
Abstract
Tanacetum parthenium (feverfew), is a well-known herb for the prophylactic treatment of migraine. The primary objective was to show a dose-response of a new stable extract (MIG-99) reproducibly manufactured with supercritical CO2 from feverfew (T. parthenium). Furthermore, the study should provide data on the safety and tolerability of MIG-99. In a randomized, double-blind, multicentre, controlled trial with an adaptive design, the clinical efficacy and safety of three dosages of MIG-99 (2.08 mg; 6.25 mg; 18.75 mg t.i.d.) were compared with placebo. The patients (n = 147) suffered from migraine with and without aura according to International Headache Society (IHS) criteria and were treated with one of the study medications for 12 weeks after a 4-week baseline period. The primary efficacy parameter was the number of migraine attacks during the last 28 days of the treatment period compared with baseline. Secondary endpoints were total and average duration and intensity of migraine attacks, mean duration of the single attack, number of days with accompanying migraine symptoms, number of days with inability to work due to migraine as well as type and amount of additionally taken medications for the treatment of migraine attacks. The design of the study included a pre-planned adaptive interim analysis for patients with at least four migraine attacks within the baseline period. With respect to the primary and secondary efficacy parameter, a statistically significant difference was not found between the overall and the confirmatory intention-to-treat (ITT) sample in the exploratorily analysed four treatment groups. The frequency of migraine attacks for the predefined confirmatory subgroup of patients (n = 49) with at least four migraine attacks during the baseline period decreased in a dose-dependent manner (P = 0.001). The highest absolute change of migraine attacks was observed under treatment with 6.25 mg t.i.d. (mean ± SD = x1.8 ± 1.5 per 28 days) compared with placebo (-0.3 ± 1.9; P = 0.02). Overall, 52 of 147 (35%) patients reported at least one adverse event (AE). The incidence of AEs in the active treatment groups was similar to that in the placebo group, and no dose-related effect was observed in any safety parameter. MIG-99 failed to show a significant migraine prophylactic effect in general. Accordingly, in the ITT analysis a dose-response relationship could not be observed. MIG-99 was shown to be effective only in a small predefined subgroup of patients with at least four attacks during the 28-day baseline period where the most favourable benefit-risk ratio was observed with a dosage of three capsules of 6.25 mg MIG-99 extract per day. Because of the low number of patients, these findings need to be verified in a larger sample. The incidence of AEs was similar for all treatment groups.
Introduction
Migraine is one of the most frequent kinds of headache (1). The 1-year prevalence of migraine ranges from 6% in men to 15% in women (2). The highest frequency of migraine attacks is observed in 35–45-year-old patients. Prevalence increases throughout childhood, adolescence and early adult life, peaks in the early 40s and then declines (3). Several studies suggest that migraine prevalence is steadily increasing (4, 5). Concerning migraine therapy there is, despite some very effective new substances for the treatment of the acute attack, still a need for a migraine prophylactic drug which combines good efficacy, tolerability and safety. To extend the spectrum of therapeutic alternatives the present clinical phase II study investigated the migraine prophylactic effect of a CO2 extract from Tanacetum parthenium (MIG-99) popularly called feverfew.
Tanacetum parthenium is a herbal medication whose potential medical properties and notable headache relief were recognized even as far back as medieval times (6). The pharmacological role of feverfew in the migraine pathophysiology is not completely understood (7). Feverfew extract enables the release of serotonin(5-hydroxytryptamin) from platelets induced by a variety of aggregation agents (8). Furthermore, extracts of feverfew and pure parthenolide inhibit the synthesis of prostaglandins. Parthenolide also markedly interferes with both contractile and relaxant mechanisms of blood vessels (9).
A number of clinical trials demonstrated a good benefit–risk ratio for feverfew in the prophylactic treatment of migraine. Concerning safety and tolerability of feverfew, there is no evidence of serious adverse effects in clinical trials and to self-reporting long-term users. Nonetheless, up to now the clinical efficacy of feverfew in the prevention of migraine has not been established beyond reasonable doubt (10).
Therefore, the aim of this clinical trial was to investigate a possible dose–response relationship of MIG-99 in migraine prophylaxis in a placebo-controlled study which, in addition, should also provide data on its efficacy, safety and tolerability.
Patients and methods
The present study was a randomized, multicentre, double-blind comparison of four parallel groups: three dosages of a special CO2 extract of T. parthenium (MIG-99) vs. placebo. A 4-week baseline period without migraine prophylaxis was followed by a 12-week active treatment phase with MIG-99 or placebo. There are several preparations on the market containing the dried pulverized drug. The daily doses range from homeopathic dilutions to approximately 1.25 g. We decided to use a dose of 1.05 g, equivalent to 6.25 mg extract, which is the traditional effective dose, and additionally 1/3, or a three-fold dose of this preparation. Accordingly, during the active treatment period patients received either 2.08 mg MIG-99 (corresponding to 0.17 mg parthenolide), 6.25 mg MIG-99 (0.5 mg parthenolide), 18.75 mg MIG-99 (1.5 mg parthenolide) or placebo. The study medications were prepared as soft gelatine capsules, which were identical in appearance, size, weight, taste and smell. Each patient received one capsule t.i.d. Study enrolment took place at visit 1 (day −28) beginning with a 4-week screening period without prophylactic drug therapy. After randomization at visit 2 (day 0) the patients were seen at three control visits every 28 days up to the regular visit 5 (day 84). Patients had to keep a special headache diary and were instructed to document each migraine attack with duration, intensity, accompanying migraine symptoms, inability to work due to migraine, duration of confinement to bed and intake of additional medication. The intake of migraine preparations for treatment of acute attacks was permitted, but had to be stable during the trial. A switch to another acute migraine therapy, however, did not result in premature study termination. The completed 4-week diaries were returned to the study centre at each control visit and collected in the case record form (CRF). Patients' compliance was registered at the study visits 3–5 by counting the returned capsules and blisters. Only patients with an intake between 75% and 125% of the expected amount were eligible for the per-protocol (PP) analysis.
The occurrence of adverse events (AEs) was assessed at the study visits 2–5, vital signs (systolic/diastolic blood pressure and heart rate) at visits 1–5, and ECG recordings and laboratory investigations at visits 1 and 5. All disturbances of well-being, deteriorations of subjective or objective symptoms or signs of known diseases, newly occurred diseases or accidents as well as occurrence of clinically relevant laboratory values outside the reference ranges were regarded as AEs, irrespective of a possible causal relationship to any other drugs administered during the trial. AEs which occurred during the baseline and the follow-up phases were also taken into account (Table 1).
Study flow chart

Inclusion criteria were as follows. Male or female out-patients between 18 and 65 years of age with a diagnosis of migraine with or without aura according to International Headache Society (IHS) criteria (12); migraine attacks for at least 1 year and age at onset <50 years; an average of two to six migraine attacks per month within the last 3 months prior to study entry; two to six migraine attacks within the 4-week baseline period; a total of at least 36 h with migraine during the baseline period; stable drug treatment regimen of migraine attacks; patients' ability to distinguish between migraine and other headaches; no prophylactic migraine treatment within 4 weeks prior to screening.
Exclusion criteria were: hypersensitivity to study medication; pregnancy; intake of analgesics, ergot preparations or other established drugs for the acute migraine attack on >10 days per month, the use of antidepressants, neuroleptics, tranquillizers, medications for headache prophylaxis, medications with headache as side-effect, magnesium-containing drugs as well as additional non-drug therapies for migraine; >10 days with headaches other than migraine per month; experience with more than three different migraine prophylactic drugs in the past; drug misuse or dependency; expected lack of compliance; psychiatric disorders according to DSM-IV; confirmed diagnosis of gastrointestinal or cardiovascular complaints; other severe diseases including hepatic, renal, gastrointestinal, pulmonary, cardiac, metabolic or endocrine disorders; participation in other clinical trials within the last 3 months or simultaneous participation in another clinical investigation.
Patients were informed about the study prior to screening and according to the requirements of Good Clinical Practice (GCP), the Declaration of Helsinki in its latest version, and legal requirements. An informed consent form had to be signed by the patients before study entry.
The patients' allocation to one of the four different treatment groups was performed by randomization after the 4-week baseline period and after another check of the inclusion and exclusion criteria. For the 4-week baseline period prior to randomization, case report forms without randomization numbers were provided. Randomization took place in centre-specific blocks on the basis of a randomization code generated by Alphamed, Göttingen, by order of Schaper & Brümmer, using the validated program Rancode Plus (IDV, Gauting). The assignment of random numbers to the patients was carried out in consecutive order according to the time of enrolment into the study starting with the lowest number available within each centre. Patients and investigators remained ‘blinded’ throughout the whole study. All other persons active in the study, including the biometrician, remained blinded until the database was locked. Investigators were provided with sealed envelopes containing a code break for emergencies. All envelopes remained sealed throughout the entire study.
The primary efficacy parameter was the total number of migraine attacks during the individual last 28-day period (weeks 13–16) of therapy compared with baseline (prophylaxis free). Migraine headaches which occurred after an interval of ≥24 h without headache were defined as a new migraine attack. The number and duration of migraine attacks were arithmetically adjusted to exactly 28 days if the baseline or a treatment period was documented with 28±7 days. In case of a duration of >35 days the last 28 days of the baseline period or the last 28 days under treatment were evaluated according to the patient's diary. The Jonckheere–Terpstra test was used to test for a positive dose–response relationship. It was expected that the difference between verum and placebo regarding the number of migraine attacks was greater than zero with advantages of higher doses compared with lower doses. The following hypotheses were tested with the Jonckheere–Terpstra test (one-sided):
H0: F 1=F 2=F 3=F 4
H1: F 1≤F 2≤F 3≤F 4
where at least one of the inequalities is strict, with F 1=placebo, F 2=MIG-99 2.08 mg, F 3=MIG-99 6.25 mg, F 4=MIG-99 18.75 mg. The significance level was set at α=0.05.
The findings of the last 28-day period under migraine prophylaxis were compared with the findings during baseline and were calculated primarily as an absolute and secondarily as a relative (percent) change. In cases of premature discontinuation of treatment later than 28 days after beginning of therapy patients entered the statistical evaluation with their data of the last 28 days under prophylaxis (last-observation-carried-forward method). In cases of study discontinuation within a period of <28 days no change in comparison with baseline was assumed.
Secondary parameters were the total and average duration and intensity of migraine attacks, the additional assessment of overall migraine severity, the number of days with accompanying migraine symptoms, the number of days with inability to work due to migraine, the mean duration of confinement to bed due to migraine, the mean duration of the single attack and the type and amount of additionally taken preparations for the treatment of attacks. In addition, the following response criterion was calculated: number of patients with an improvement of approximately 50% in the average number of migraine attacks within 4 weeks. For comparisons across all dosages, the Jonckheere–Terpstra test was used. For two sample comparisons, the Wilcoxon test (one-sided) was used in case of continuous data and Fisher's exact test (one-sided) in case of binomial data. Resulting P-values were interpreted as exceeding probabilities in the exploratory sense. In addition, a test for interactions between study centres and treatment effects was performed.
Primarily, an intention-to-treat (ITT) and additionally a PP analysis were performed. The final determination of the PP sample was carried out after closure of the database and prior to opening of the random code. A blinded meeting was dedicated to the assessment of which violations of inclusion and exclusion criteria or deviations from the study protocol should be rated as major or minor. Only patients with major protocol violations were to be excluded from the PP analysis. In addition, patients with premature study discontinuation definitely not related to the study medication were to be excluded from the PP analysis.
An adaptive interim analysis was performed according to Bauer and Röhmel (12) following discussions with the regulatory authority. The final confirmatory analysis of the primary efficacy parameter focused on the subset of patients with at least four attacks per 28 days during the baseline period. The final sample size was calculated with the data of the adaptive interim analysis scheduled at 100 patients (α1=0.0087, α =0.05, β =0.1; two-stage Bauer and Köhne method (14)).
Results
Patients were recruited for this trial in 10 centres in Germany. The number of patients per centre ranged from seven to 28. A total of 167 patients were included into the study screening period between visit 1 (day −28) and visit 2 (day 0). Of these, 147 patients (120 women (81.6%) and 27 men (18.4%)) aged 43±11 years (mean ± SD) were enrolled and randomly assigned to one of the three different doses of study medication or placebo. Thirty-eight patients suffered from migraine with aura (25.9%). For demographic characteristics see Table 2. The study was completed regularly in 112 (76.2%) of 147 patients. The reasons for premature termination of 35 patients (23.8%) are presented in Table 3. The entire study population, i.e. 147 patients, was regarded as the overall ITT sample. With regard to the adaptive design and the results of the interim analysis, this investigation focused on the confirmatory subset of the ITT patients who had had at least four migraine attacks per month at baseline (n=49). The PP sample was derived from ITT by excluding patients with major protocol violations and/or premature study termination due to reasons not related to the study drug.
Demographic characteristics of the confirmatory subset of patients (and all patients)

Main reasons for premature study termination

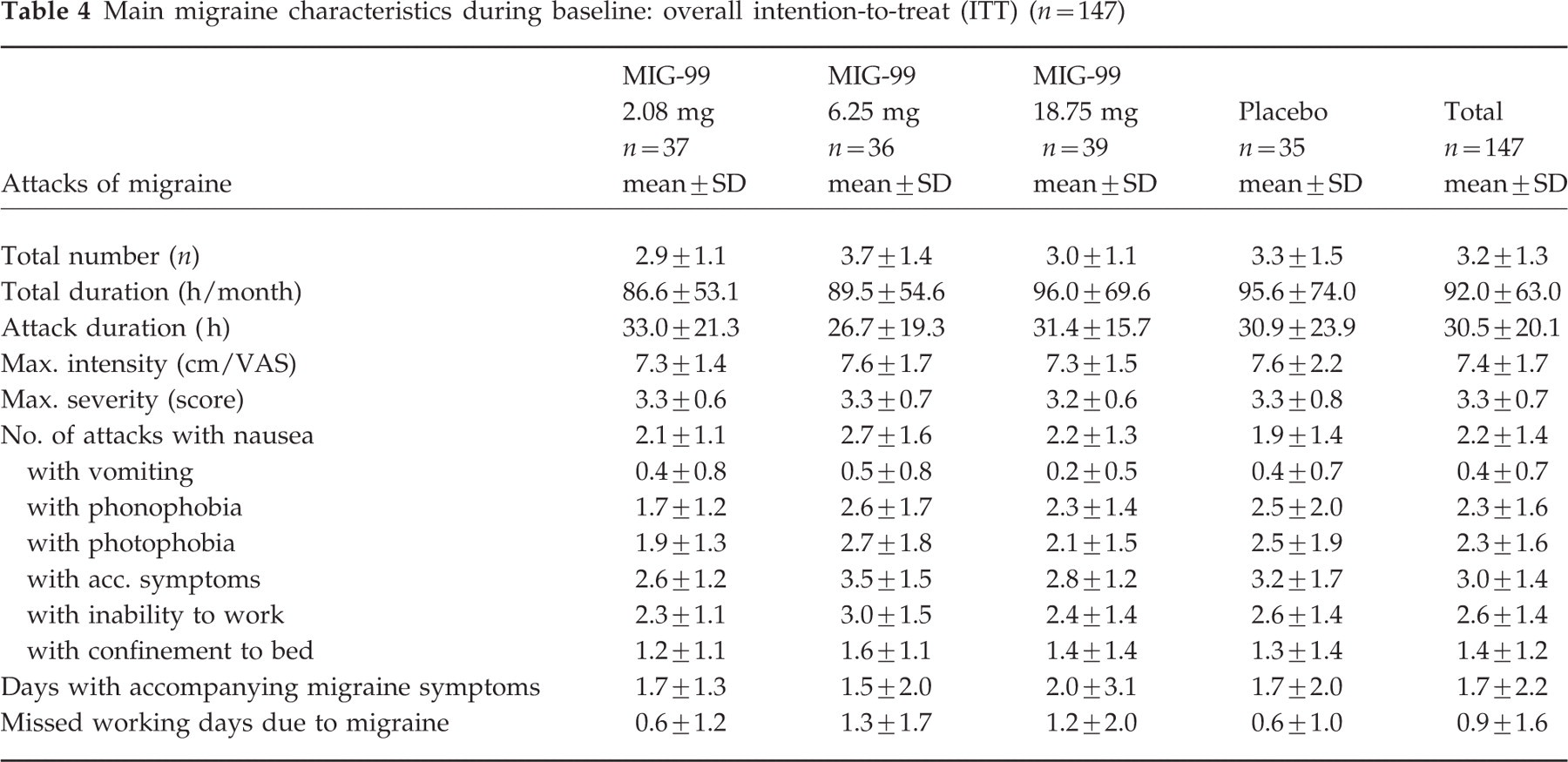
Table 4 shows the main migraine characteristics during the 4-week baseline in the overall ITT sample and Table 5 in the confirmatory ITT. In summary, the analysis of demographic and other baseline conditions revealed no clinically relevant differences between the study groups, and no selection bias was found regarding the single subsets.
Main migraine characteristics during baseline: overall intention-to-treat (ITT) (n=147)
Main migraine characteristics during baseline: confirmatory intention-to-treat (ITT) (n=49)

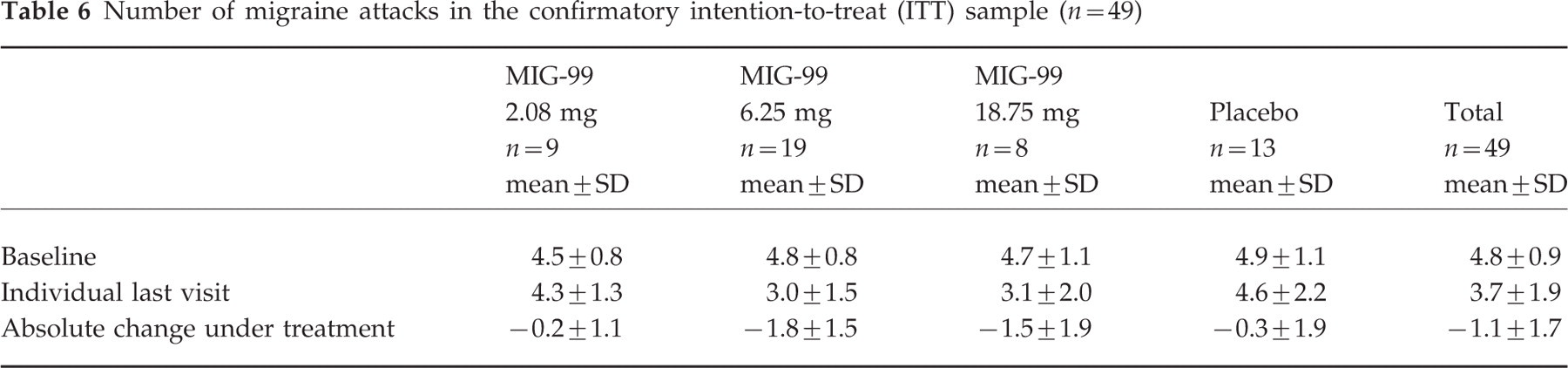
Table 6 gives the total number of migraine attacks within 28 days during the individual last 28-day period compared with the number of attacks during the 28-day baseline phase for the confirmatory ITT sample, Table 7 for the overall ITT sample. Differences between the treatment groups were tested using the Jonckheere–Terpstra test. For the absolute change under treatment including placebo, the P-value resulting for the interim sample was P 1= 0.0024 (n=31). According to the adaptive design of the study, this had to be multiplied with the P-value resulting for the disjoint set of patients enrolled between interim analysis and final analysis (P 2=0.4183, n=18). The product of the P-values (P 1×P 2=0.0010) was compared with the adjusted significance level of cα=0.0087, indicating a significant dose–response relationship. The most marked decrease in the number of migraine attacks occurred in the group receiving MIG-99 6.25 mg (see Fig. 1). The 95% confidence intervals (CIs) for the mean decreases were as follows: MIG-99 2.08 mg: (−0.60, 1.08), MIG-99 6.25 mg: (1.07, 2.49), MIG-99 18.75 mg: (−0.05, 3.15), placebo: (−0.81, 1.46).
Number of migraine attacks in the confirmatory intention-to-treat (ITT) sample (n=49)
Number of migraine attacks in the overall intention-to-treat (ITT) sample (n=147)
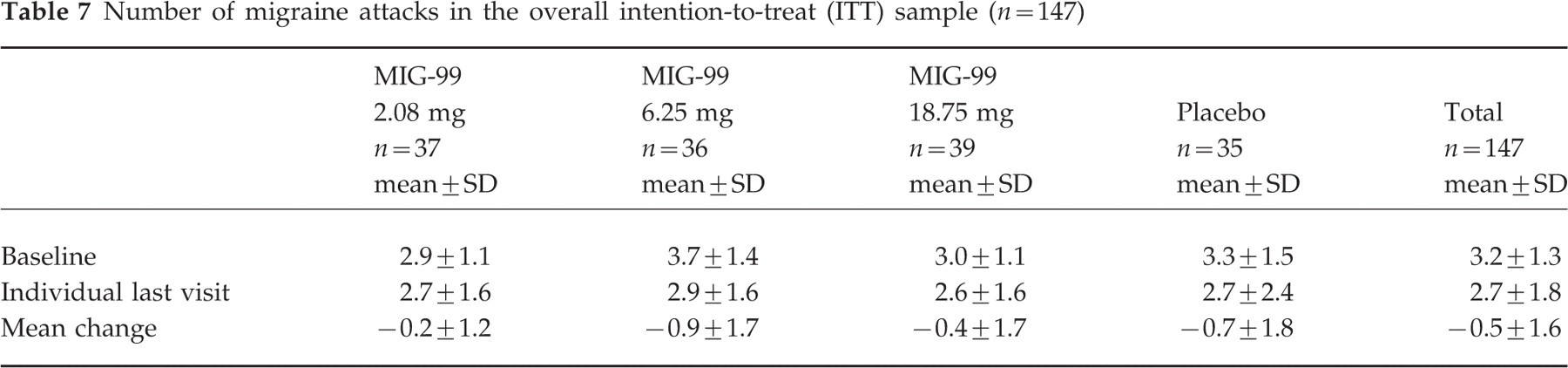

Migraine frequency and dose response relationship of MIG-99 in the confirmatory and overall ITT sample.
An exploratory comparison of the placebo and the MIG-99 6.25-mg group using the Wilcoxon rank sum test established a superiority of the active study treatment. In the overall ITT sample the P-value for the absolute change under treatment including placebo did not support a trend (upward slope) across doses. Again, the clearest decrease in the number of migraine attacks was found for the group receiving MIG-99 6.25 mg. The analysis of the primary efficacy parameter comprised supplementary evaluations. As shown in Table 8, the figures derived for the overall PP sample were almost similar to the results of the overall ITT analysis. Again, the decrease was clearest in the group treated with MIG-99 6.25 mg; 95% CIs for the mean decreases are: MIG-99 2.08 mg: (−0.18, 0.60); MIG-99 6.25 mg: (0.29, 1.46); MIG-99 18.75 mg: (−0.11, 0.97); placebo: (0.07, 1.30).
Number of migraine attacks in the overall per-protocol (PP) sample (n=110)

With respect to the secondary efficacy criteria (duration of attacks, maximum intensity of migraine attacks (VAS), maximum severity of migraine attacks, mean duration of the single attack, number of days with accompanying migraine symptoms, missed working days due to migraine attacks and type and amount of additionally taken medications for the treatment of acute migraine attacks), there were no relevant differences between the four treatment groups and between the overall and the confirmatory ITT sample. Further benefit of MIG-99 is shown in Table 9. The maximum intensity of migraine attacks, the number of migraine attacks with confinement to bed and number of missed working days decreased with increasing dosage in the confirmatory ITT sample.
Some secondary efficacy parameters: absolute change from baseline to end of treatment in the confirmatory intention-to-treat (ITT) sample (n=49)
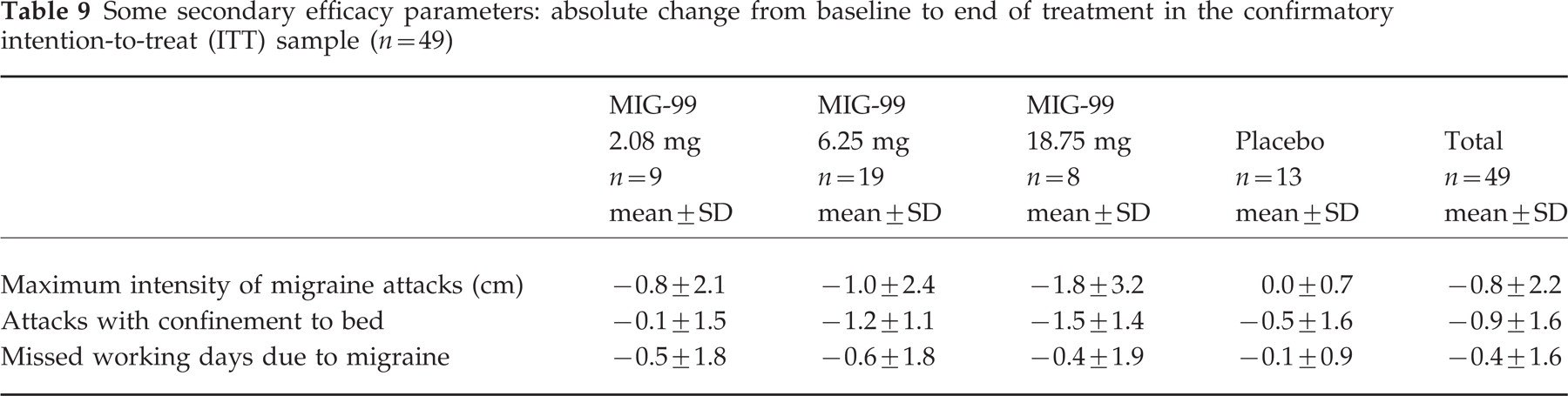
According to study protocol another secondary efficacy criterion was the response rate specified as the number of patients with an improvement of approximately 50% regarding the average number of migraine attacks. The achieved data for the overall and the confirmatory ITT samples are shown in Table 10.
Number of patients with >50% improvement of migraine attack frequency

The global assessment of efficacy revealed a difference between MIG-99 6.25 mg and placebo in the confirmatory ITT sample. A good or very good efficacy was reported in 63% vs. 8% of these patients when assessed by the investigator (P=0.003) and in 53% vs. 23% when assessed by the patients (P=0.15). However, a clear trend to a better tolerability of MIG-99 6.25 mg compared with placebo in the confirmatory ITT subset was observed for the assessments of both investigator and patient, but this was just not statistically significant (both P=0.058).
The subgroup analysis of this study includes a comparison of the patients treated with triptans in case of acute attacks with those not receiving triptans during the study. There was a tendency for a more pronounced reduction of the number of migraine attacks in patients without triptan medication during the study. As this slight difference concerned the groups obtaining MIG-99 as well as the placebo group, assumptions of a possible interaction between MIG-99 and triptans were not justified.
All enrolled 147 patients entered the analysis of safety parameters. Overall, 134 AEs were documented in 52/147 (35.4%) study patients. In total, 64 different kinds of events/symptoms were reported by the 52 patients with AEs. Only one event was reported by 23, two events by 16, three events by four, and four, five and six events were reported by three patients, respectively. The proportion of patients with at least one AE was lowest in the group of patients receiving the highest dose (28.2%) and highest in the low-dose group (40.5%). In the placebo group 34.3% of patients reported at least one AE. A similar result was obtained when the analysis was restricted to AEs for which the causal relationship was not excluded according to the investigator's assessment, i.e. rated as definite, probable, possible, or unlikely. In this analysis the proportion of patients with suspected adverse drug reactions ranged between 12.8% (MIG-99 18.75 mg) and 30.6% (MIG-99 6.25 mg), and was 28.6% in the placebo group. AEs were stated as the main reason for premature termination in nine patients, four receiving MIG-99 2.08 mg, two receiving MIG-99 6.25 mg, one receiving MIG-99 18.75 mg, and two receiving placebo. The only serious AE occurred in the placebo group and concerned an ovarian cyst leading to hospitalization. The relationship of this event to study medication was assessed as unlikely by the centre-specific investigator and as not related by the principal investigator (see Table 11).
Frequency of adverse events (AEs)

∗Not otherwise specified.
Compliance was rated as ‘good’ in the majority of the study patients (n=109 (74.1%)). There were no relevant differences during therapy with regard to mean and median values between the four treatment groups at baseline for any laboratory parameter. Weight, heart rate and blood pressure remained constant throughout the study, without differences between the treatment groups.
Discussion
In this dose–response study, efficacy of MIG-99 in migraine prophylaxis was investigated by comparing three doses of CO2 extract of MIG-99 (2.08 mg, 6.25 mg, 18.75 mg) with placebo. The primary efficacy criterion was defined as the number of migraine attacks during the third 28-day period of therapy vs. baseline. The exploratory testing of the overall ITT results by the Jonckheere–Terpstra test did not support differences between the three different doses and placebo. From the analysis of the secondary efficacy criteria, only the maximum migraine intensity and severity and the number of attacks with confinement to bed were reduced by MIG-99. The review of all parameters reveals that they reflected characteristics of the acute migraine attack. Thus, the most relevant influence on these parameters had to be assumed for each patient's individual treatment of the acute attack, but not for the substance given as migraine prophylaxis.
In agreement with the regulatory authority, an additional confirmatory primary efficacy analysis was performed in a subgroup of 49 patients with at least four attacks during baseline. The sample size estimation was adjusted correspondingly. The most pronounced reduction was obtained with MIG-99 6.25 mg t.i.d. and the trend across doses (upward slope) was confirmed by the Jonckheere–Terpstra test (P=0.0010). The average decrease of 1.8±1.5 attacks in the MIG-99 6.25 mg group during the study course has to be classified as clinically relevant. Nevertheless, these findings should be regarded with caution, due to the small number of patients.
Regarding the safety of MIG-99, the incidence of adverse events in the active groups was similar to placebo, and no dose effect was observed. Laboratory investigations and recordings of vital signs indicated no undesired effects, either.
In conclusion, MIG-99 failed to reveal a significant migraine prophylactic effect in general. It seemed to be safe and effective with at least 6.25 mg t.i.d. only in a small subgroup of patients with a minimum of four attacks per month. Accordingly, in a current placebo-controlled study this effect is investigated in migraine patients with four or more attacks per month.
Footnotes
Acknowledgements
The following centres participated in the study: H.-C. Diener and A. Gendolla, Essen; G. Arnold, Berlin; I.W. Husstedt, Münster; V. Pfaffenrath, München; M.J. Ribbat, Mainz; Z. Taneri, Duisburg; A. Beckmann-Reinholdt and I. Mierck, Königstein; M. Föh, Fulda; P. Donat and I. Neuber, Duisburg; D. Backhaus, Hildesheim.