
Abstract
We evaluated telmisartan 80 mg for migraine prophylaxis. Migraine patients (n = 95) with three to seven migraine attacks in 3 months were randomized, double-blind to telmisartan or placebo. The primary end-point was the reduction in the number of migraine days (i.e. a day with ≥ 1 h of symptoms) between the 4-week baseline period and the last 4 weeks of the 12-week treatment period. A responder was recorded when there was a symptom reduction of ≥ 50% in these 4-week baseline and treatment periods. The reduction in migraine days was 1.65 with telmisartan and 1.14 with placebo (P > 0.05). Post hoc analyses adjusting for baseline and centre showed a 38% reduction in migraine days with telmisartan vs. 15% with placebo (P = 0.03), and a borderline significant difference in responders (40% vs. 25%, P = 0.07). The incidence of adverse events was similar between treatments. This study indicates that telmisartan might be effective in migraine prophylaxis.
Introduction
Worldwide, approximately 240 million people have an estimated 1.4 billion migraine attacks each year (1). The effect of migraine on society has been characterized by the World Health Organization using the Global Burden of Disease scale, which includes severe migraine in the highest disability class (together with active psychosis, dementia and quadriplegia), emphasizing that this illness represents a serious health problem both for individuals and for society (2).
Triptans given orally or by nasal spray are a major contribution to migraine management. However, only about 50–60% of patients consistently respond to this type of medication (3, 4), and in many it provides only partial relief. Frequent use of triptans can lead to medication overuse headache (5, 6). Therefore, prophylactic migraine treatment is indicated if acute treatment is inadequate and if patients experience three or more migraine attacks per month (7, 8). The prophylactic agents available today have limited efficacy and their poor specificity contributes to their adverse event profile, which in turn limits adherence and long-term use. Thus, there is a need for more effective prophylactic drugs that have good efficacy and are better tolerated.
In a small crossover trial, the angiotensin-converting enzyme (ACE) inhibitor lisinopril was shown to be an effective prophylactic treatment for frequent migraine attacks (9). Angiotensin II receptor blockers provide specific blockade of the renin–angiotensin system by competing directly with angiotensin II at its receptor, blocking the effects of angiotensin II produced by both ACE and non-ACE pathways. Since angiotensin II receptor blockers do not interfere with bradykinin, substance P or tachykinin metabolism, the usual potential adverse events associated with ACE inhibitors, such as coughing (10) and angioneurotic oedema, are greatly reduced. Recently, the type I angiotensin II receptor antagonist, candesartan, has also been shown to be effective as a migraine prophylactic agent (11). These studies indicate that inhibition of the production of angiotensin II or blocking of its actions on the type I angiotensin II receptor offer new targets for migraine prophylaxis. We therefore undertook a Phase II pilot study to investigate the prophylactic efficacy of telmisartan in migraine prophylaxis.
Telmisartan (MICARDIS®; Boehringer Ingelheim, Ingelheim, Germany) is an orally active, non-peptide type I angiotensin II receptor antagonist that lowers blood pressure with once-daily dosing (12). In subjects with normal blood pressure, the decrease of blood pressure while taking telmisartan is small. Telmisartan monotherapy has been evaluated for safety in > 10 900 patients in 50 clinical trials, including 1026 patients treated for ≥ 1 year and 636 patients treated for ≥ 2 years. Adverse experiences have generally been mild and transient in nature, and have only infrequently required discontinuation of therapy. In placebo-controlled trials of various doses of telmisartan (20–160 mg) monotherapy for up to 12 weeks, the overall incidence of adverse events was comparable to placebo (data on file; Boehringer Ingelheim).
Methods
Patients
The aim of this Phase II study was to assess the efficacy and safety of telmisartan as a migraine prophylactic agent. We followed as closely as possible the guidelines of the International Headache Society for controlled trials in migraine (13). The primary efficacy end-point was the reduction in the number of migraine days between the 4-week baseline period on single-blind placebo and the last 4 weeks of the 12-week double-blind treatment period (Fig. 1).

Study design.
A migraine day was defined as a calendar day with ≥ 1 h of migraine symptoms, irrespective of intake of medication to treat a migraine attack. Secondary end-points included hours with migraine per 4 weeks, days with any headache, hours with any headache, doses of triptans and doses of analgesics. Responder analysis was determined for each efficacy measure; a responder was recorded when the number of migraine days was reduced by ≥ 50% during the treatment period when compared with baseline. Analysis of responders was performed for the last 4 weeks of the 12-week treatment period. Safety was assessed by recording adverse events, vital signs (i.e. systolic and diastolic blood pressure, and pulse rate), physical examination and laboratory assessments. Inclusion and exclusion criteria are shown in Table 1
Inclusion and exclusion criteria

Study design
Telmisartan 80 mg (MICARDIS®; Boehringer Ingelheim) was provided as tablets for oral administration. Placebo tablets, matching telmisartan 80 mg, were used as the comparator. Patients were assessed during the screening period to determine if they were eligible to participate in the study. After completion of all screening procedures and on meeting the appropriate inclusion and exclusion criteria, patients underwent a 4-week single-blind baseline period on placebo (Fig. 1). At the end of the baseline period patients returned to the clinic. Patients meeting the inclusion criteria for the number of migraine days required by the study were randomly assigned on a 1:1 basis to telmisartan or placebo. Patients and physicians were both blinded to study medication during this phase of the study (Fig. 1).
Patients recorded headache occurrence, type, intensity, autonomic symptoms, duration and acute medication use in a diary. Use of analgesic, ergotamine and triptan medication for rescue treatment of migraine attacks was allowed, and documented in the patient diary.
The trial was carried out in compliance with the protocol, the principles laid down in the Declaration of Helsinki (October 1996 version), in accordance with the ICH Harmonised Tripartite Guideline for Good Clinical Practice and applicable regulatory requirements. The trial was reviewed and approved by a local institutional review board or an independent ethics committee. Prior to participation in the trial, written informed consent was obtained from each subject.
Statistics
Previous studies in migraine prophylaxis suggested that the standard deviation of the change from baseline in the number of migraine days per 4 weeks is approximately 3 days. Using this estimate, a sample size of 40 patients per treatment group would have provided 80% power at the 5% (two-sided) level of significance to detect a difference of two migraine days between treatments.
The primary objective of the study was to determine if telmisartan is efficacious in migraine prophylaxis compared with placebo. The reductions in the number of days with migraine per 4 weeks were compared for both treatment groups. Reductions were evaluated as changes between the last 4 weeks of randomized treatment and baseline, where baseline was the last 4-week time period of the placebo run-in phase. Testing of the null hypothesis was planned to be performed using a two-sided Wilcoxon rank sum test at the α level (type I error rate) of 0.05. Rejection of the null hypothesis would show that telmisartan is different from placebo. The analysis was performed on all patients who had an evaluable baseline period, were randomized, received at least one dose of study medication and had an evaluable final period.
After unblinding, it was apparent that the baseline value for the number of migraine days was different between treatment groups, and that reductions in migraine days were not consistent across centres. Therefore, a post hoc analysis of covariance (
The secondary end-points were descriptively evaluated using the same statistical methods described for the primary end-point for continuous variables, and by use of logistic regression for response variables. These were adjusted for treatment, centre and baseline effects, if appropriate.
Baseline demographics and baseline disease data were summarized for each treatment. The safety evaluation was performed on all patients having received at least one dose of active treatment and was based on all adverse events observed during the study. For all efficacy variables, when the observation periods at baseline and end of randomized treatment were < 4 weeks, the number of migraine days was extrapolated to the full 4 weeks. The commercial program ClinPro/LBL version 5.2 (running on a PC under Windows 95; Clinical Systems Inc., Garden City, NY, USA) was used to create the 1:1 randomization lists. Randomization was stratified by study centre. The analysis was run on
Results
We randomized 95 patients, of whom 84 finished the study period with evaluable data and were included in the efficacy analysis. Patient disposition is shown in Table 2.
Patient disposition
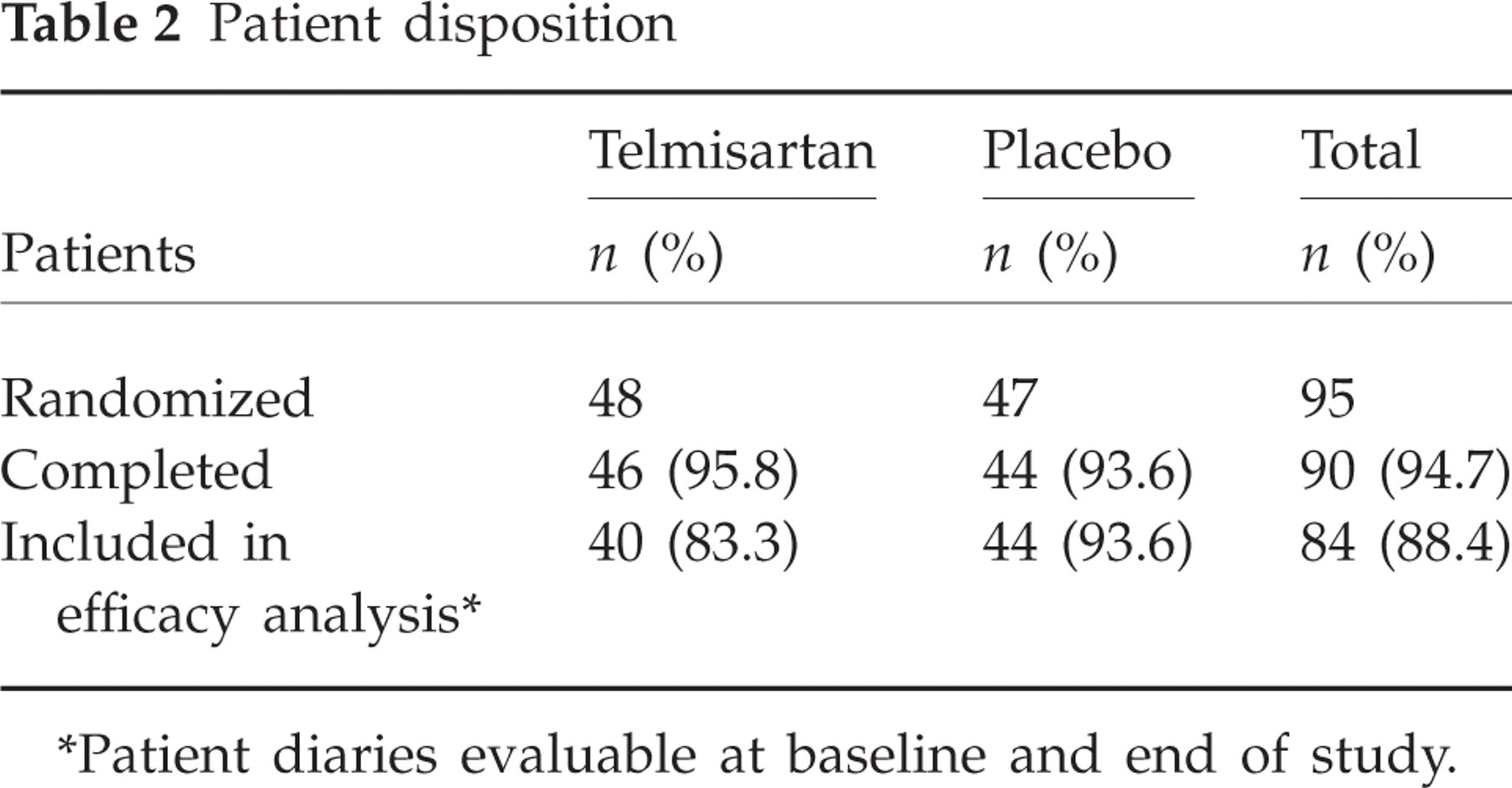
∗Patient diaries evaluable at baseline and end of study.
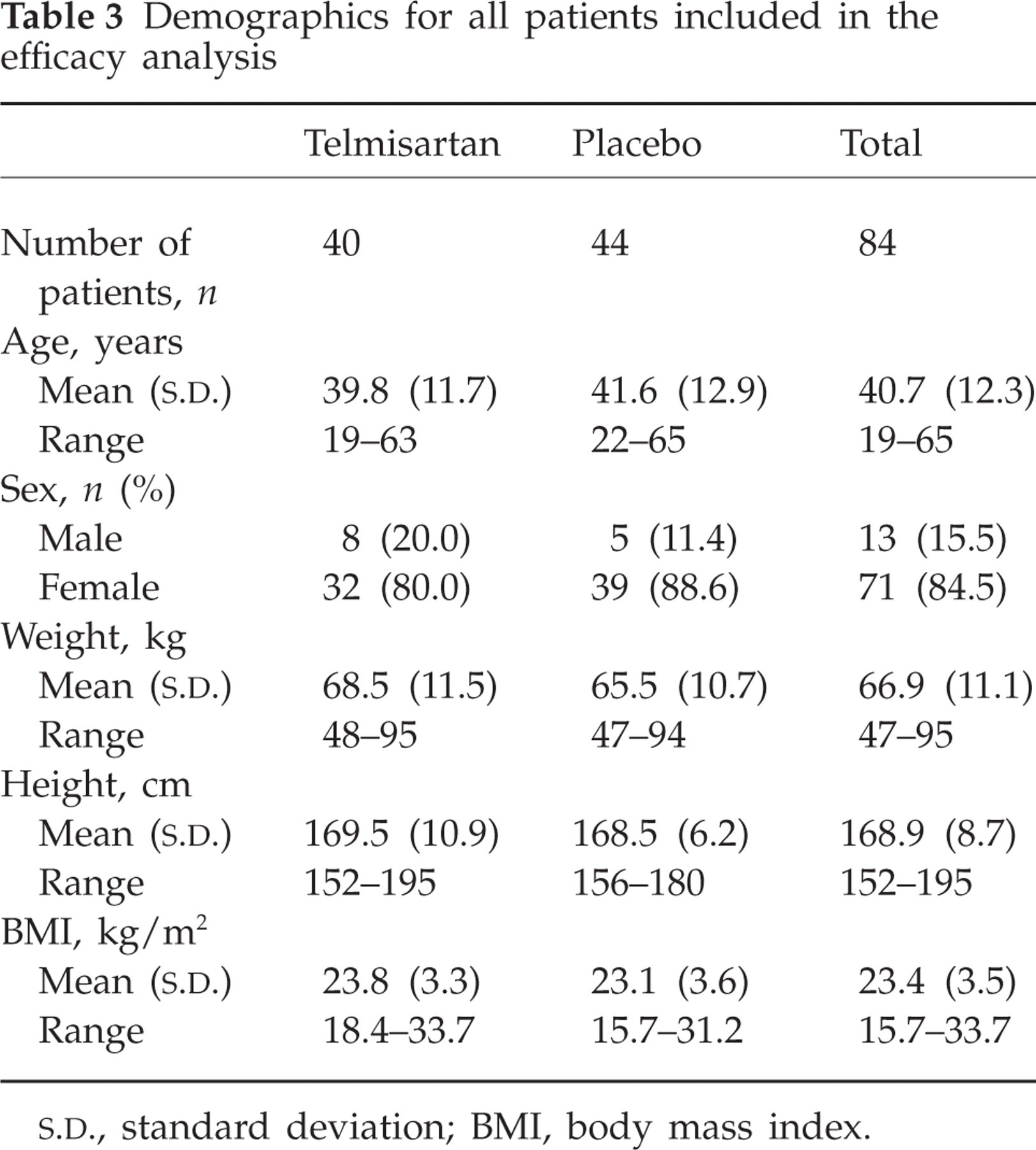
Baseline demographics were comparable between the two treatment groups (Table 3). The majority of patients were female.
Demographics for all patients included in the efficacy analysis
The mean number of headache days was different between the treatment groups (Table 4). Post hoc statistical comparison revealed that the number of migraine days in the placebo group (1.4 days more than in the telmisartan group) in the baseline period was close to being significant (Wilcoxon rank sum test; P = 0.09). As higher baseline values often correlate with higher reductions in migraine days (more room for improvement), post hoc analysis adjusting for baseline migraine days and centre effects was performed.
Baseline values for all patients included in the efficacy analysis

Table 5 shows the number of migraine days in the baseline period, and the reduction in migraine days with telmisartan and placebo. The reduction in migraine days was numerically higher with telmisartan, but was not statistically significant in the preplanned analysis (Wilcoxon test). After adjusting for baseline differences and centre, the percent change from baseline showed a significant 38% reduction with active drug compared with placebo (15%; P = 0.0262). However, it should be noted that there was also a significant treatment-by-centre interaction (P = 0.0242), indicating that the efficacy of telmisartan was not consistent across all participating centres.
Analysis of migraine days

∗Adjusted for baseline and centre, data log-transformed.
The analysis of secondary end-points is shown in Table 6. Telmisartan was always numerically superior to placebo, but none of the differences was significant.
Analysis of secondary end-points

∗Logistic regression, adjusted for baseline migraine days and centre.
†
CI, confidence interval.
As expected, telmisartan resulted in a drop in blood pressure (4.6 mmHg) (Table 7). The tolerability of telmisartan was not different from that of placebo (Table 8). One patient in each of the treatment groups discontinued due to side-effects. For the patient in the placebo group, the adverse events that led to discontinuation from the study drug (nausea, stomach ache, loss of appetite) were considered to be drug related.
Blood pressure at baseline and end of study

∗Based on 46 patients.
Adverse events during the 12-week treatment period

AE, adverse event.
In conclusion, telmisartan did not result in a statistically significant reduction in migraine days in a 12-week treatment period. Telmisartan was well tolerated and had an adverse event profile similar to that of placebo.
Discussion
This Phase II study has shown that telmisartan numerically reduced primary and secondary end-points in this placebo-controlled study, but failed to reach statistical significance in the prespecified test. When we corrected post hoc for imbalances in migraine days at baseline and also took into account the effect of centres, the difference became significant in favour of telmisartan. The percent reduction in migraine days (−38%) and the responder rate of 40% are in the range of effects seen in other trials with migraine-preventive drugs (8, 14, 15). Considering the imbalances in migraine days at baseline, we have to admit that the preplanned testing strategy and sample size calculation were not adequate. The tolerability was excellent and the drop-out rate due to side-effects was much lower than for other drugs used in migraine prevention (14, 16, 17).
The strength of our study is its state-of-the-art trial design, including randomization and placebo control, but the major shortcoming is the inadequate sample size. Therefore, confirmatory conclusions about the efficacy of telmisartan migraine prophylaxis cannot be drawn from this pilot study and can be reached only when the trial is repeated with a higher number of patients.
Competing interests
H.C.D. received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from: Addex Pharma, Allergan, Almirall, AstraZeneca, Bayer Vital, Berlin Chemie, CoLucid, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Grünenthal, Janssen-Cilag, Lilly, La Roche, 3M Medica, MSD, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper and Brümmer, SanofiAventis, and Weber & Weber. Financial support for research projects was provided by Allergan, Almirall, AstraZeneca, Bayer, GSK, Janssen-Cilag, Pfizer. Headache research at the Department of Neurology in Essen is supported by the German Research Council (DFG), the German Ministry of Education and Research (BMBF) and the European Union. H.C.D. has no ownership interest and does not own stocks of any pharmaceutical company. A.G. has received honoraria for contributions to advisory boards and oral presentations from Addex Pharma, Allergan, Almirall, Astra Zeneca, Bayer Vital, Berlin Chemie, BMS, Janssen Cilag, 3MMedica, and MSD. S.E. received honoraries and research funding from the following companies: Addex Pharma, AGA Medical Corporation, Allergan, AstraZeneca, Berlin Chemie, Boehringer Ingelheim, CoLucid, Desitin, Eisai, GlaxoSmithKline, Ipsen, Janssen-Cilag, Merz, MSD, Novartis, Pfizer, Reckitt-Benckiser, and UCB. A.S. received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from: Addex Pharma, Allergan, Almirall, Boehringer Ingelheim, GlaxoSmithKline, Grünenthal, Janssen-Cilag, MSD, Novartis, Johnson & Johnson, and Pfizer. Furthermore, scientific projects were supported by Allergan and Almirall. H.S. and G.D. are employees of Boehringer Ingelheim. A.F. has no conflict of interest.
Footnotes
Acknowledgements
The authors would like to acknowledge the contributions of the Study Group investigators and their staff for their work in enrolling patients in this trial. The members of the Study Group were: Guy Arnold, Department of Neurology, University Hospital Charite, Berlin; Annette Beckmann Reinhold, Headache Clinic, Königstein; Bettina Classen, Pain Specialist, Bochum; Stefan Evers, Department of Neurology, University of Münster; Hartmut Göbel, Kiel Pain Clinic, Kiel; Eleonore Jakobasch, Department of Neurology, University Hospital Dresden; Klaus Lengler, Pain Specialist, Erkelenz; Pia Laudan, West German Headache Centre, Essen; Volker Pfaffenrath, Neurologist, Munich; Guenther Schuhmann, Neurologist, Bochum; Andreas Straube, Department of Neurology, Ludwig Maximilians University Munich. The study was designed by H.C.D. and G.D. and executed by H.C.D. and the Essen Headache Centre. Data management and statistical analysis was performed by A.F. and H.S. Boehringer Ingelheim provided unrestricted grants, but had no influence on the planning, conduct or interpretation of the study. Editorial support was provided by PAREXEL MMS Europe Ltd.