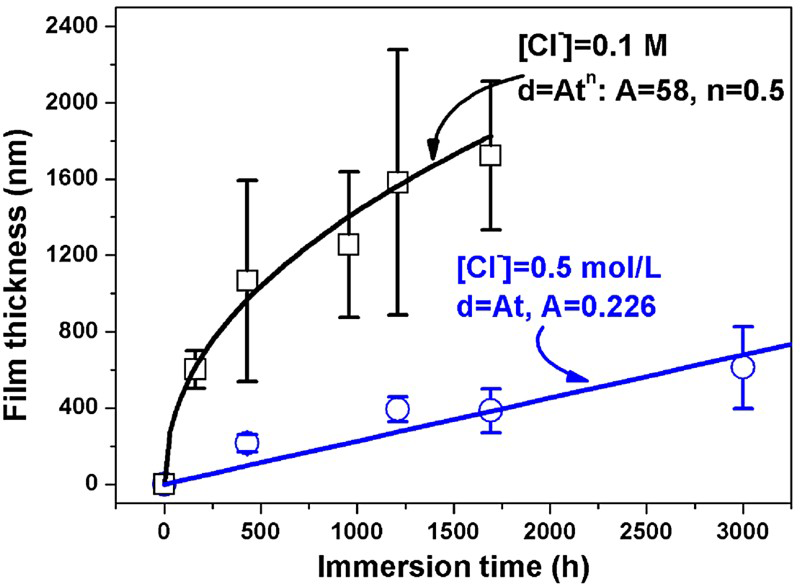
When the [Cl−] was increased to 0.5 molL−1, the Cu2S film grew linearly with immersion time. The linear fit to the data in Figure 3 yields values for A of 0.226, with a fitting dependency of >0.98. According to our previous work [8], this corresponds to an average film growth rate of ∼1.98 μm/year. The linear increase in film thickness with immersion time indicates that the corrosion film was not protective over this time period (∼125 days), with the interfacial reaction rate being larger than the diffusive SH− flux. Under this condition, the Cu content in solution was below the detection limit, indicating that Cu degraded by sulphide is completely sequestered as corrosion product deposited on the Cu surface. Thus, the decrease in film growth rate despite the increased film porosity may be attributable to the ability of Cl− to displace adsorbed SH− from the exposed Cu surface at the base of the pores, the essential first step in the overall corrosion process. A detailed electrochemical investigation of the anodic reaction is under way.
Conclusions
The properties and structure of sulphide films formed by corrosion on Cu surfaces were investigated in stagnant, anaerobic 10−3 molL−1 sulphide solutions containing different chloride contents in the range 0.1-5.0 mol L−1. The main conclusions drawn were as follows:
At low [Cl−] (0.1 mol L−1), the Cu2S film was protective and exhibited a parabolic growth law. At a higher [Cl−] of 0.5 molL−1, EIS data indicate the Cu2S film was porous. This was confirmed by micrographs on FIB cut cross-sections. The film grew linearly with time indicating it was not protective. At a very high [Cl−] (5.0 molL−1), the Cu2S film grew two-dimensionally with extensive porosity. The film growth process appeared to be controlled by a combination of SH−diffusion in the bulk of the solution and CuCl2 − in the pores of the cellular sulphide film. Solution analyses confirmed that Cu was released to the bulk solution. Chloride influences the Cu2S film growth process in a number of ways: (a) it displaces adsorbed SH− from the Cu surface thereby inhibiting the critical first step in the corrosion process; (b) it induces and maintains porosity in the film; (c) at extremely high concentrations it facilitates transport of Cu into the solution possibly by transport as CuCl2 − in the pores in the film and as Cu-sulphide complex/clusters in the bulk solution.
Footnotes
Acknowledgements
The authors would like to thank Dr T. Simpson for his help with SEM/FIB, and Christina Lilja (SKB) and Fraser King (Integrity Corrosion Consulting, Nanaimo, BC, Canada) for many helpful discussions.
Disclosure statement
No potential conflict of interest was reported by the authors.