
Abstract
Despite evidence emerging from the experimental model of nitroglycerin-induced headache, the endogenous increase in nitric oxide (NO) production during migraine attacks is only speculative. It has been hypothesized that there is a close relationship between activation of the
Keywords
Introduction
Activation of the trigeminovascular system and release of vasoactive peptides from trigeminal sensitive endings supplying cerebral circulation are believed to be the final pathway which mediates migraine attacks. The evidence supporting trigeminovascular system activation in migraine furnishes a potential explanation for vasodilatation of large cerebral blood vessels and cerebral blood flow changes which have been described during migraine crises, but does not completely clarify the onset and maintenance of head pain. First evidence suggest that calcitonin gene-related-peptide (CGRP) and neurokinin A (NKA), the two neuropeptides which were found to be increased in the extrajugular blood and in the peripheral venous blood, respectively, during migraine attacks, are not able to induce pain when infused intravenously or locally in the temporal muscle, nor are they able to potentiate the nociceptive effects of other algogenic substances (1–4). More recent research, in contrast, demonstrated that CGRP may induce headache when infused to migraineurs (5) and this finding prompted the development of a CGRP antagonist for their potential use in migraine (6).
Further mechanisms have been advocated to explain pain mechanisms in the ictal period. In the last few years interest has been focused on several substances with vasoactive properties produced by endothelium, which can play a fundamental role in mediating variations in the vascular tone and cerebral blood flow, as well as induction and maintenance of head pain during migraine crises.
Nitric oxide (NO) is one of the most important substances with vasodilatatory effects produced by the endothelium. This intra- and intercellular messenger, with a very short half-life (5–15 s) is a highly reactive free radical, which is rapidly converted into its stable metabolites, nitrites and nitrates. NO-synthase (NOS) is the enzyme which catalyses the synthesis of NO from nitrogen of guanidino nitrogen atoms of
Olesen et al. (10) believed that NO could be the ‘key molecule’ in the pathogenesis of migraine, suggesting that the activation of the pathway involved in its synthesis represents a common mechanism in experimental headache models, such as nitroglycerin- and histamine-induced headaches (11).
The mechanism which mediates pain induction during nitroglycerin infusion is even now a matter of controversy. It has been hypothesized that NO released from nitroglycerin can be responsible for head pain in the early phases. This radical could act both on cerebral vessel endothelium and on perivascular sensitive endings, or induce the release of histamine from endothelium and mast cells, even though this last mechanism has not yet been confirmed (12). It has also been suggested that another mechanism could be involved in the late stages of nitroglycerin-induced headache, consisting not only of the activation of the endothelial
If the experimental model of nitroglycerin-induced headache suggests the occurrence of a hypersensitivity to exogenous NO (furnished by NO donors), an up-regulation of the endogenous
Jugular blood provides much more information than peripheral blood regarding the neurotransmitter and biochemical changes which characterize migraine attacks. In this regard internal jugular blood can more exactly reflect intracranial neurotransmitter and biochemical variations, whereas the external jugular blood can express neurotransmitter and biochemical variations in both intra- and extracranial cerebral structures.
No studies have been carried out until now on the variations of NO markers in the internal jugular blood of migraine patients, or on the relationship between their modifications and those of algogenic and vasosactive prostaglandins, as well as vasoactive peptides from the trigeminovascular system. It is possible to study the temporal variations in NO markers during attacks but it is impossible to distinguish their endothelial or neural origin. A prevailingly endothelial origin of NO can be hypothesized, but release from non-adrenergic, non-cholinergic (NANC) fibres, or from the same trigeminal fibres supplying cerebral vessels, cannot be excluded.
The present research was aimed at verifying the changes in levels of the stable metabolites of NO, the nitrites, and those of its intracellular mediator, cGMP, in the internal jugular blood of migraine in-patients examined during attacks at the Neurological Clinic at the University of Perugia. The determination of these levels was carried out at different times during attacks and their variations were compared with those of some prostaglandins with algogenic and vasoactive effects, i.e. prostaglandin E2 (PGE2) and 6 keto PGF1α (stable product of PGI2). Variations in the intracellular mediator cyclic adenosine 3′5′ monophosphate (cAMP) and the levels of vasoactive peptides from the trigeminovascular system, CGRP and NKA, were also assessed at the same time.
Patients and methods
Patients
The study protocol was approved by the local ethical committee.
After written informed consent, five migraine without aura patients were admitted to the Neurological Clinic at the University of Perugia as in-patients, and assessed during their first spontaneous, non-induced attack occurring during the period of hospitalization (mean length of Hospital stay=7 days). Diagnosis was made using the current International Headache Society (IHS) criteria (16). Exclusion criteria for patients joining the study were smoking habit, migraine below 2 h, no systemic or comorbid pathologies (renal diseases, cardiovascular diseases including hypertension, epilepsy) and pregnancy.
None of the patients was treated with prophylactic drugs in the previous 2 months. They had been on a low nitrite diet in the last week. The characteristics of the patients are reported in Table 1.
Characteristics of migraine without aura patients

For the central venous access procedure, the internal jugular vein was first examined with an hand-held sonography transducer to determine the size, patency and location of internal jugular vein (17). Access to the right or left internal jugular vein was subsequently performed (depending on the laterality of head pain: in four patients right jugular vein, in the remaining patient left jugular vein) under sterile conditions, using sonographic guidance. The internal jugular vein was punctured just cephalad to the clavicle by using the 21 G needle for peripherally inserted catheter (PICC) kits (18). Once venous access was obtained, a 0.018-in wire was passed through the needle with fluoroscopic guidance (19). The needle was then removed. A 4 or 5 peel away sheath (from the PICC kit) was inserted over the wire. The wire was then removed. Only one puncture was required for four patients, two punctures for the other migraine patient undergoing central vein access procedure. The common carotid artery was not inadvertently punctured in any case. No periprocedural complications occurred. Post-procedural chest radiographs showed no evidence of pneuromotorax, haemothorax or mediastinal haematoma in any of the migraine patients undergoing central venous catheter insertion.
Internal jugular blood was taken within 30 min after the onset of the attack (T0), at the first hour (1st h), at the second (2nd h), at the fourth (4th h), at the sixth (6th h) from the beginning of the attack and within 2 h after the end of the attack, when patients were just headache-free. Within the first hour from the catheter insertion the head pain was severe enough to be regarded as migraine (score=3 in pain intensity) in all migraine patients examined.
No symptomatic drugs were administered to patients during migraine attacks to stop the migraine attack. Details of migraine attacks of each patient are reported in Table 2.
Details of individual migraine attacks with particular reference to pain intensity at each time point
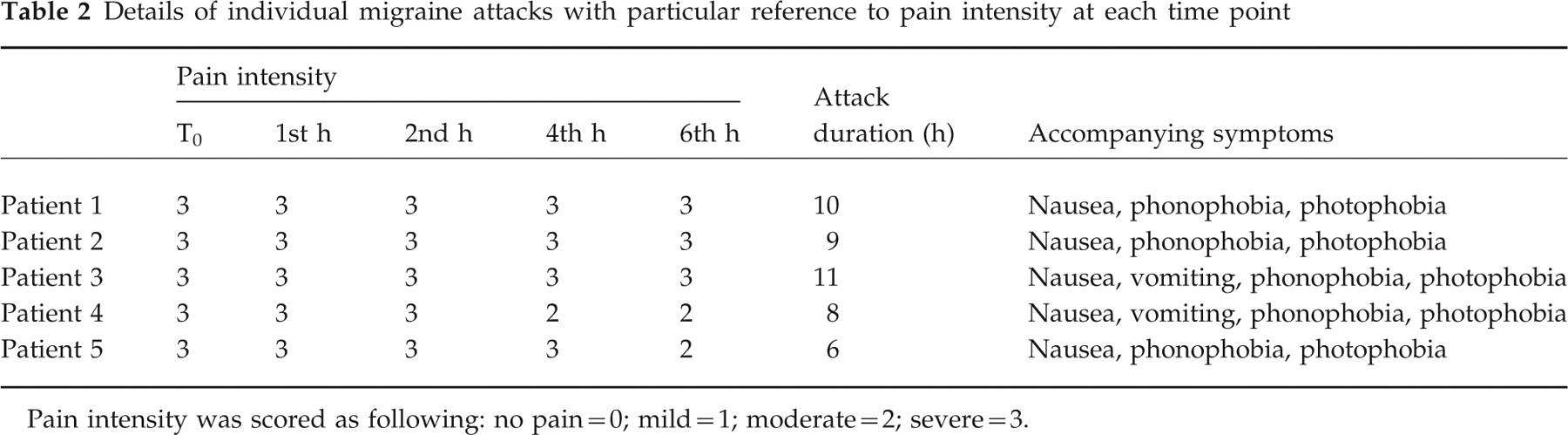
Pain intensity was scored as following: no pain=0; mild=1; moderate=2; severe=3.
To discriminate whether the modification in biochemical variables observed in our migraine patients was the response to the insertion of the catheter through the endothelium of the internal jugular vein, we determined the same parameters in the internal jugular vein blood from four patients having elective internal jugular vein catheters placed for post-operative management in intensive care. Even if the timing of sampling was not exactly the same, these measures could help us to answer the difficult question whether what was found was the result of trauma and not associated with migraine attack.
Methods
Determination of NO2− plasma levels
Jugular venous blood (2–3 ml) drawn at the times fixed in the study protocol was collected in polypropylene tubes, centrifuged and stored at −80°C for 3–5 days until determination.
Oxidation products of NO, into which this radical is rapidly converted at intra- and extracellular levels, were determined by high performance liquid chromatography (HPLC). The NO3− ion, which represents the greater amount of NOx−, was measured as NO2− following its enzymatic reduction with nitrate-reductase in plasma samples. After reduction of NO3− to NO2−, proteins were precipitated and the extract derivatized with hydralazine at acid pH and directly analysed. This method is reasonably sensitive and reproducible, even though some compounds such as cromophores can interfere with the absorption spectra. With the aim of overcoming this problem, an automated analysis system was developed, consisting of the pretreatment of extracted sample with an ionic exchanger to remove interfering substances. Concentrations of nitrites were determined from a linear standard curve obtained from sodium nitrite under the same experimental conditions. Detection limit of the method is 0.3–0.5 pmol of NO2− in the column.
Data were expressed as μmol/l.
Although NO3− and NO2− are produced at high levels in the gut, and even though care was taken by keeping the patients on a nitrite-restricted diet, control samples of plasma were taken from the antecubital fossa to determine their levels in the peripheral circulation, at the same time points as blood drawing from the internal jugular vein.
Determination of CGRP, NKA, cGMP, cAMP, PGE2 and 6 keto PGF1α
Jugular venous blood (10 ml) drawn at the times fixed in the study protocol were collected in polypropylene tubes containing EDTA (1 mg/ml) and callicreine (500 U/ml).
CGRP, NKA, PGE2, cGMP, cAMP, and 6 keto PGF1α were eluted with 60% acetonitrile in 0.1% trifluoroacetic acid in SEP-C18 columns activated with 0.1% trifluoroactic acid and 60% acetonitrile in 0.1% trifluoroacetic acid. Eluates were associated with a centrifuge concentrator (Supervap PL-CC-180). Residues were dissolved in radioimmunoassay (RIA) buffer and determined with RIA kits (Peninsula Labs GMBH, Belmont, CA). Data were expressed as pmol/l for neuropeptides and PGs and as nmol/l for both cyclic nucleotides.
Standards for the above substances were dissolved in 0.1 mol/l phosphate buffer pH 7.5, containing 0.1% bovine albumin, 0.01% sodium azide and 500 kU/ml callicreine.
CGRP and NKA human antisera were obtained from rabbit and are specific for the C-terminal end for both neuropeptides.
CGRP antiserum showed a cross-reactivity with rat and chicken CGRP (100%) but not with other neuropeptides. The intra- and interassay variabilities were 3% and 6%, respectively. The detection limit of the assay was < 1 pmol/l.
NKA antiserum cross-reacts with kassidin (100%), porcine neuropeptide K (100%) and neurokinin B (80%), but shows a cross-reactivity of < 0.05% and 0.02% with substance P and physalaemin, respectively. No cross-reactivity for the same antiserum is present with neuropeptide Y (porcine), neuromedin B (porcine), ACTH (human) and Arg8 vasopressin.
The minimum amount of immunoreactive NKA detectable by the assay is < 1 pmol/l. The intra- and interassay variabilities for NKA radioimmunoassay are 5% and 8%, respectively.
Much evidence supports that NKA and SP come from the same preproneuropeptide, and that these two neuropeptides are co-stored and released together, cooperating in the mediation of neurogenic inflammation in response to the activation of the trigeminovascular pathway.
Despite this, we did not determine SP in jugular blood of our migraine patients, while previous studies on this topic showed only changes in CGRP and NKA but not SP in the peripheral blood of adult and young migraine patients examined ictally (1, 2).
There are also data showing differences in pharmacokinetic properties between the tachykinins. SP is rapidly degraded in the plasma by peptidases, whereas NKA, like NPK, has a longer half-life and is metabolized in other compartments (20, 21). This could at least in part explain the finding of no significant variation in their levels either in the peripheral or jugular blood of migraine patients assessed ictally.
PGE2 and 6-keto-PGF1α human antisera were obtained from the rabbit. The intra- and interassay coefficients of variations were 6% and 8%, respectively, for both PGs tested. The antibody for PGE2 cross-reacts with PGE1 (70%), PGF2α (3%), PGA1 (1%), and PGA2 (1%). The antibody for 6-keto-PGF1α cross-reacts with PGE2 (3%), PGF2α (1%). The detection limits for PGE2 and 6-keto-PGF1α were 0.1 and 0.2 pmol/l, respectively.
cGMP and cAMP human antisera were obtained from the rabbit. The intra- and interassay variation coefficients were 5% and 7% and 4% and 6%, respectively, for the two cyclic nucleotides. The anti-cAMP antibody does not cross-react with 5′-AMP, 5′-ADP, 5′-ATP or 3′5′-cGMP, whereas anti-cGMP does not show cross-reactivity with 5′-AMP, 5′-ADP, 5′-ATP 3′5′-cAMP, 5′-GMP, 5′-GDP, 5′-GTP. The detection limit for both cGMP and cAMP was < 1 pmol/l.
Specificity data on CGRP, NKA, PGE2 and 6-keto-PGF1α cGMP and cAMP were provided by Peninsula Laboratories.
Statistical analysis
All values were expressed as mean±2 SD. One-way within-subjects
Results
Nitrite levels in the internal jugular blood of migraine patients assessed ictally reached the highest values at the first hour, then they tended to decrease progressively to the levels detected at the time of catheter application (T0) in the venous jugular samples collected after the end of migraine attack (Table 3) (Fig. 1a,b).
Mean (± 2 SD) of the nitrites (μmol/l), cyclic guanosine 3′,5′-monophosphate (cGMP) (nmol/l), calcitonin gene-related peptide (CGRP) (pmol/l) and neurokinin A (NKA) (pmol/l) levels, found at the various stages of attacks, in the internal jugular venous blood of migraine without aura patients examined
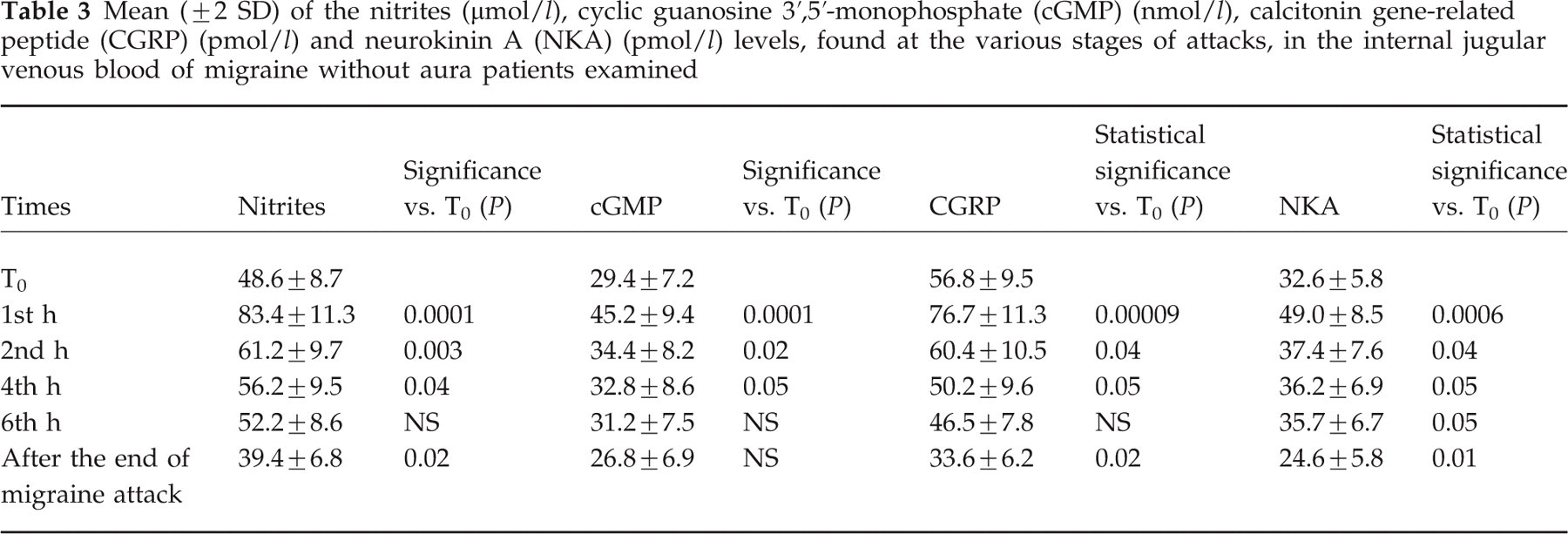

(a) Scatter plot of mean ± SD and SE of nitrite (NO2−) (μmol/l) levels in the internal jugular blood of migraine patients examined within 30 min from beginning of the attack (T0), at the 1st (T0), 2nd, 4th hours and after the end of the ictal period (headache-free period). (b) The curves of nitrite levels for each patient included in the study at the same times.
Levels of nitrites in the antecubital fossa of migraine patients taken at the different times of the study are shown in Table 4. A slight but significant increase in nitrite levels was found in the peripheral blood, especially at the first hour, but a broader range of variation in peripheral nitrite levels emerged compared with their levels in the jugular blood.
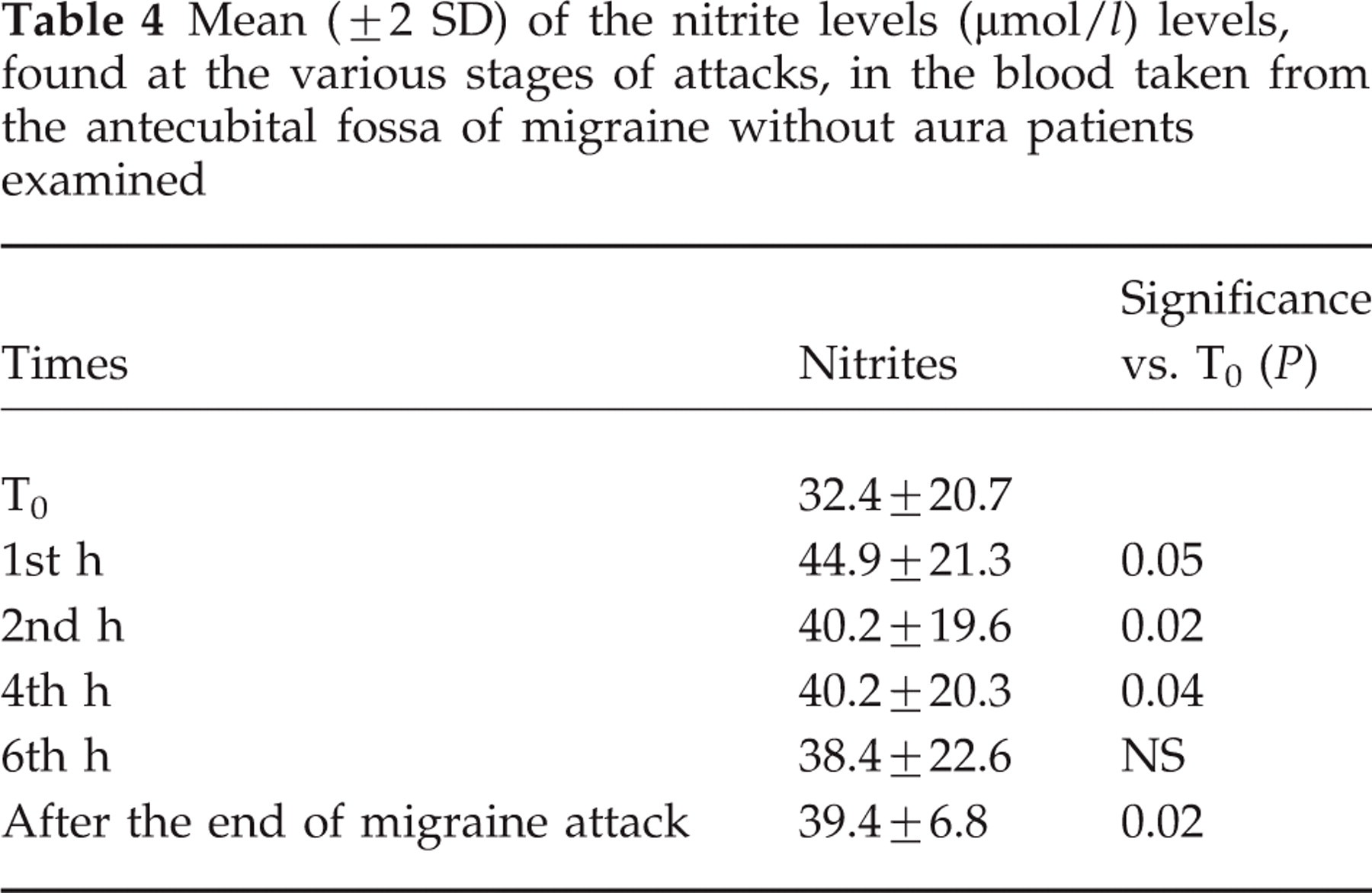
Mean (± 2 SD) of the nitrite levels (μmol/l) levels, found at the various stages of attacks, in the blood taken from the antecubital fossa of migraine without aura patients examined
A parallel increase in the levels of NKA and CGRP was observed, with the highest value being detected at the first hour. The levels of these vasoactive peptides tended to decrease in the following hours. The values detected after the end of attack were significantly lower than those detected at T0 time in the internal jugular venous blood sample (

(a,b) Scatter plot of mean ± SD and SE of nitrite (NO2−) (μmol/l), calcitonin gene-related peptide (CGRP) (pmol/l) and neurokinin A (NKA) (pmol/l) ((a) and (b), respectively) levels in the internal jugular blood of migraine patients examined within 30 min from beginning of the attack (T0), at the 1st (T0), 2nd, 4th hours and after the end of the ictal period (headache-free period).
PGE2 levels, and to a lesser extent 6-keto PGF1α levels, significantly increased at the 1st h compared with those measured at T0, but they reached their peak at the 2nd h. These highest values were maintained till the 4th h and 6th h, and then decreased at the end of attack, reaching values lower than those measured immediately after the catheter positioning at T0 (
Mean (± 2 SD) of prostaglandin E2 (PGE2), 6-keto PGF1α (all expressed as pmol/l) and cyclic adenosine 3′5′ monophosphate (cAMP) levels (nmol/l) detected at the indicated times, during migraine attacks, in the internal jugular venous blood of the migraine without aura patients examined


(a,b) Scatter plot of mean ± SD and SE of prostaglandin E2 (PGE2) (pmol/l) and 6-keto PGF1α (pmol/l) levels ((a) and (b), respectively) in the internal jugular blood of migraine patients examined within 30 min from beginning of the attack (T0), at the 1st, 2nd, 4th hours and after the end of the ictal period (headache-free period).
The increase in the value of the stable metabolite of NO, NO2−, was accompanied by a similar rise in the levels of cGMP which even reached a peak at the first hour (

(a,b) Scatter plot of mean ± SD and SE of cyclic guanosine 3′,5′-monophosphate (cGMP) (nmol/l) and cyclic adenosine 3′5′ monophosphate (cAMP) (nmol/l) levels ((a) and (b), respectively) in the internal jugular blood of migraine patients examined within 30 min from beginning of the attack (T0), at the 1st, 2nd, 4th hours and after the end of the ictal period (headache-free period).
Variations in cAMP levels in the internal venous jugular blood appeared instead to vary in time, as was observed for the levels of the two prostaglandins examined, which displayed the highest values at the 4th h and 6th h, therefore later than what was found for the values of the other intracellular messenger, cGMP (Fig. 4b). cAMP values measured after the end of attack were also significantly lower than those detected at T0 (
The levels of nitrites, neuropeptides from trigeminovascular system and prostaglandins and also intracellular messengers cGMP and cAMP did not differ significantly from those detected at the end of attacks in all migraine patients (when they were headache-free).
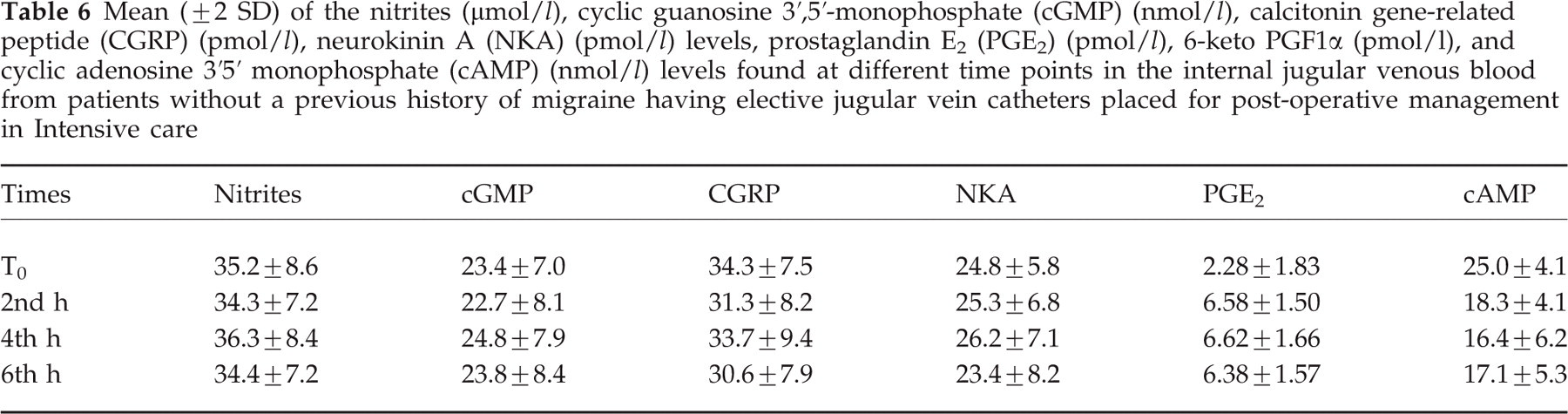
The fact that no variations were found in the levels of trigeminal peptides, nitrites and prostaglandins in the blood of patients having elective internal jugular vein catheters placed for post-operative management in intensive care, allowed us to exclude that changes found in jugular blood of the migraine patients examined were due to trauma of catheter insertion and not associated with migraine attack (Table 6).
Mean (± 2 SD) of the nitrites (μmol/l), cyclic guanosine 3′,5′-monophosphate (cGMP) (nmol/l), calcitonin gene-related peptide (CGRP) (pmol/l), neurokinin A (NKA) (pmol/l) levels, prostaglandin E2 (PGE2) (pmol/l), 6-keto PGF1α (pmol/l), and cyclic adenosine 3′5′ monophosphate (cAMP) (nmol/l) levels found at different time points in the internal jugular venous blood from patients without a previous history of migraine having elective jugular vein catheters placed for post-operative management in Intensive care
Discussion
Nitric oxide has been considered the putative ‘key molecule’ of migraine. However, nitroxergic system markers in migraineurs have been studied only in peripheral blood, both ictally and interictally, and studies carried out in this regard are conflicting and inconclusive.
One of the major difficulties encountered in the study of NO in migraine is the inherent nature of this molecule, particularly its short half-life. NO is in fact rapidly converted to nitrogen dioxide (NO2), which forms the more stable metabolites, nitrites (NO2−) and nitrates (NO3−). The search for these end-products of NO in the peripheral blood furnished inconsistent results.
The first findings of a slight increase in the peripheral levels of NO, concomitant to a rise in the endothelin-1 levels, emerged from research by Nattero et al. on migraine patients examined during migraine attacks (22). A recent study carried out by our group showed that only in some patients examined ictally was there a significant rise in nitrite levels in peripheral blood compared with interictal periods, with a broad range of inter-individual variations (23). There is also evidence of an increase in nitrite levels in peripheral blood of migraine patients examined interictally (24).
As far as peripheral nitrite levels are concerned, one of the most important factors which also acts to induce inter-individual variations is haemodilution. Dietary habits can also influence serum and plasma levels, therefore a nitrite-restricted diet is recommended when studying endogenous variations in NO production.
That nitrites can exactly express the biological effects of NO is also a matter of controversy. It should be remembered that NO biological effects can be mediated by active S-nitroso-thiols that possess potent vasorelaxant and platelet inhibitory properties in vitro. In particular, sulphydrilic species, such as acetylcysteine, even in physiological conditions, can react with NO giving nitroso-thiols which have a longer half-life than NO and preserve the same properties of endothelium-derived relaxing factor (EDRF) (25). Thiol groups of proteins, especially albumin, can explain the long-term effects of NO, both as a vasodilatatory agent and platelet anti-aggregant. No evidence of modifications in the nitroso-thiols levels is available for the blood of migraineurs assessed during attacks. It must also be remembered that haemoglobin and other haeme-containing proteins show high affinity for NO, contributing to scavenger NO, preventing NO-induced activation of soluble guanylate cyclase.
Despite these observations, nitrites can be considered a marker of NO production in migraine, even if indirect, particularly if their concentration is measured in the internal jugular blood during attacks.
The present study confirms the increase in the levels of the stable metabolites of NO, nitrites, in the internal jugular venous blood of patients affected by migraine without aura, examined during a spontaneous migraine attack, supporting previous findings obtained in the peripheral blood. This rise reached a peak at the 1st h and persisted in the subsequent hours of attack. This increased nitrite production was accompanied by a parallel rise in the levels of cGMP.
The major source of NO found in our study could be the endothelium of the cranial vessels, even if another potential source of NO may be attributed to NOS immunoreactive perivascular nerve fibres. Activated platelets and also mast cells, as suggested by the model of the plasma extravasation, could also be involved in the release of NO during migraine attacks in the internal jugular blood. No evidence is available at the moment on the involvement of immune cells as a potential source of NO via activation of inducible NOS during migraine attacks.
It is well known that NO binds to the iron of the haeme group of soluble guanylate cyclase, the enzyme which catalyses the formation of cGMP, being its intracellular messenger. The search for this intracellular messenger could therefore indirectly reflect NO production.
A recent study was carried out by Stepien & Chalimoniuk on cGMP levels in the peripheral blood of patients affected by migraine (26). In the 37 migraine patients examined by the authors during the crises, significantly higher levels of cGMP were found which correlated with pain intensity. These levels tended to dramatically decrease after sumatriptan administration. Our findings therefore support the evidence of increased cGMP production also in internal jugular blood of migraine patients.
From our research a significant increase in the plasma levels of the trigeminal vasoactive peptides, CGRP and NKA, also emerged, which was greater at the 1st h but also persisted at the 2nd, 4th and 6th h.
A significant increase in CGRP levels has already been demonstrated by Goadsby et al. (1) in the external jugular blood of patients affected by migraine examined in the ictal period. In a subsequent study carried out by our group (Gallai et al. (2)), a significant rise in both CGRP and NKA levels emerged in the peripheral blood of young migraine patients evaluated during the attacks. No changes in SP were found in the above studies, and this has been attributed to the fact that the cerebral circulation has a preferential CGRP innervation from the trigeminal ganglion (27). According to the authors of the present study, release in SP cannot be excluded, taking into account the observation of SP-immunoreactive fibre innervation of the large cranial arteries, cranial veins and also brain microvessels, and the relevance of this neuropeptide to the model of neurogenic inflammation. One possible explanation for the negative findings concerning SP is its short half-life due to its rapid degradation by plasma peptidases.
In any case, the only positive finding obtained until now, on vasoactive peptide changes in cranial vessels of migraine without aura patients assessed ictally, concerns release of CGRP from extracranial structures (measured in the external jugular vein blood), whereas it was not possible to measure increased release of any vasoactive peptides from the brain itself (as measured in the internal jugular blood) (28). The findings of the present study of increased levels of CGRP and NKA in internal jugular blood counteract the current suggestion that the ictal events of migraine are strictly connected to extracranial structures via trigeminal nerve and that CGRP released to extracranial jugular vein indicates that the actual pain arises extracranially. Changes in vasoactive peptides and NO in intrajugular pain found in our study could reflect intracranial cerebrovascular events occurring during migraine attacks, in particular dilatation of large intracranial arteries which are known to be innervated by nociceptive fibres projecting to the trigeminal ganglion and potentially be activated during a migraine attack (29, 30).
To exclude the possibility that variations in the above neuropeptides, as well as those of the other biochemical variables assessed, were due not to pathogenic events of migraine, but rather to the trauma subsequent to catheter insertion, we measured the same parameters in the blood of patients having elective jugular blood vein catheters placed for management in Intensive Care. We did not found significant variations between their values and those measured in internal jugular blood of patients after attack (therefore when they were completely headache-free).
On the basis of the above findings, we therefore strictly confirmed the release of these two vasoactive peptides from trigeminovascular system also into the internal jugular venous blood in the course of migraine crises, further supporting the activation of the trigeminovascular system during the ictal period. Variations in CGRP and NKA levels in the internal jugular venous blood of migraine without aura patients seem to follow the variations in the nitrites observed.
The mechanisms involved in the production of NO in relation to the release of vasoactive peptides from the trigeminovascular system are still controversial. Some experimental evidence showed that the potent vasodilatatory effect of CGRP is endothelium-independent and does not necessarily involve NO generation (31). Moreover, two additional papers demonstrated that NO responses are independent of CGRP, because they are identical even after blockage of CGRP receptors or after depletion of CGRP by capsaicin (32, 33). The effect of CGRP seems to be prevailingly mediated by stimulation of cAMP, which induces the hyperpolarization of endothelial cells followed by the activation of calcium-activated potassium channels (BKCa) and ATP-sensitive potassium channels (KATP) (34, 35). In our study, however, the increase in cAMP levels seemed to follow that of prostanoids rather than that of CGRP, making the occurrence of a CGRP-mediated activation of endothelial adenylate cyclase less probable. On the other hand, it should be pointed out that BKCa channel activation is responsible for NO generation, and this potential mechanism could explain our observation of the parallel changes in nitrite and CGRP levels in the internal jugular venous blood of migraine patients during the crises (36). There are, however, further experimental data suggesting that the vasodilation induced by CGRP and substance P, at least in the pial arteries of rats, is abrogated by endothelial injury, therefore supporting the role of NO as a final mediator of vasodilatatory effects of these vasoactive peptides from the trigeminal system (37). No data are available in this regard for NKA, which seems to interact directly with its own receptors, NK2 (38).
Conversely, there is also evidence demonstrating that NO stimulates CGRP release from trigeminal endings. Nitroglycerin trinitrate applied locally to the pial surface induced release of this vasoactive peptide from perivascular nerve fibres, and hence the observed increase in concentration of CGRP during migraine attacks may be secondary to NO formation (14).
Although an increase in stable metabolites in the internal jugular venous blood during migraine attacks has been demonstrated, it is unknown at the moment if, and how, NO induces head pain during migraine ictal periods, independently of its putative relation with the release of vasoactive peptides from trigeminal endings. Additional mechanisms can be advocated to explain its increase in the internal jugular venous blood of migraine patients.
Neurotransmitter and biochemical fluctuations in the perivascular milieu, both concerning sensitive endings and blood stream, could stimulate the formation of nitric oxide by cerebral endothelium. Increased flow velocity and the subsequent increase in the shear stress in endothelial cells may also stimulate eNOS (39). It cannot be excluded that NO can be released from NOS immunoreactive neurones from non-adrenergic, non-cholinergic (NANC) fibres supplying cerebral vessels. The contribution of parasympathetic fibre activation to the increase in NO, as suggested by much experimental evidence (40–42), cannot be excluded.
NO release in migraineurs could be responsible for cerebrovascular changes and also headache, on the grounds of a hypersensitivity. It is impossible to speculate at the moment on the contribution of neural NOS to the modification in the levels of nitrites found by our group in the internal jugular blood of migraine without aura patients examined during attacks. As mentioned above, the NO radical itself could be responsible for the activation of the trigeminovascular system. In addition, its formation (together with that of CGRP) could account for the increase in cerebral blood flow accompanying spreading depression, which is considered the pathophysiological substrate of migraine aura (43). This aspect was not investigated in our study, in which we could assess only migraine patients without aura, but it is the first time that attacks have been investigated in progress.
The present study also demonstrates that PGE2 levels, and to a lesser extent 6-keto PGF1α levels, tended to increase in the internal jugular venous blood of migraine patients examined during attacks, but reached a peak later than those of nitrites, exactly at the 2nd hour, and maintained increased values during the migraine attack.
Although it is impossible to identify, on the basis of the results obtained, the causative relationship between increase in NO and that of the vasoactive and algogenic prostanoids observed in internal jugular venous blood in the course of a spontaneous migraine attack, it can be hypothesized that early activation of NOS pathways and a late increase in prostanoid synthesis (peraphs from endothelium) could account for maintenance of head pain and cerebrovascular changes.
This increase could be permitted by cross-talking between the NOS and COX systems (44). This relationship is well known in the cells of the immune system due to proinflammatory cytokines and growth factors, but has been less investigated in cerebral vessels.
Some in vitro data suggest a strong relationship between NOS1 and COX in endothelial cells. NO production could contribute to activate endothelial PG synthase which is followed by an increased production of PGE2 and PGI2. This could explain our findings in internal jugular venous blood in migraine patients examined ictally.
We cannot speculate at the moment which type of COX is preferentially activated in the endothelium of the cerebral vessels as a consequence of increased NO synthesis. COX-2 activity is induced in endothelial cells, condrocytes, fibroblasts and also cells of the immune system, by proinflammatory cytokines and growth factors. Its intervention in migraine seems less probable. To clarify its real involvement in migraine crises, selective inhibitors of COX, particularly those of COX-2, could be helpful (44).
The putative intervention of PG synthase activation is supported by the finding of a later rise in the levels of cAMP which paralleled the increased production of PGE2 and PGI2. Through the activation of the pathways involved in the synthesis of intracellular messengers, cGMP and cAMP (the former early, the latter late), NO and PGE2 and PGI2, respectively, could contribute to decrease in cerebrovascular tone in the large cerebral arteries, induction of modifications in regional cerebral blood flow and mediation of head pain during migraine attacks.
At present, the literature is divided with respect to whether NO activates or inhibits PG production. A recent study was sought to determine whether conflicting observations could be accounted for by divergent effects of NO on the cyclooxygenase isoforms. In this study NO seems to strongly stimulate PGE2 release in COX-2-deficient cells, by activating COX-2 isoenzyme. In contrast, NO appeared to inhibit COX-2-derived PGE2 production both in lipopolysaccharide (LPS)-stimulated macrophages and COX-1 knock-out cells. This inhibition was associated with both decreased expression and nitration of COX-2 (45). These findings, confirming divergent effects of NO on PG production, further support the hypothesis of a complex NOS/COX cross-talk depending on the local environment in which their pleiotropic mediators are produced.
The complex relationship between NOS and COX systems emerged also in migraine from a previous study from Stirparo et al. (46). This study in fact demonstrated that NO production seems to inhibit and not stimulate PGE2 release from mononuclear cells of patients affected by migraine. In this regard, it remains to be clarified if immune system cells represent a reliable and valid peripheral model for studying the relationship between NOS and COX systems in migraine.