
Abstract
Attention deficit hyperactivity disorder (ADHD) is a genetically based [1] neurobiological disorder characterized by hyperactivity, impulsivity and difficulties in sustaining attention [2]. Its pathophysiology is thought to involve alterations in dopaminergic and noradrenergic pathways [3] and affects 3% to 10% of school-aged children, with persistence of symptoms into adulthood in up to 60% of patients [4, 5]. The disorder is associated with large number of comorbidities, such as learning disabilities, conduct disorder, oppositional defiant disorder (ODD), depression and anxiety, all of which are associated with poorer outcomes later in life [6, 7].
Treatments for ADHD include pharmacological and non-pharmacological interventions. The Multimodal Treatment Study of Children with ADHD (MTA), conducted by the US National Institute of Mental Health, showed the superiority of pharmacotherapy in symptom reduction when compared with psychosocial interventions [8]. Traditionally, pharmacotherapy of ADHD has involved the use of psychostimulants (e.g. methylphenidate hydrochloride and dextroamphetamine sulphate), which are believed to produce their beneficial effects by potentiating the effects of synaptically released catecholamines through blockade of re-uptake transporters [9]. Although effective and in use for more than half a century [10], these therapies have limitations (including the potential for abuse and diversion [11]) and may be problematic for patients with comorbidities such as anxiety and tics [12]. Moreover, because these drugs have relatively short half-lives, multiple daily doses have to be administered, which may cause insomnia after late afternoon or evening administration [13]. Nonetheless, about 10% to 30% of patients do not respond to, or are intolerant of, psychostimulant therapy [14]. These concerns about the tolerability and abuse liability of psychostimulants have created the need for effective alternative non-stimulant therapies. Despite the strong need for non-stimulant treatments for ADHD, none of the currently available non-stimulant alternatives (e.g. clonidine, guanfacine or the antidepressants desipramine and bupropion) has proved to be clinically viable [15, 16].
To date, atomoxetine, a selective noradrenergic re-uptake inhibitor, is the only effective non-stimulant treatment for ADHD in both children and adults and has been approved in the United States and Australia, as well as countries within Europe, South America, Asia, Middle East and Africa. It can be administered as a single daily dose or split into two evenly divided doses [17]. It carries negligible risk of abuse or diversion and is not considered a controlled substance [18].
Several placebo-controlled trials have shown atomoxetine to be markedly superior to placebo in reducing symptoms of ADHD [19–23]. Data from these trials also have shown atomoxetine to be safe and well tolerated. However, head-to-head comparison data on atomoxetine and psychostimulants (particularly methylphenidate) are still lacking. To our knowledge, only two trials have directly compared atomoxetine and methylphenidate in the treatment of ADHD [24, 25], with somewhat contradicting results. The multicentre, randomized, open-label study of African-American children with ADHD [24] showed that both atomoxetine and methylphenidate significantly improved ADHD symptoms from baseline, with methylphenidate being associated with significantly greater improvements in total ADHD symptoms, inattentiveness and global improvement. The incidence of adverse events was similar in both groups. A prospective, randomized, open-label study in US children with ADHD [25] showed similar efficacy, as well as similar safety and tolerability, of the two therapies.
T o obtain additional head-to-head data for atomoxetine and methylphenidate, we conducted a multicountry, multicentre, randomized, double-blind, approximately 8 week study in children and adolescents with ADHD to test the hypothesis that atomoxetine is non-inferior to methylphenidate on conventional ADHD symptom measures. The study also investigated the tolerability profiles of atomoxetine and methylphenidate.
Method
Patients
Eligible participants included outpatient children and adolescents, 6–16 years of age, weighing between 20 and 60 kg, who met the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV [4]), criteria for ADHD as assessed by clinical interview and confirmed by structured diagnostic interview using the Kiddie Schedule for Affective Disorders and Schizophrenia for School-Aged Children-Present and Lifetime Version (K-SADS-PL) [26]. All patients were required to meet the following symptom severity thresholds: a score of ≥25 for boys or ≥22 for girls, or >12 for a specific subtype, on the Attention Deficit Hyperactivity Disorder Rating Scale-IV-Parent Version: Investigator-Administered and -Scored (ADHDRS-IV-Parent: Inv [27]), as well as a Clinical Global Impressions-Attention Deficit Hyperactivity Disorder-Severity (CGI-ADHD-S [28]) score of ≥4.
Exclusion criteria included any history of bipolar, psychotic or pervasive developmental disorders; suicidal risk; or ongoing use of psychoactive medications other than the study drug. Patients with motor tics, a diagnosis or family history of Tourette's syndrome or those who met DSM-IV criteria for anxiety disorder as assessed by the investigator and confirmed by the K-SADS-PL were also excluded from participating.
After the oral and written explanation of the study, each patient, as well as patient's parent or guardian, provided written informed consent to participate. This study was approved by each site's institutional review board and was conducted in accordance with the 1975 Declaration of Helsinki, as revised in 2000.
Study design
The study was a multicountry, multicentre, randomized, double-blind study conducted between January and October 2004 at six study sites in China, four sites in Korea and three sites in Mexico. The study consisted of three periods: (i) screening and medication washout (5–33 days); (ii) double-blind treatment (∼8 weeks); and (iii) discontinuation period (∼1 week). This report presents the results of the double-blind period.
After the initial screening and medication washout period, patients were randomly assigned in 1:1 ratio to atomoxetine or methylphenidate treatment for approximately 8 weeks. Patients randomly assigned to receive atomoxetine began therapy at 0.8 mg kg−1 day−1 administered once daily in the morning, which was titrated to 1.2 mg kg−1 day−1 on Day 5, and could be either maintained or titrated upward or downward within the final range of 0.8–1.8 mg kg−1 day−1. Patients randomly assigned to methylphenidate began therapy at 0.2 mg kg−1 day−1 administered twice daily (in the morning and at lunch), which was titrated to 0.4 mg kg−1 day−1 on Day 5, and could be either maintained or titrated upward or downward within the final range of 0.2–0.6 mg kg−1 day−1. To maintain blinding, a placebo was administered to the atomoxetine group at lunch. The dosage of methylphenidate was chosen on the advice from the Chinese thought leaders during the design of this study, based on usual practice in China.
The use of over-the-counter medications was to be kept to a minimum during the study. In addition, any health-food supplements that might have central nervous system activity (e.g. St. John's Wort or melatonin) were excluded.
Assessments
The primary outcome measure was the response rate, defined as a ≥40% reduction from baseline to end point in the ADHDRS-IV Parent: Inv Total [27] score.
Secondary efficacy assessments included ADHDRS-IV-Parent: Inv Total and Inattention and Hyperactivity/Impulsivity subscales; ADHD Index, Oppositional, Cognitive Problems/Inattention and Hyperactivity subscales of the Conners’ Parent Rating Scale-Revised: Short Form (CPRS-R:S) [29]; and the CGI-ADHD-S scale [28].
Tolerability measures included the extent of exposure to the study drug (dosage and duration), the assessment of treatment-emergent adverse events (TEAEs; data on TEAEs were collected via open-ended questions), as well as monitoring of vital signs, electrocardiograms and clinical laboratory tests (chemistries, haematology and urinalysis).
Statistical analyses
This study was designed to randomly assign approximately 330 patients between the atomoxetine and methylphenidate treatment groups (165 per treatment group). This sample size was intended to provide approximately 95% power to assess overall non-inferiority of atomoxetine to methylphenidate across the countries, under the assumption that the two treatment groups would have equal response rates.
The primary efficacy analysis was a comparison of response rates between atomoxetine and methylphenidate groups for all randomly assigned patients who had both a baseline and a post-baseline score, and had no more than one visit during which less than 80% of the randomized study drug was taken. If the one-sided 95% lower confidence limit of the difference in response rate (atomoxetine minus methylphenidate) was greater than −18%, atomoxetine would be considered non-inferior to methylphenidate. The delta of 18% is approximately one-half the estimated difference between methylphenidate and placebo (data on file; Eli Lilly and Company) and is also between one-half and two-thirds of the one-sided 80% lower confidence limit in the difference of response rates between methylphenidate and placebo, based on pooled data from two identically designed, double-blind, placebo- and active comparator-controlled studies involving both atomoxetine and methylphenidate [21]. A similar analysis was conducted for all intended-to-treat patients to ensure the robustness of the primary efficacy analysis.
Secondary efficacy analyses assessed changes (from baseline to each visit) in the ADHDRS-IV-Parent: Inv Total score using a mixed-model repeated measures approach with terms for treatment, country, investigator (nested within country), visit, baseline and treatment-by-visit interaction. Other secondary efficacy analyses assessed changes (from baseline to last-observation-carried-forward end point) in the ADHDRS-IV-Parent: Inv Total and subcale scores, the CPRS-R:S scores and the CGI-ADHD-S scores, evaluated using an ANCOVA model with independent effects for treatment, country, investigator (nested within country) and baseline score. When added to the ancova model for the change from baseline to end point in ADHDRS-IV-Parent: Inv Total scores, treatment-by-investigator interaction was not statistically significant (p = 0.621), indicating no significant differences in treatment effects across investigators.
Patient characteristics were compared across treatment groups using Fisher's exact test for categorical data and an ANOVA model, with treatment as the independent effect for quantitative data.
Subgroups of patients for efficacy analyses were based upon investigational site, prior-stimulant-use strata, ADHD subtype, sex, age and comorbid ODD. The subgroup analyses were conducted using an anova model, including terms for the treatment-by-subgroup interaction. If the interaction was statistically significant at the 0.10 level, then the potential causes of the interaction were to be investigated.
Tolerability analyses included all patients who took at least one dose of the study drug. Tolerability analyses for continuous measures were assessed using an ANOVA, with country, investigator (nested within country) and treatment as independent effects in the model. Treatment differences in binary measures were assessed using Fishers’ exact test.
All statistical tests were performed using a two-sided, 0.05 significance level. All statistical analyses were conducted using SAS software (version 8.2; SAS Institute, Cary, NC, US).
Results
Patient characteristics
Of the 361 patients screened, 330 (China, n = 242; Korea, n = 60; Mexico, n = 28) were randomly assigned to receive either atomoxetine (n = 164) or methylphenidate (n = 166) treatment. All patient characteristics (except for age) and baseline symptom measures were not significantly different between treatment groups (Table 1). Approximately 83% of the children enrolled were male, and the participants’ mean age was 9.7 years. The majority of children in both groups met the criteria for the combined ADHD subtype (Table 1). The most common concurrent DSM-IV diagnosis was ODD (atomoxetine, 40/164 [24.4%]; methylphenidate, 29/166 [17.5%]; p = 0.137); all other concurrent diagnoses were experienced by less than 5% of randomly assigned patients. The mean (SD) baseline CGI-ADHD-S scores for the atomoxetine and methylphenidate groups were 5.3 (0.8) and 5.3 (0.9), respectively, corresponding to the ‘markedly ill’ symptom severity.
Baseline demographic and clinical characteristics of paediatric patients with ADHD randomly assigned to atomoxetine or methylphenidate
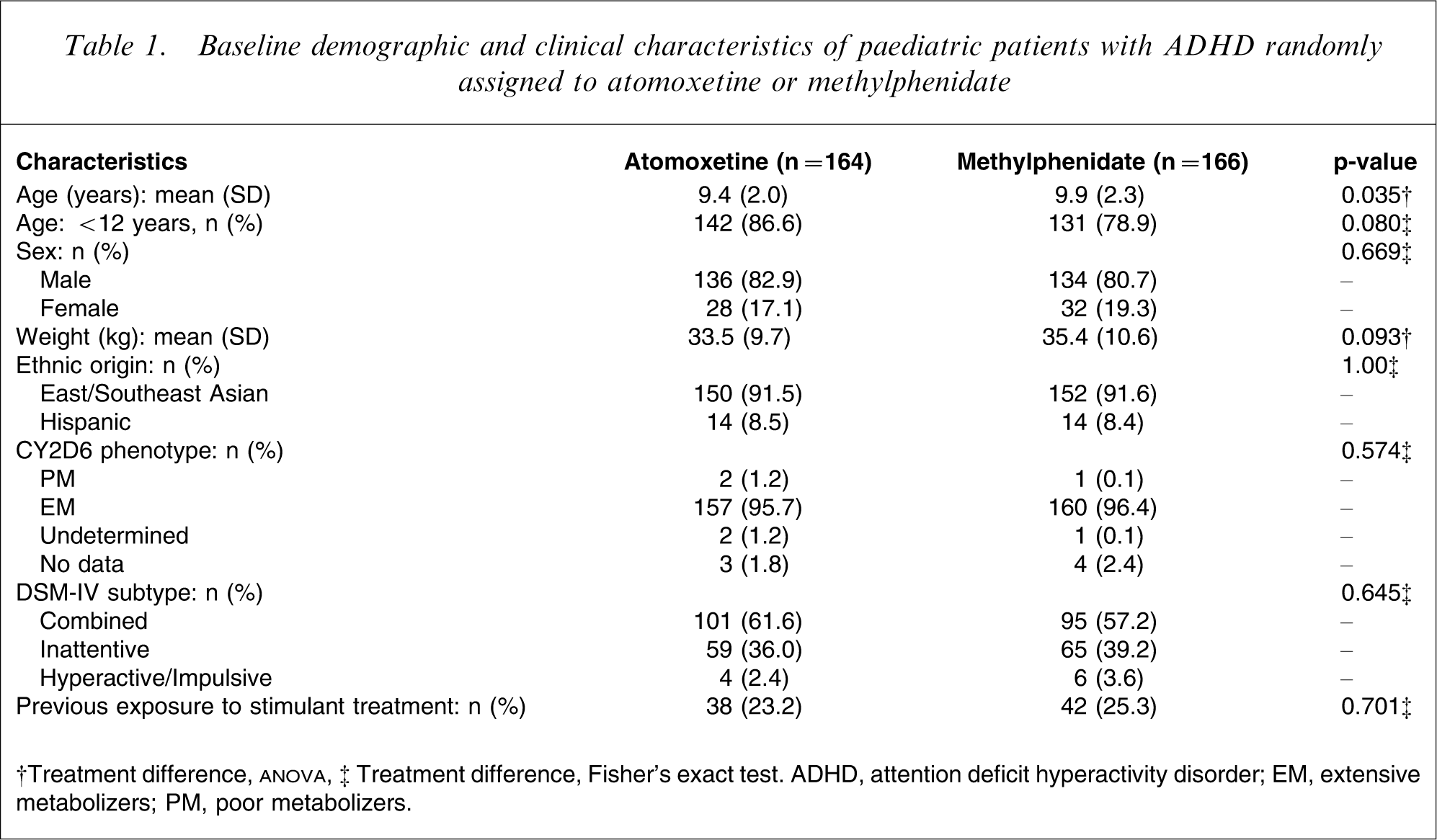
†Treatment difference,
The overall study completion rate was high (87.9% [290/330]), and the methylphenidate group had a significantly higher completion rate compared with the atomoxetine group (91.6% [152/166] vs. 84.1% [138/164]; p = 0.044). Significantly more atomoxetine-treated patients discontinued because of TEAEs compared with methylphenidate-treated patients (p = 0.011). Eighteen patients (11.0%) in the atomoxetine group discontinued because of TEAEs (anorexia: 5; decreased appetite: 2; nausea: 2; abdominal pain: 2; chest pain: 1; constipation: 1; dermatitis medicamentosa: 1; rash: 1; simple partial seizure: 1; somnolence: 1; varicella: 1) and six patients (3.6%) in the methylphenidate group discontinued due to TEAEs (anorexia: 1; decreased appetite: 2; nausea: 1; dizziness: 1; palpitations: 1).
The usage of concomitant medications did not significantly differ between the treatment groups; none of the concomitant medications used during the study affected the patients’ central nervous system or the metabolic activity of the study drugs.
Efficacy
The primary efficacy analysis showed that atomoxetine was non-inferior to methylphenidate in improving ADHD symptoms, based on response rates (atomoxetine, 77.4% [123/159]; methylphenidate, 81.5% [128/157]; one-sided 95% lower confidence limit = − 11.7%; p = 0.404). The result was consistent with a similar analysis on the intent-to-treat population (atomoxetine, 75.9% [123/162]; methylphenidate, 81.1% [133/164]; one-sided 95% lower confidence limit = − 12.8%; p = 0.282).
The first secondary analysis of efficacy, the analysis of mean changes from baseline to each visit in ADHDRS-IV-Parent: Inv Total scores, showed a similar improvement between the two treatment groups. However, patients in the atomoxetine group experienced a significantly greater reduction in these scores from baseline at Week 1 (p = 0.018; Figure 1). Additional secondary efficacy analyses showed that whereas patients within each of the treatment groups experienced clinically meaningful and statistically significant improvements from baseline to end point in all the measures (ADHDRS-IV-Parent: Inv, CPRS-R:S and CGI-ADHD-S; all p < 0.001), the between-group improvements in scores were numerically similar and not statistically significant (Table 2).
LS mean (MMRM) change in ADHDRS-IV-Parent: Inv total score. LS, least squares; MMRM, mixed-model repeated measures; ADHDRS-IV-Parent: Inv, Attention Deficit Hyperactivity Disorder Rating Scale-IV-Parent Version: Investigator-Administered and -Scored; BL, baseline. atomoxetine, methylphenidate.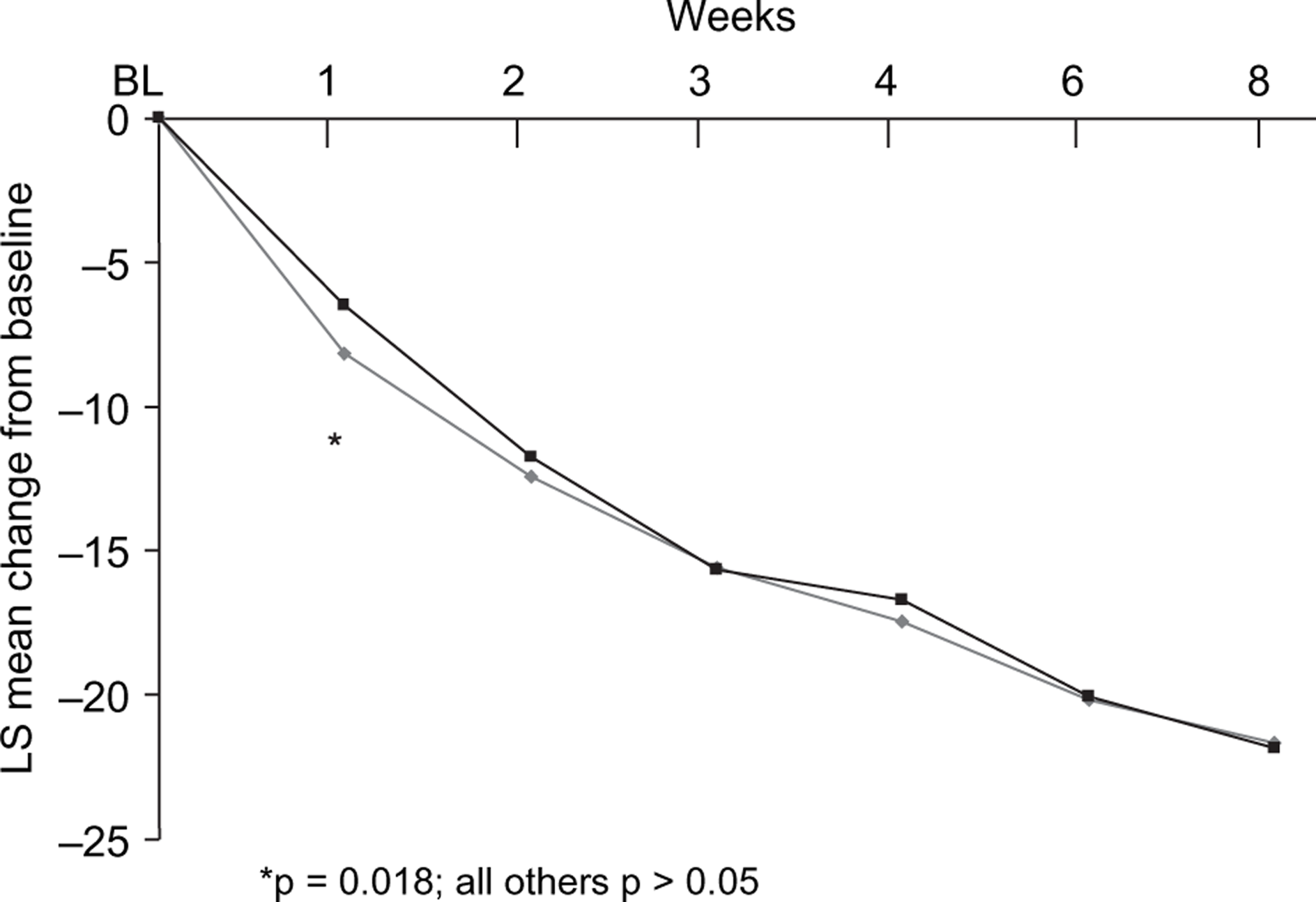
Changes from baseline to end point on secondary efficacy measures
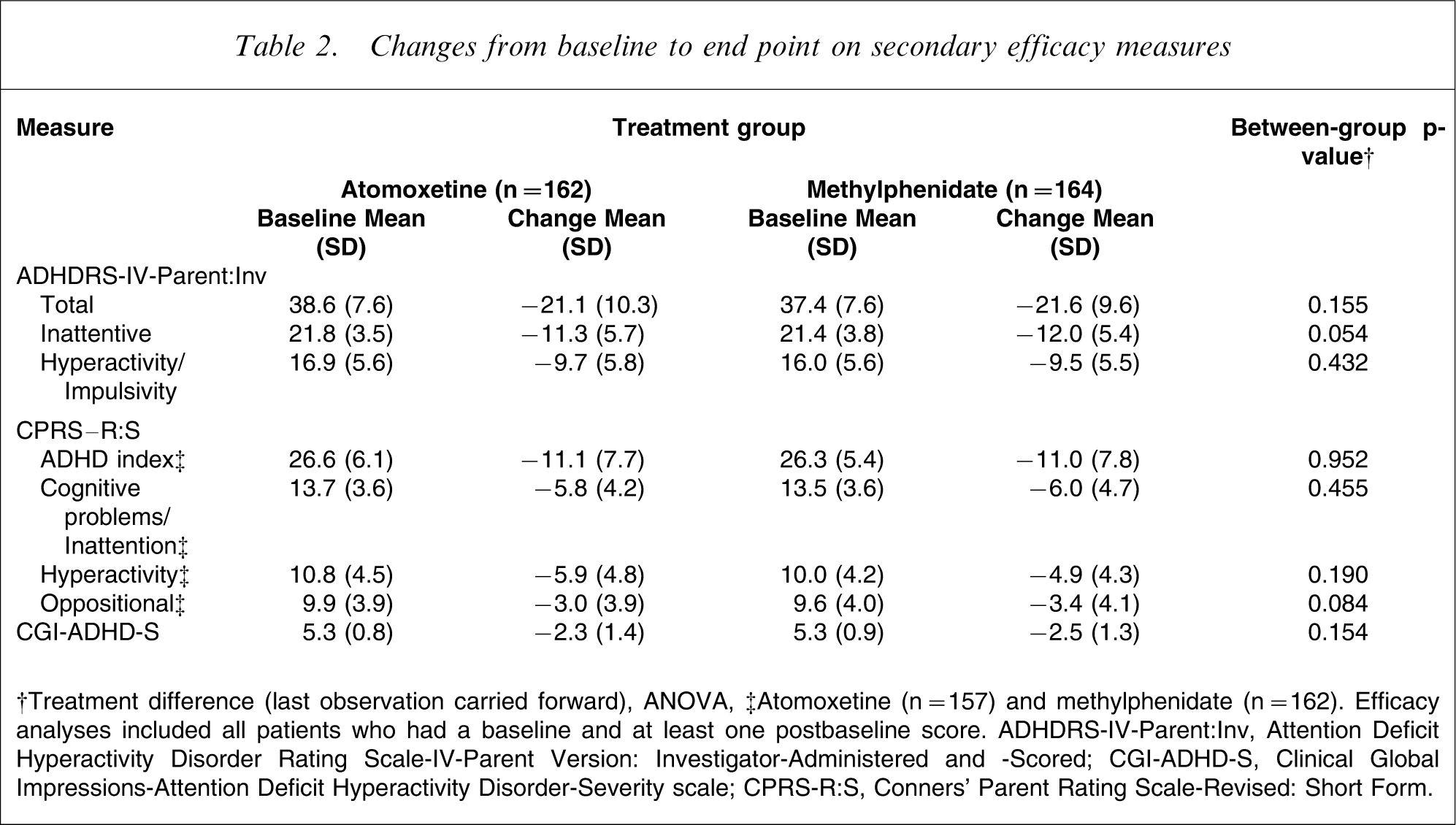
†Treatment difference (last observation carried forward), ANOVA, ‡Atomoxetine (n = 157) and methylphenidate (n = 162). Efficacy analyses included all patients who had a baseline and at least one postbaseline score. ADHDRS-IV-Parent: Inv, Attention Deficit Hyperactivity Disorder Rating Scale-IV-Parent Version: Investigator-Administered and -Scored; CGI-ADHD-S, Clinical Global Impressions-Attention Deficit Hyperactivity Disorder-Severity scale; CPRS-R:S, Conners’ Parent Rating Scale-Revised: Short Form.
No significant interactions between treatment and subgroups (ADHD subtype, sex, age [<12 years vs. ≥12 years], previous stimulant use and comorbid ODD) were observed in this study.
Tolerability
Among all patients randomly assigned to treatment, the mean duration of therapy was 59.9 days and was similar in both treatment groups (atomoxetine, 58.2 days; methylphenidate, 61.6 days). The mean final prescribed dosage of atomoxetine was 1.37 mg kg−1 day−1 (44.4±15.7 mg day−1) and of methylphenidate, 0.52 mg kg−1 day−1 (17.8±7.1 mg day−1).
A significantly greater percentage of patients in the atomoxetine treatment group experienced TEAEs compared with the methylphenidate treatment group (86.6% [142/164] vs. 67.5% [112/166]; p < 0.001). Most TEAEs were reported to be mild or moderate in severity (Table 3). The majority of TEAEs reported by patients in the atomoxetine group occurred in the first 3 weeks of the double-blind treatment period; these TEAEs then subsided over time and were generally of mild or moderate severity. Among TEAEs, anorexia (p = 0.024), nausea (p = 0.014), somnolence (p < 0.001), dizziness (p = 0.024) and vomiting (p = 0.007) were reported significantly more often among atomoxetine-treated patients compared with methylpheni-date-treated patients (Table 3). No deaths were reported during the study. One serious adverse event, a simple partial seizure, was reported for a patient randomly assigned to the atomoxetine group; this event led to discontinuation from the study.
Treatment-emergent adverse events reported by ≥5% of patients in either treatment group
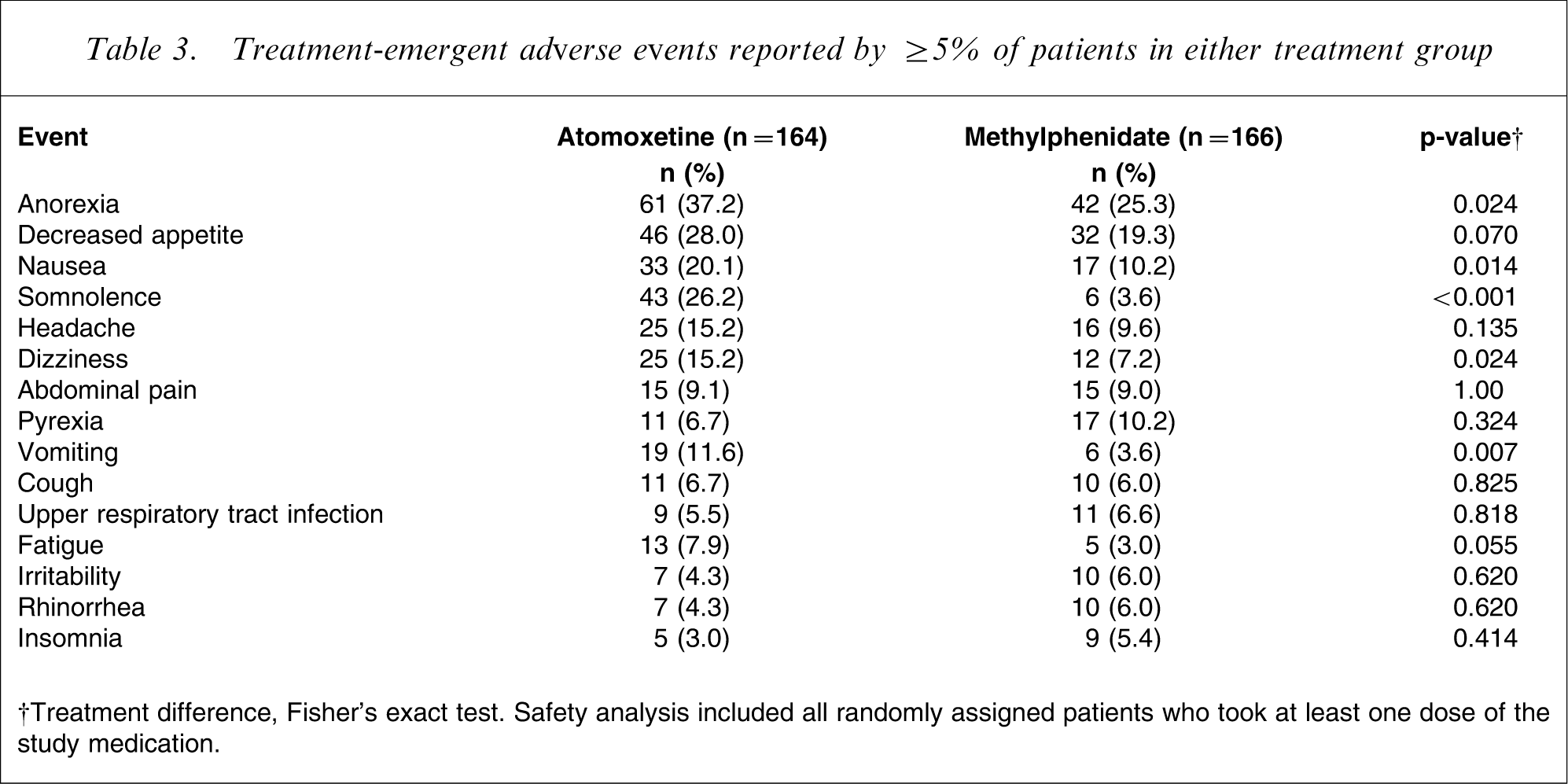
†Treatment difference, Fisher's exact test. Safety analysis included all randomly assigned patients who took at least one dose of the study medication.
Patients in the atomoxetine treatment group experienced a small but significantly greater mean weight loss compared with the methylphenidate treatment group (atomoxetine, −1.2 kg vs. methylphenidate, −0.4 kg, p < 0.001), with 49.4% (80/162) of atomoxetine-treated patients losing at least 3.5% of initial body weight compared with 25.6% (42/164) of methylphenidate-treated patients (p < 0.001). No significant between-group differences were observed in blood pressure or corrected QT intervals. Patients in both treatment groups experienced increases in heart rate, with a significant difference between the groups (mean increases from baseline to end point: atomoxetine, 8.51 bpm; methylphenidate, 4.76 bpm; p = 0.005).
No clinically relevant differences in laboratory analytes were observed between the treatment groups. Significant between-group differences were observed only in mean baseline-to-end point changes in alkaline phosphatase (mean±SD: atomoxetine,−6.4±10.7 U L−1; methylphenidate, −2.4±10.6 U L−1; p < 0.001) and creatine phosphokinase (mean±SD: atomoxetine, −7.9±39.7 U L−1; methylphenidate, 7.9±151.2 U L−1; p = 0.041). Because the mean decreases from baseline to end point in alkaline phosphatase levels were small in both treatment groups, and no patients had values below the lower reference limit (24.7 U L−1), the treatment differences observed were not considered clinically relevant. Of note, more methylphenidate-treated patients had abnormally high levels of creatine phosphokinase values than patients treated with atomoxetine; however, this difference was not significant (11/122 [9.0%] vs. 17/129 [13.2%]; p = 0.322). No other significant changes in laboratory values were observed between the treatment groups (including changes in bilirubin, alanine aminotrans-ferase, aspartate aminotransferase or gamma-glutamyltransferase).
Discussion
The results of the present study show that atomoxetine is non-inferior to methylphenidate in the improvement of ADHD symptoms based on a comparison of the response rates derived from the ADHDRS-IV-Parent: Inv Total score after approximately 8 weeks of treatment. Both atomoxetine and methylphenidate treatments were associated with clinically meaningful improvements in ADHD symptoms, including inattention and hyperactivity/impul-sivity. These results are consistent with those previously published and support atomoxetine's efficacy in reducing ADHD symptoms [19, 20, 30–32]. They also provide further comparative information for the efficacy of atomoxetine and methylphenidate in reducing ADHD symptoms in children [24, 25]. Symptom severity decreased to ‘mildly ill’ for both treatment groups (CGI-ADHD-S score at end point: atomoxetine, 2.97; methylphenidate, 2.80; p < 0.001 within each group), suggesting that both atomoxetine and methylphenidate were efficacious in reducing the severity of ADHD symptoms.
The age difference between the treatment groups was unlikely to have had any impact on efficacy findings. In addition, other subgroup-specific data did not suggest any subgroup effect on efficacy (however, this study was not powered to detect significant differences in subgroups).
In this study, a vigorous and rapid-dose titration schedule for atomoxetine was used, which made it possible to observe the early onset of treatment effects. Improvements on the primary efficacy measure were observed in both treatment groups as early as 1 week after randomization (with atomoxetine-treated patients showing significantly greater improvements at Week 1) and continued at each weekly visit until end point. Thus, the current study also replicates previous data showing the efficacy of once-daily dosing of atomoxetine, as well as the onset of drug effect within the first week of treatment [17, 20, 23].
The tolerability of atomoxetine in this study was also consistent with observations from previous studies, except for some gastrointestinal adverse events such as anorexia and decreased appetite [17, 20, 23]. Atomoxetine is associated with a slightly different adverse event profile than stimulant medications, particularly increased somnolence and gastrointestinal symptoms. These patterns of adverse events and effects on vital signs observed for atomoxetine are consistent with increased noradrenergic tone [13]. Treatment-emergent adverse events in the atomoxetine treatment group were primarily related to gastrointestinal symptoms and decreased appetite, and were of mild or moderate severity. They occurred more frequently during the first 3 weeks of the double-blind treatment period, and their incidence tended to decline over time. Acute treatment with atomoxetine was well tolerated in this study population, despite the use of a more aggressive and shorter titration schedule (starting dosage of 0.8 mg kg−1 day−1, titrating to 1.2 mg kg−1 day−1 on Day 5) than is generally now recommended (starting dosage of 0.5 mg kg−1 day−1, titrating to 1.2 mg kg−1 day−1 after 7–10 days [19]). A similar rapid-dose titration was used in a previous study of atomoxetine in children [17], but the incidence of discontinuations due to adverse events (4.5%), as well as the incidence of TEAEs (decreased appetite, 17.6%; nausea, 11.5%; and somnolence, 14.5%), were much lower as compared with the present study. Hence, the present study population appears to be more sensitive to atomoxetine treatment than the US population. This observation may be due in part to cultural differences in how the drug was administered, before breakfast or after the meal.
The timing of adverse events observed in the meth-ylphenidate group was more sporadic across visits. Discontinuations due to adverse events were significantly greater in the atomoxetine group (11.0%) compared with the methylphenidate group (3.6%; p = 0.011). Eleven atomoxetine-treated patients versus four methylphenidate-treated patients discontinued because of gastrointestinal adverse events. These differences, as well as the relative lack of reports of insomnia in children receiving methylphenidate, may be due to both the very low dose of methylphenidate and/or the lack of a late afternoon dose of this drug used in this study. In previous studies of methylphenidate, insomnia was reported in approximately 60% of patients treated with this drug when administered either twice or thrice daily at a mean maximum dosage of 30±0.1 mg kg−1 day−1, and a 7% incidence of poor sleep was reported for the immediate-release methylphenidate preparation when administered thrice daily at an average dose of 0.88±0.32 mg kg−1 day−1[33, 34].
As seen in previous studies [19–23], atomoxetine-treated patients had a modest mean decrease in weight as compared with those treated with methylphenidate. However, this is unlikely to be of any serious clinical concern because in long-term studies, weight loss has generally been observed early in treatment with atomoxetine, with weight re-gain during treatment periods greater than 6 months [35, 36].
Changes in vital signs (heart rate and blood pressure) consistent with a noradrenergic-mediated increase in autonomic tone were observed in the atomoxetine-treat-ment group, but for most patients these changes were modest and asymptomatic. No QTc interval or laboratory value changes of clinical relevance or statistical significance were observed with either medication.
Study limitations
We believe that this study has two major limitations. First, the final mean daily dosage of methylphenidate was only 17.8±7.1 mg day−1. This methylphenidate dosage was lower than previously recommended [37] and was administered only twice daily (in the morning and at lunchtime, with no afternoon dose) instead of three times daily. In the MTA study, to prevent untoward side effects in small children, the MTA protocol limited the highest methylphenidate dose to the ‘15 mg condition’ (0.8 mg kg−1 per dose) for those weighing 25 kg or less, and it was given as three daily doses of 15 mg, 15 mg and 5 mg (total daily dose, 35 mg) [37]. Larger children weighing more than 25 kg received the ‘20 mg condition’, which was three daily doses of 20 mg, 20 mg and 10 mg (total daily dose, 50 mg) [38]. Because of the relatively low final mean dosage of methylphenidate used in this study (17.8±7.1 mg day−1), which was half of the lowest dosage tested in the MTA study, the rates of certain adverse events in the methylphenidate treatment group may have been lower than would be expected.
Second, the current trial lacked a placebo control group. Parents and investigators knew that all patients received an active medication, which may have contributed to the large overall response rates (methylphenidate, 81.5%; atomoxetine, 77.4%) despite the fact that patients treated with methylphenidate received only two doses per day at less than half the maximal dose previously used in the MTA study [37].
Conclusions
The results of this study suggest that atomoxetine (0.8–1.8 mg kg− day−1) is non-inferior to methylphenidate (0.2–0.6 mg kg−1 day−1) in the improvement of ADHD symptoms in paediatric outpatients after 8 weeks of acute treatment. Although both of the drugs were well tolerated, atomoxetine was associated with a higher incidence of TEAEs than methylphenidate (86.6% vs. 67.5%, respectively).
Footnotes
Acknowledgements
The authors thank Simrat Sohal, MD, and Christopher Carlson, PhD, for their assistance in writing and preparing this article. In addition, the authors thank Prof Tong Xu, Prof Jian Yang and Prof Linyan Su for their assistance in planning and conducting the study.