
Abstract
The purpose of the present study was to determine if there is evidence to support the hypothesis that schizophrenia is a human-specific disorder associated with the need for highly complex central nervous system (CNS) development. A review was therefore undertaken of published literature relevant to the identification of human-specific CNS development. There was no clear evidence found at the macroscopic, microscopic or molecular level that suggests unique changes have occurred in the evolution of the human CNS. Rather, highly significant changes in the size of the frontal lobe, increases in numbers of specific cell types, changes in gene expression and changes in genome sequence all seem to be involved in the evolution of the human CNS. Human-specific changes in CNS development are wide ranging. The modification in CNS structure and function that has resulted from these changes affects many pathways and behaviours that appear to be also affected in subjects with schizophrenia. Therefore there is evidence to support the hypothesis that schizophrenia is a disease that develops because of derangements to human-specific CNS functions that have emerged since our species diverged from non-human primates.
It has been argued that language is a ‘sapiens-specific capacity’ that arose from an evolutionary split between Homo sapiens and other hominid species (Figure 1) [1]. Furthermore, the development of language must have required the human central nervous system (CNS) to be able to accurately separate thoughts from actual speech and auditory inputs. Derangements in the capacity of the CNS to carry out these functions could clearly result in symptoms such as auditory hallucinations. The argument that schizophrenia is a disease unique to humans has also been made based on data from CNS gene expression studies [2]. From these studies a hypothesis has been proposed that posits that there is a great demand for energy by the CNS to maintain human cognitive ability. The hypothesis goes on to argue that, in some individuals, a failure of the CNS to produce adequate amounts of energy to maintain normal levels of functioning results in the onset of the symptoms of schizophrenia.
Late evolutionary human phylogenetic tree.
The hypotheses that schizophrenia is a disease unique to humans is intriguing and, if correct, focusing on human-specific CNS capabilities might accelerate the unravelling of the complex paradigms underlying the pathology of the disorder. Hence, this review will attempt to identify mechanism that may make the human CNS a unique organ and consider how derangements in these mechanisms could result in the onset of the symptoms of schizophrenia.
Macroscopic features unique to human CNS function
Language is one of many complex vocalization skills in the animal kingdom. This premise has underpinned a comparison of the auditory processing areas in the CNS of humans and bats, another species capable of complex vocalization. These studies suggest that the lateralization of language processing may be a unique way in which humans manage to process and produce complex vocalizations [3]. This postulate, however, is not supported by studies showing that lateralization of auditory processing occurs in primates [4]. A more defensible position is that auditory lateralization is more pronounced in humans compared to primates, for example the lateralization of mini-columns in human auditory cortex is absent in primates [5].
The CNS pathways underpinning language have been extensively explored in humans. One particularly relevant finding has emerged from the use of positron emission tomography (PET) to study CNS activation in individuals with glossolalia [6]. Subjects with glossolalia vocalize incomprehensible language that, to them, has great personal meaning. During vocalization subjects with glossolalia have been shown to underactivate the prefrontal cortex (PFC) compared to vocalizing non-glossolalia subjects. These data suggest that the PFC may be particularly important in linking thought processes to comprehensible speech.
In bringing together comparisons of auditory processing in primates and humans with the data on glossolalia, it could would appear that changes in the lateralization of auditory processing and altered PFC functioning during vocalization could be involved in the pathology of schizophrenia. This argument has merit because it has long been acknowledge that abnormal PFC functioning is apparent in subjects with schizophrenia [7]. In addition, it has been shown that monozygotic twins discordant for schizophrenia have altered language processing lateralization when compared to healthy monozygotic twins [8]. In particular, the unaffected twins have increased right hemisphere activation. Hence, recent findings seem to support the hypothesis that at least some of the human-specific changes in CNS structure/function that have evolved to accommodate the development of language may also be involved in the genesis of some of the symptoms of schizophrenia.
Humans as a species have a relatively large CNS, but studies in other animals have shown that a large CNS per se is not associated with high levels of CNS function [9]. Significantly, a characteristic of non-primates with large CNS is a relatively low neuron density in the cerebral cortex. This would suggest that the advancement of higher cognitive abilities may be linked to both large CNS and increased neuronal density in the cortex. Indeed, it has been proposed that complex CNS function in humans has been able to evolve solely because of an increase in the number of neurons throughout the human cortex [10].
Focusing further on the human cortex, one of the most notable differences in the CNS of humans and non-human primates is the relative size of the frontal lobe. This region of the CNS has increased dramatically, both in terms of absolute size and proportion of whole CNS in humans compared to non-human primates [11]. A better understanding of the functional role of the frontal lobe (Brodmann's area 10) is emerging as a result of neuroimaging studies that have shown this area to be important in episodic memory [12], risk benefit assessment [13], moral conundrums [14] and reward-based learning [14]. Given that episodic memory and reward-based learning are affected in subjects with schizophrenia [15, 16], it would seem possible that functional abnormalities in the frontal lobe could be contributing to the symptoms of schizophrenia.
The notion that the frontal lobe is affected in schizophrenia is supported by data from neuroimaging studies that suggest that there are both structural and functional changes in that CNS region in subjects with the disorder [17, 18]. Moreover, neuroimaging studies are now clearly showing that there are complex interactions between the frontal lobe and neighbouring cortical regions [19]. Understanding how these interactions may differ in humans and non-human primates may shed light on whether deficits in cortical–cortical interactions may also underlie the symptoms of schizophrenia.
Dividing the cortex on cytoarchitectural grounds has allowed progress to be made on understanding which regions of this complex structure contribute to different CNS functions [20]. More recently, neuroimaging studies are beginning to localize functions of the human brain to specific CNS regions and, in the cortex, such studies have localized functions such as action monitoring, self-knowledge, person perception, mentalizing and outcome monitoring to specific areas of the medial frontal cortex [21]. These studies do not necessarily localize such functions to a single cytoarchitectural region. Of course one of the issues in determining if CNS function is localized to analogous regions of human and non-human primate CNS relates to whether primates can complete equivalent tests to those completed by humans. This is clearly important with regards to cognitive functions because there are well-defined differences in cognitive abilities in humans and non-human primates [22]. This could severely limit the information that will be able to be obtained on the unique activation of the human CNS during the completion of complex tasks. Recent data, however, suggest that certain complex behaviours that were thought to be unique to humans, such as cognitive dissonance, are detectable in non-human primates [23]. Therefore there may be opportunities to better understand the foci of the cognitive deficits associated with schizophrenia [24] by identifying human-specific CNS regions involved in maintaining cognitive function.
Cellular findings unique to human CNS
There do not appear to be many clear-cut differences in the cellular content of human and primate CNS, other than that the human CNS contains much higher neuronal densities [9]. But there are large, spindle-shaped cells in layer 5 of the anterior cingulate that appear to be unique to the primate CNS [11]; and these cells are larger and more abundant in the human CNS. Intriguingly, spindle cells are apparent in the anterior cortex of primate CNS from embryonic day 224 [25] but they do not become apparent in human CNS until birth, after which they appear to migrate into laminae V of the anterior cortex over several months [11]. This suggests that the late development of spindle cells is a human-specific phenomenon and it may be related to the anterior cingulate developing more slowly in humans. Significantly, laminae V of the anterior cingulate also contains another primate-specific cell type: pyramidal cells that contain the calcium-binding protein calretinin [26]. Given the presence of primate CNS-specific cell types in laminae V of the anterior cingulate, and the recognized importance of this CNS region in the pathology of schizophrenia [27], a study focusing on laminae V of the anterior cingulate would be worthwhile in subjects with schizophrenia.
The most dramatic event in the maturation of the human CNS is the rapid growth in synaptic numbers in the first 2 years of life [28]. This event is followed by the dramatic loss of synaptic number and a smaller loss in neuronal number, which proceeds until late adolescence. Intriguingly, modelling of this process suggests that pruning may occur to ensure that the energy requirements for the maintenance of synaptic function are able to be sustained in the face of limited CNS metabolic resources [29]. In contrast, another suggestion is that high synaptic density in the developing mammalian CNS is critical to allow the intense learning that is required in that phase of development [30]. In this latter model, synaptic pruning may reflect a decrease in the need for the CNS to support such a dramatic rate of learning later in life. Importantly, in all mammals the CNS appears to have a period of synaptic stability early in life, this being longest in humans, lasting to approximately 5 years of age compared to approximately 2 weeks in the macaque [29]. Similarly, the process of synaptic pruning is longest in humans and closely precedes the peak age of onset of schizophrenia [31], with synaptic pruning likely to be ongoing during the prodromal phase of the illness. If the argument that schizophrenia results from an abnormality in human-specific CNS functions is viable, then, as has been suggested previously [32], it would seem that abnormalities in synaptic development and/or pruning could underlie the disorder.
The human CNS has approximately 10-fold the number of neuroglia compared to the number of neurons, hence the neuroglia/neuron ratio is highest in humans [33]. In primates and humans there are unique populations of astrocytes [34], one of which originates in cortical laminae I, extending long fibres into other laminae of the cortex. The other are polarized astrocytes in cortical lamina V and VI that project distinctive long processes and in humans are much more developed with more complex structures. Neuroglia also seem to have critical roles in maintaining contact between themselves, neurons and the capillary cells of the CNS [35], with this connectivity being most complex in humans [34]. In addition, it has become clear that many synapses require a close interaction between pre- and post-synaptic neurons, as well as enveloping neuroglia, to maintain their normal function [36]. At present there seem to be no data on whether these synapses, termed ‘tripartite synapses’, are particularly over- or under-abundant in the CNS of humans compared to primates. What is becoming clear is that neuroglia are providers of energy substrates to neurons [37] and any derangement in this relationship could contribute to a state of energy deficiency that is proposed to contribute to the pathology of schizophrenia [2].
At a human neuroglia-specific level, it appears that many more human cortical microglia express the SIGLEC11 gene compared to chimpanzees and orang-utans [38]. SIGLEC11 is involved in sialic acid biology and therefore these data could suggest that human microglia must have uniquely harnessed sialic acid pathways in recent evolutionary times. This argument is supported by the presence of human-specific mutations in genes in sialic acid pathways [39]. Because sialic acid pathways are involved in the immune response the presence of an increased number of microglia expressing sialic biology-related genes may indicate that human microglia have more diverse mechanisms for responding to immune or inflammatory challenges. Independent of functional consequences, however, the uniquely human expression characteristics of SIGLEC11 make it a gene of interest in schizophrenia. This notion is supported by studies on a closely related gene, SIGLEC4A or MAG, which has already lead to this gene being cited as a potential risk gene for schizophrenia [40].
Molecular findings unique to human CNS
Having entered the era of ‘omics’ it is now possible to begin to identify variations in genomes across species. With the availability of the draft human and chimpanzee genomes differences in nucleotide sequences between ourselves and our closest species can be readily identified. These differences suggest that human genome-specific sequence variations have contributed to the development of habitual bipedality, a greatly enlarged CNS and complex language [41, 42], the latter two features being of particular significance to schizophrenia.
The completion of the human and chimpanzee genome has shown that at the level of single nucleotide polymorphisms (SNPs), the mean rate of sequence difference between the genomes is 1.23% [43–45]. In context, even at this low per cent variation, there must be >1 million SNPs that differ between chimpanzees and humans. This means that variation in gene sequences will exist between humans and non-human primates and that these variations could well contribute to changes in CNS development. Extending this hypothesis, the level of human–chimpanzee divergence is significantly higher on Y chromosomes and less on the X chromosome [46]. This could be interpreted as making the Y chromosome of particular importance with respect to human evolutionary divergence. Care needs to be taken, however, in assessing divergence in humans and non-human primates because it has been suggested that changes in the Y chromosome sequence are chimpanzee driven, and are related to female chimpanzees mating with multiple partners. This female behaviour has led to changes in chimpanzee Y chromosome genes that relate to spermatogenesis and sperm motility in a way that would confer advantages in the face of polygamous mating. The idea that changes in genes on the Y chromosome may not be human specific and therefore are not related to the potential to develop diseases such as schizophrenia is supported by the lack of data to suggest that the genes on the human Y chromosome are of particular importance with regards to susceptibility to developing the disorder (http://www.schizophreniaforum.org/res/sczgene/default.asp).
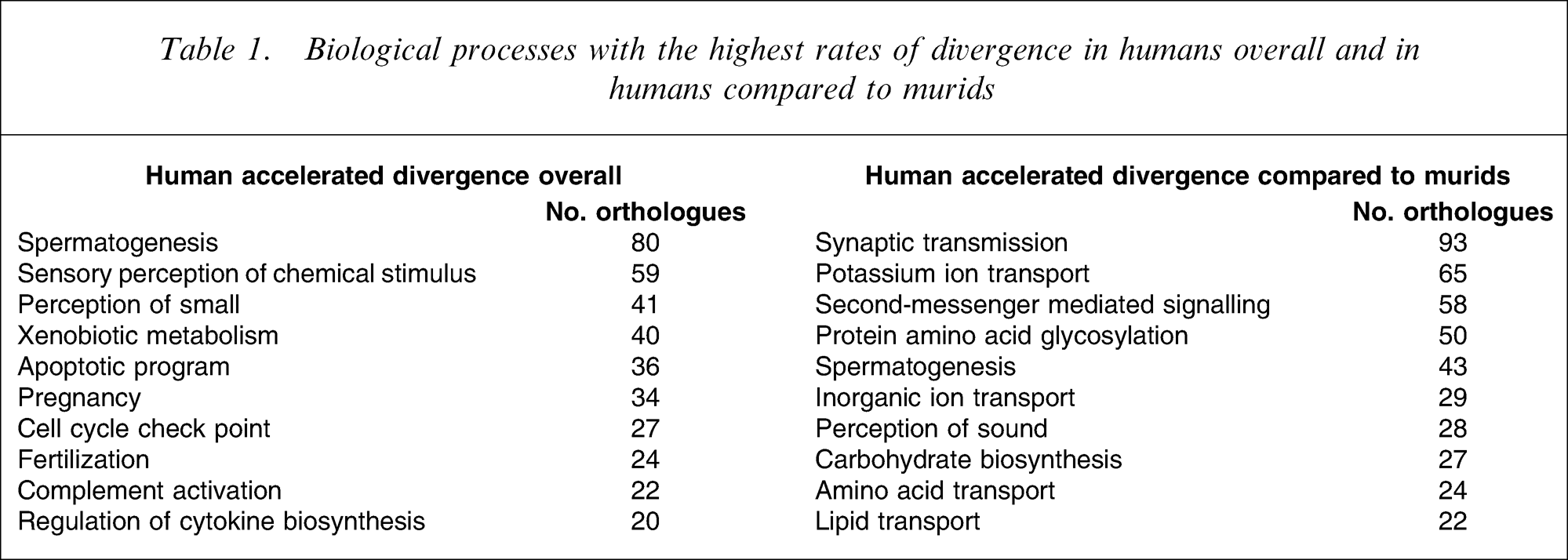
It is now becoming increasingly clear that variation in nucleotide sequence only constitutes the base level of comparison of inter-species genomes and that insertions, deletions and gene duplication add at least another 1.5% to genome variability [45]. This takes the overall variation in the genome of humans and chimpanzees to at least 2.7%. Significantly, the categorization of genes that have shown the highest rates of divergence or are showing accelerated divergence in humans suggests that human-specific evolution has occurred in a wide range of functions (Table 1). Significantly, functions such as perception of smell, apoptotic programming, pregnancy, lipid transport, second messenger signalling, perception of sound and synaptic transmission have all been reported to be affected by the pathology of schizophrenia [47–57]. Therefore, this relatively superficial comparison of the human and chimpanzee genomes tends to support the notion that schizophrenia could have arisen in humans as a consequence of species-specific variation in the human genome. It is also of interest that the largest biological process displaying differential sequence divergence between humans and murids is synaptic transmission. Because the family muridae contains mice and rats, two of the commonest species used in laboratory research, genomic data would suggest caution in freely interpreting data from such animal models to what may be occurring in primates.
Biological processes with the highest rates of divergence in humans overall and in humans compared to murids
A comparison of genomes across species that particularly focused on DNA duplication in the human and chimpanzee genome has reported a bias for human-specific duplications in regions of chromosomes 5 and 15 [58]. Significantly, there is a recent report of a significant association between copy number variation deletions on chromosome 15 (15q13.2) and an increased risk of schizophrenia [59]. In addition, there are 44 genes associated with an altered susceptibility for schizophrenia associated with chromosome 5, and 16 genes on chromosome 15 (http://www.schizophreniaforum.org/res/sczgene/default.asp). Thus, it would seem probable that gene sequence changes on chromosomes 5 and 15 will be linked to an altered susceptibility for schizophrenia.
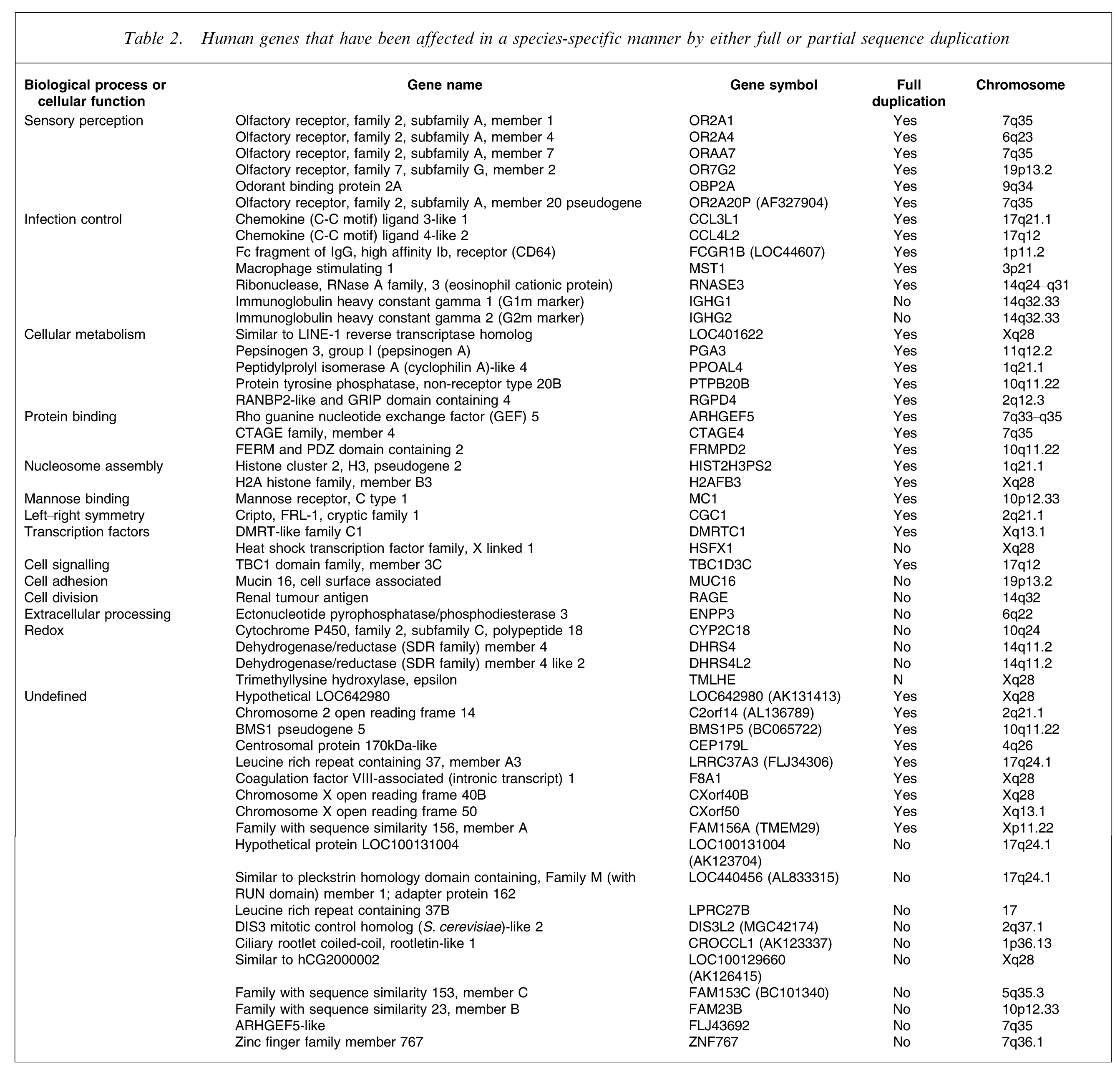
It has been suggested that ‘genomic triangulation’ produces more information on species-specific genome modifications than simple between-species comparisons [60]. This approach requires a comparison of three genomes, two of which form a monophyletic group, and looks for species-specific breakpoints in and unrooted phylogenic tree (a tree in which all branches are not connected to a common ancestor). By using this approach, which is more focused on identifying sequence insertions, deletions and inversions of nucleotide sequences, 58 human genes have been identified as being affected by insertions, with full gene copies being included in 36 insertions. In addition, 36 genes were shown to have undergone full gene duplication. These affected genes are involved in sensory perception, infection control, cellular metabolism, protein binding, nucleosome assembly, mannose binding, left–right CNS symmetry, transcriptional regulation, redox activity, stimulus response, signalling cascades, cell adhesion, kinase activity, transferase activity, ribonuclease and guanosine triphosphatase-mediated activity (Table 2). Perhaps, not unexpectedly, many of these diverse pathways and functions have been suggested to be affected in schizophrenia.
Human genes that have been affected in a species-specific manner by either full or partial sequence duplication
To add to the complexity of genome-based comparisons, it has been shown that the affects of ageing on CNS gene expression are dramatically different in humans and non-human primates [61]. These findings have implications for the suitability of different animal models for the study of CNS ageing, if the effects of ageing on gene expression are not consistent across species. This observation may also be relevant to schizophrenia, in which changes in gene expression profiles differ between subjects with schizophrenia early and late in the disease process [62, 63]. These data could be interpreted as suggesting that differential age-related changes in gene expression occur in subjects with schizophrenia, and this may underpin the long-recognized evolving symptom profile that is associated with the disorder [64]. These data also raise the possibility that changes in human-specific age-related gene expression may be involved in the pathology of schizophrenia, rather than simply global disease-specific changes.
It is now clear that much of the genome that was thought to be non-coding sequence, and therefore considered of lesser significance, may encode microRNAs, which are critical in regulating human gene expression [65]. MicroRNAs suppress gene expression by binding to the cis-regulatory site of transcribed mRNA to repress protein translation. It has now been shown that many microRNAs are primate specific [66] and it has been estimated that approximately 14% of all microRNAs in humans will be species specific. Given that 1 to 2% of the human genome encodes for microRNA [67], there will be a large number of human-specific microRNAs and therefore human-specific mutations in the sequence for microRNAs could underlie schizophrenia. It has been suggested that microRNAs might act as a lock-down mechanism to stabilize an already differentiated state [65]. Therefore human-specific microRNAs could be involved in stabilizing expression changes in the human CNS; their role in the pathology of disease states may be to stabilize changes in gene expression resulting from either gene mutations or gene–environment interactions. In such a situation, understanding the outcomes of changes in microRNAs that have been reported in the CNS from subjects with schizophrenia [68–70] could be an important pointer to changes in the expression of critical genes conferring altered susceptibility to developing the disorder. A much more extensive knowledge, however, of this new area of gene expression regulation will be required to identify microRNAs that may be involved in diseases such as schizophrenia. This is especially the case given that individual microRNAs have minimal control of protein expression [71, 72], and that the targeting of one gene product by multiple different microRNAs is required for any potent effect on protein expression.
It is now increasingly acknowledged that schizophrenia presents in subjects with genetic predisposition after they encounter as-yet-to-be-identified environmental factors [51]. Thus, it could be argued that the ability of the human CNS to modulate gene expression and how that is affected by specific environmental factors could be central to understanding the symptoms of schizophrenia. Relevant to this review, attempts have been made to identify human-specific gene expression patterns in CNS using microarrays. Hence, one study has compared gene expression at the level of mRNA in the CNS (Brodmann's area 9 or homologues) from humans, chimpanzees and orang-utan [73]. Cluster analyses of these data showed that inter-species variation in gene expression was substantial and was much greater in the CNS compared to liver. Another study using a ‘transcriptomic triangulation’ approach using CNS from humans, chimpanzee and macaques reported that 91 of 169 across-species differentially expressed genes were human specific [74]. In that latter study the differential expression observed in human CNS was absent in heart and liver and, for some genes, differential CNS expression was confirmed using in situ hybridization and immunohistochemistry. Genes with altered human CNS-specific expression were reported to be involved in glutamatergic neurotransmission, signal transduction, neurotransmitter release, cell chaperones and cell structural integrity, all of which have been reported to be affected in studies of gene expression in CNS from subjects with schizophrenia [75]. Moreover, some of the genes showing human-specific CNS expression [74] have already been suggested to be candidate genes for schizophrenia, unipolar depression [76], bipolar disorder (all CAMK2A) [77] and amyotrophic lateral sclerosis (KIF3A and DCTN1) [78, 79].
Genes and environment
The mechanisms by which genes and environment can interact are becoming increasingly understood, which for a disorder such as schizophrenia that is proposed to result from the interactions of these two variables [51], is of paramount importance. The processes by which environmental factors can modulate gene expression are known as epigenetic mechanisms [80]. DNA methylation, probably the most investigated epigenetic mechanism, involves the methylation of cytosine at position C5 in CpG dinucleotides, which affects the ability of a gene to be transcribed [81]. To better understand the impact of varying environments on a standardized genetic background during a human lifetime one study has examined differential DNA methylation in tissue (lymphocytes, epithelial skin cells, muscle and abdominal tissue) from monozygotic twins at various ages [82]. That study found approximately 5000 differentially methylation sites in monozygotic twins at 3 years of age compared to approximately 75 000 differences between 50-year-old monozygotic twins. That study clearly shows that lifetime environmental exposure can impact dramatically on gene expression, even in individuals who have inherited a highly homologous genome.
Given that there is a significant level of within-species variation in gene methylation, comparison of epigenetic measures across species must be considered with caution. Preliminary data, however, from a comparison of gene methylation in humans and non-humans primates suggest that there are differences in methylation that far exceed any likely within-species variation [83]. This suggests that there will be human-specific changes in gene methylation that could represent a species-wide response to certain environmental factors. This is not a surprising outcome given that species-specific responses to environmental stressors exist in unicellular organisms [84]. It is also notable that levels of gene methylation are higher in the CNS compared to other tissue in both humans and primates and that levels of gene methylation are higher in CNS from humans compared to non-human primates [83]. Given that the role of gene methylation is to reduce gene expression, these data would suggest that a more extensive dampening of gene expression by epigenetic mechanisms is a feature of human CNS development.
As a relatively new area of research it will be some time before human-specific epigenetic mechanisms are fully understood. The impact of epigenetics on mammalian development is, without doubt, profound and is at work at all stages of life [80]. Moreover, there is clear evidence for an involvement of altered epigenetic regulation in psychiatric disorders [85]. Whether the alterations in epigenetic mechanisms in schizophrenia are due to changes in human-specific mechanism remains open to conjecture. However if there are human-specific modifications in gene methylation, identifying these changes in gene methylation may help direct research that has already shown alterations in gene methylation in the CNS of subjects with the disorder [86–90].
Summary and conclusions
The search for unique features by which the CNS of humans could be separated from that of all other species has been long pursued, with structures such as the hippocampus minor being one of the more contentious structures reported as being unique to the human CNS [91]. Current data at the macroscopic, microscopic, cellular and gene expression level seem to now favour the argument that the human CNS has certain features that have developed far beyond what can be observed in non-human primates. Therefore it must be the combination of these more evolved factors that gives the CNS of Homo sapiens its unique capabilities, and existing data suggest that many of these factors could be involved in the pathology of schizophrenia. Hence, it could well be argued that schizophrenia is a disorder unique to humans that has emerged because of the need for a complex CNS. Pursuing this argument, it seems important to conclude by further exploring the two hypotheses that have been put forward as to why schizophrenia may be a disease limited to humans [1, 2].
The first of these hypotheses argued that changes in the CNS needed to support the development of language also introduced the pathways in the CNS that, when they become deranged, cause the symptoms of schizophrenia [1]. Relevant to this hypothesis is the finding that deletions or mutations in the FOXP2 gene are associated with developmental verbal dyspraxia [92, 93], a symptom of which is severe language impairment. These data suggested that FOXP2 could be a gene of interest in schizophrenia and it has been reported that there is an association between a FOXP2 haplotype and an increased risk of schizophrenia involving auditory hallucinations [94]. Furthermore, increased levels of FOXP2 protein have been reported in the offspring of mice exposed to viral infection [95], suggesting that changes in the expression of FOXP2 may be an outcome of gene–environment interaction. This would appear to be good supportive data for the hypothesis that changes in the human CNS that allowed the development of language could also have led to the emergence of schizophrenia as a disease of the human CNS. The recent finding, however, that FOXP2 protein is decreased in the CNS of birds during increased periods of singing [96] suggests that this gene may be involved in complex vocalization, rather than language per se. This would suggest that the association between changed expression of FOXP2 and complex vocalization precedes language development in humans. Although this does not preclude a role for FOXP2 expression in the pathology of schizophrenia, it would mean that such changes are not likely to be associated with language development and would therefore not support the hypothesis that this was the critical event in human evolution that saw the emergence of schizophrenia as a disorder of our species.
The second hypothesis, based on gene expression profiling in the CNS of subjects with schizophrenia, suggests that a reduced capacity of the CNS to generate energy to maintain cognition results in the cognitive impairments associated with the disorder [2]. A reduced capacity to utilize available metabolic energy due to a decrease in the activity of metabolic pathways would be predicted to decrease the capacity of a tissue to utilize available glucose [97]. Such a decrease in energy utilization has been shown using PET, where decreased glucose utilization was observed in the PFC of subjects with schizophrenia [17, 98, 99]. Thus it is possible that human-specific changes in gene expression may have limited the ability of the CNS in humans to generate energy [60], and that this evolutionary process, taken to the extreme, means that in some individuals energy generation is so limited that the symptoms of schizophrenia develop. Relevant to this finding is the argument that the extended period of cortical synaptic pruning in human CNS is necessary to reduce energy demand to a level that can be sustained by the human cortex [29]. If that argument is correct then an increased level of synaptic pruning would be required in subjects in whom metabolic pathways were most suppressed. Therefore the combination of relative energy deficiency and a resulting over-pruning of synapses could be a development in the human CNS that underlies the symptoms of schizophrenia. Supporting this argument are recent findings that show that surrogate markers of synaptic density are consistently decreased in the cortex of subjects with schizophrenia [100, 101]. Thus, there are data that support the hypotheses that schizophrenia occurs in humans because of modifications in human CNS development required to accommodate a species-specific reduction in energy generation capability.
In summary, it is difficult to identify characteristics that can be argued to be an absolute marker of human CNS-specific development but there are CNS structures, cells, gene expression and gene sequence variations that are clearly most highly developed in the human CNS. Moreover, there are data suggesting that such species-specific CNS developments could underpin the emergence of schizophrenia as a species-specific disorder. Therefore ongoing research that continues to identify human-specific changes in CNS development should be of particular interest to those interested in understanding the possible mechanisms that underlie schizophrenia.
Footnotes
Acknowledgements
BD is an NHMRC Senior Research Fellow.