
Abstract
In this double-blind study, the efficacy and tolerability of a single dose of almotriptan (6.25 or 12.5 mg) was compared with placebo in the treatment of three consecutive migraine attacks of moderate or severe intensity. Of 1013 randomized patients, 722 evaluable patients completed the study. The total number of attacks relieved (severe or moderate pain reduced to mild or no pain) at 2 h post-dose was significantly higher (P < 0.001) after treatment with almotriptan 6.25 or 12.5 mg compared with placebo (60% and 70% vs. 38%, respectively). Moreover, a consistent response was achieved across and within patients for almotriptan 6.25 or 12.5 mg compared with placebo (pain relief in at least two out of three attacks within 2 h for 64% and 75% vs. 36%, respectively) and less than one-third of the patients relapsed within 24 h. Almotriptan was well tolerated with no significant differences between the almotriptan and placebo treatment groups in the percentage of patients reporting adverse events. Overall, the 12.5-mg dose was associated with the most favourable efficacy/tolerability ratio and is, therefore, the recommended dose.
Keywords
Introduction
Migraine is a common condition, with a prevalence of approximately 15–20% in women and 6% in men (1–4). The prevalence is highest in 35–45-year-olds and is often associated with moderate to severe disability, accounting for its high impact in the workplace (5).
Although the mechanisms involved in the pathogenesis of migraine remain unclear, pre- and post-synaptic 5-hydroxytryptamine (5-HT) receptors are thought to play a role in the pathophysiology of migraine pain. The potential of 5-HT agonists as migraine treatment was based on the initial observation that intravenous tryptophan significantly improved the clinical course of migraine compared with patients who received placebo treatment (6). Of the seven classes of 5-HT receptor identified to date, including five subtypes of5-HT1 (7), 5-HT1B/1D receptors have been implicated in the pathophysiology of migraine (8, 9).
Sumatriptan was the first 5-HT1B/1D receptor agonist to be developed for the treatment of migraine (10). However, it is associated with a number of disadvantages, including headache recurrence, poor oral bioavailability and, rarely, cardiovascular complications (11).
Almotriptan is a 5-HT1B/1D receptor agonist developed by Almirall Prodesfarma for the acute treatment of migraine with or without aura (Fig. 1). Almotriptan has a well-defined pharmacological profile with high and specific affinity for 5-HT1B/1D receptors. Moreover, almotriptan acts very selectively at human cranial vessels, having little activity at peripheral human arteries (12). Almotriptan also has a markedly higher oral bioavailability than sumatriptan, 70% compared with only 14% ((11), Almirall Prodesfarma, data on file). These favourable pharmacological and pharmacokinetic properties of almotriptan could theoretically result in therapeutic benefits compared with sumatriptan treatment.

Chemical structure of almotriptan.
A phase II dose-finding study indicated that a single oral dose of 12.5 mg almotriptan would provide the optimum efficacy/safety ratio (13). Furthermore, a double-blind, placebo-controlled phase III trial has demonstrated that almotriptan 12.5 mg and 25 mg have equivalent efficacy to sumatriptan 100 mg in terms of amelioration of pain 2 h after administration, but the 12.5-mg dose in particular had a significantly improved tolerability profile, comparable to that of placebo treatment (14).
The aims of this study were to investigate further the efficacy and tolerability of 6.25- and 12.5-mg doses of almotriptan compared with placebo in patients with migraine during three consecutive migraine attacks and to analyse the consistency of the therapeutic effect.
Patients and methods
This randomized, double-blind, parallel-group, placebo-controlled trial was conducted in 133 centres in nine European countries: Belgium, Czech Republic, Estonia, Germany, Hungary, Poland, Spain, The Netherlands, and the UK. The protocol was approved by the appropriate local ethics committees for each of the study centres and the study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All patients provided written informed consent before entering the study.
Patient population
Patients between 18 and 65 years of age suffering from migraine with or without aura, according to the criteria established by the International Headache Society (15), were recruited. In addition, patients had suffered from migraine attacks for at least 1 year and had one to six attacks per month. Exclusion criteria included: migraine with prolonged aura, familial hemiplegic migraine, migrainous infarction or vertebro basilar migraine; clinically relevant abnormal screening results; breast-feeding or pregnant women and women of child-bearing potential without adequate contraceptive protection; any chronic physiological or neuropsychiatric illness; anti-psychotic or antidepressant medication taken during the 3 months period prior to the study; prophylactic drugs for migraine taken during the 2 weeks prior to the study; and intolerance or known hypersensitivity to sumatriptan or other related compounds.
Study design
Eligible patients were randomized 1:2:2 to receive a single dose of placebo, almotriptan 6.25 mg or almotriptan 12.5 mg during three consecutive migraine attacks of moderate or severe intensity. One group of 180 patients (placebo) and two parallel groups of 360 patients each (6.25 and 12.5 mg almotriptan) were to be included in the study in order to obtain 700 evaluable patients.
The allocation of the patients to the treatment groups was carried out electronically in blocks of five patients. The investigators received complete block(s) of patients but were not informed of the block size. The almotriptan and placebo tablets were identical in taste and appearance and packaged in boxes with identical labelling. The investigators were provided with the individual randomization codes in sealed envelopes, but the code was only to be broken if the patient had a serious adverse event requiring knowledge of treatment assignment: this occurred in only one patient. The code envelopes were to be returned unopened after the completion of the study.
The study consisted of four visits, the first screening visit to determine eligibility. At the second visit, the patient was allocated treatment and provided with the first test medication (test and relapse medication), instructions for treatment administration (to be taken at the next migraine attack), and a self-assessment booklet with instructions for its fulfilment. The patient was requested to return for the third visit within 2–6 days after the first attack. During this visit, the next two treatments and self-assessment booklets were provided and safety was assessed. Patients was withdrawn from the study if they did not suffer a migraine attack within 8 weeks of randomization. At the final visit, 2–6 days after the third treated attack, the remaining self-assessment booklets were returned and safety assessed.
If the migraine attack reappeared within 24 h of taking the test medication, an extra dose of identical relapse medication could be taken, provided the patient had not taken escape medication for the first attack. Escape medication, chosen by the investigator according to the patient's characteristics and preferences, was allowed if the migraine pain was still moderate or severe 2 h after intake of the test medication. Ergot derivatives and 5-HT1B/1D receptor agonists were not permitted as escape medication.
Efficacy and safety measurements
The primary efficacy variable was defined as the number of migraine attacks out of three that were relieved (pain reduced from moderate or severe to mild or no pain) at 2 h after intake of the test medication. Severe headache was defined as that prohibiting normal activity for which bed rest may be necessary; moderate headache was that disturbing but not prohibiting normal activity for which bed rest was unnecessary; and mild headache was that allowing normal activity. Migraine pain was assessed just before taking the study drug, and 30, 60, 90, 120 min and 24 h later using the self-assessment booklets. Other efficacy parameters included: the number of patients in which the pain disappeared within 30 min, 1 h, 90 min and 2 h of administration; time between test drug intake and the pain relief; the relief of migraine-related symptoms such as nausea, vomiting, photophobia, and phonophobia; and the number of patients who relapsed within 24 h of taking the test medication.
Safety was assessed by the occurrence of adverse events, clinical laboratory tests (haematology, biochemistry and urinalysis), vital sign measurements, ECG recordings and physical examinations.
Patients could be withdrawn from the trial due to adverse events, lack of efficacy, protocol violations, and if the patient required medications which could interfere with the test medication. All patients who received at least one dose of the study medication were included in the safety analysis. Adverse events were coded according to the WHO adverse reaction terminology dictionary. Treatment-emergent adverse events were those which were not observed before the first intake of the drug or worsened after the first intake.
Statistical analysis
Based on previous efficacy data, a sample size of 280 patients per active dose of almotriptan and 140 patients in the placebo group was calculated to give a power (α=0.05, two-sided) > 99% for the Cochran–Mantel–Haenszel (CMH) test. In addition, 600 patients treated with almotriptan were estimated to be required to demonstrate an adverse event with a probability of occurrence of at least 2% with a power of 80%. As, according to previous clinical trials, up to 20% of post-randomization withdrawals could be expected, 900 patients were to be recruited and randomized in order to obtain a total of approximately 700 evaluable patients.
The efficacy analysis was performed on an intent-to-treat (ITT) basis. A migraine attack was included in the ITT analysis if the patient recorded moderate or severe pain intensity at baseline, took the study medication according to protocol, and recorded at least one measure of efficacy. Each patient who was randomized and had at least one ITT migraine attack was included in the ITT analysis. In case of missing observations, escape or relapse medication, last observation carried forward (LOCF) data were used. If a patient had fewer than three attacks before the end of the trial the last attack was carried forward (LACF) for analysis of the number of attacks.
A per-protocol (PP) analysis was also performed for the number of migraine attacks with pain relief after 2 h. The PP analysis of the primary efficacy variable (the number of attacks out of three relieved within 2 h) included only patients who experienced three PP attacks. All PP attacks were included for the additional analysis of the separate attacks.
All tabulations and statistical evaluations were performed with the SAS system version 6.12 for Windows NT. All tests were done at a two-sided level of significance, α=0.05. Differences in the response rates between the treatment groups were tested with the CMH test for non-zero correlation by means of pair-wise comparisons.
Results
Of 1013 randomized patients, 722 completed the study: 131 (65%) in the placebo group, 287 (71%) in the almotriptan 6.25 mg group and 304 (75%) in the almotriptan 12.5 mg group (Table 1).
Trial profile

According to the final evaluation form. Four of these randomized patients did not receive study medication and so were not included in the safety sample. Although for one patient in the placebo group, lack of efficacy was given as the major reason of withdrawal on the final evaluation form, the adverse event form stated that the ‘study medication stopped’ due to the adverse events.
The most common reasons for discontinuation were ‘protocol violations’ and ‘others’, usually specified as more than 8 weeks before randomization and first attack or fewer than three attacks before the end of the study. Only eight patients withdrew due to adverse events, 5/404 (1.25%) in the almotriptan 6.25-mg group and 3/408 (0.7%) in the almotriptan 12.5-mg group.
The PP sample of patients who had three PP attacks consisted of 605 patients; 112, 242 and 251 patients in the placebo, 6.25-mg and 12.5-mg groups, respectively.
Efficacy
The ITT sample consisted of 909 patients (Table 2). There were no significant differences between treatment groups in relation to race (99.4–99.7% Caucasian), age, height, weight or BMI. However, the female/male ratio was slightly unbalanced (P = 0.049 overall), with a higher percentage of males in the placebo group compared with the almotriptan 6.25-mg group (P = 0.014).
Demographic data for intent-to-treat sample

Baseline characteristics probably prognostic for the efficacy of treatment, such as the percentage of patients taking concomitant medication, did not differ between the treatment groups, with the exception of the baseline pain intensity which tended to be more severe in the almotriptan treatment group, particularly in the 12.5-mg group, compared with the placebo group, with a statistically significant difference for the second attack (P = 0.015) (data not shown).
Consistency of response
A consistent response was achieved, with similar percentages of first and third attacks relieved at 2 h (Fig. 2). Of those patients who had three PP attacks, 35.7%, 63.8% and 74.5% in the placebo, 6.25-mg and 12.5-mg groups had pain relief of at least two out of three of the attacks within 2 h (Fig. 3). While almost 50% of patients in the 12.5-mg group had three out of three attacks relieved within 2 h, only 16% of the placebo group achieved this.

The percentage of attacks relieved at 2 h for each separate attack (per-protocol (PP) sample). ∗P < 0.001 vs. placebo; ∗∗P = 0.001 vs. almotriptan 6.25 mg.

The percentage of patients with three of three or at least two of three migraine attacks relieved at 2 h (per-protocol (PP) sample)
Pain relief
The total number of attacks with pain relief at 2 h for patients having three attacks were 129/336 (38.4%), 435/726 (59.9%) and 529/753 (70.3%) in the placebo, 6.25-mg and 12.5-mg groups, respectively (Fig. 2). This dose-dependent percentage increase was statistically significant (P < 0.001). Statistically significantly more attacks were relieved after treatment with 6.25 mg and 12.5 mg almotriptan compared with placebo (P < 0.001) with a statistically significant difference between the 6.25-mg and 12.5-mg doses (P = 0.002). The number and percentage of migraine attacks with pain relief at 2 h after intake of the test drug, per separate attack, also increased in a dose-dependent manner (P < 0.001 overall and for almotriptan 6.25 or 12.5 mg groups vs. placebo) (Fig. 2). The difference between the almotriptan 6.25- and 12.5-mg groups was approaching statistical significance for the third attack (P = 0.011, 0.169 and 0.052 for the first, second and third attacks, respectively).
Pain free
The number and percentage of attacks out of three that were pain-free at 2 h, without the use of escape medication, increased in a dose-dependent manner (P < 0.001 overall), and the pair-wise comparisons between treatment groups were statistically significant (all P < 0.001) (Table 3). Overall statistics for the first, second and third attack separately were also statistically significant (all P ≤ 0.001). Pair-wise comparisons between almotriptan 6.25 mg and placebo, and between almotriptan 12.5 mg and placebo were statistically significant for all three attacks considered separately (all P < 0.05). There was a statistically significant difference between almotriptan 6.25 mg and 12.5 mg for the first (P = 0.005) and second attack (P = 0.007) (P = 0.052 for the third attack).
Migraine attacks pain-free at 2 h (intent-to-treat (ITT) sample)∗

Including last attack was carried forward (LACF) data.
Onset of action
The drug efficacy was dose-dependent and increased with time (Fig. 4). Only 1 h after treatment, 21.6%, 30.1% and 34.5% of attacks were relieved in the placebo, almotriptan 6.25- and 12.5-mg groups, respectively, and 5.7%, 7.3% and 12.5% of attacks had no pain (Table 4). The number of attacks (out of three) that were relieved or pain-free at 1 h showed a statistically significant dose-dependent increase (both P < 0.001 overall). Statistically significant differences were found between almotriptan 6.25 mg and placebo, and between almotriptan 12.5 mg and placebo (both P < 0.01), but not between the almotriptan groups for the number of attacks (out of three) that were relieved at 1 h. Pair-wise comparisons of the number of attacks (out of three) that were pain-free at 1 h after treatment showed statistically significant differences between almotriptan 12.5 mg and placebo and between almotriptan 6.25 mg and 12.5 mg (both P < 0.01).
Migraine attacks relieved or pain-free at 1 h (intent-to-treat (ITT) sample)∗
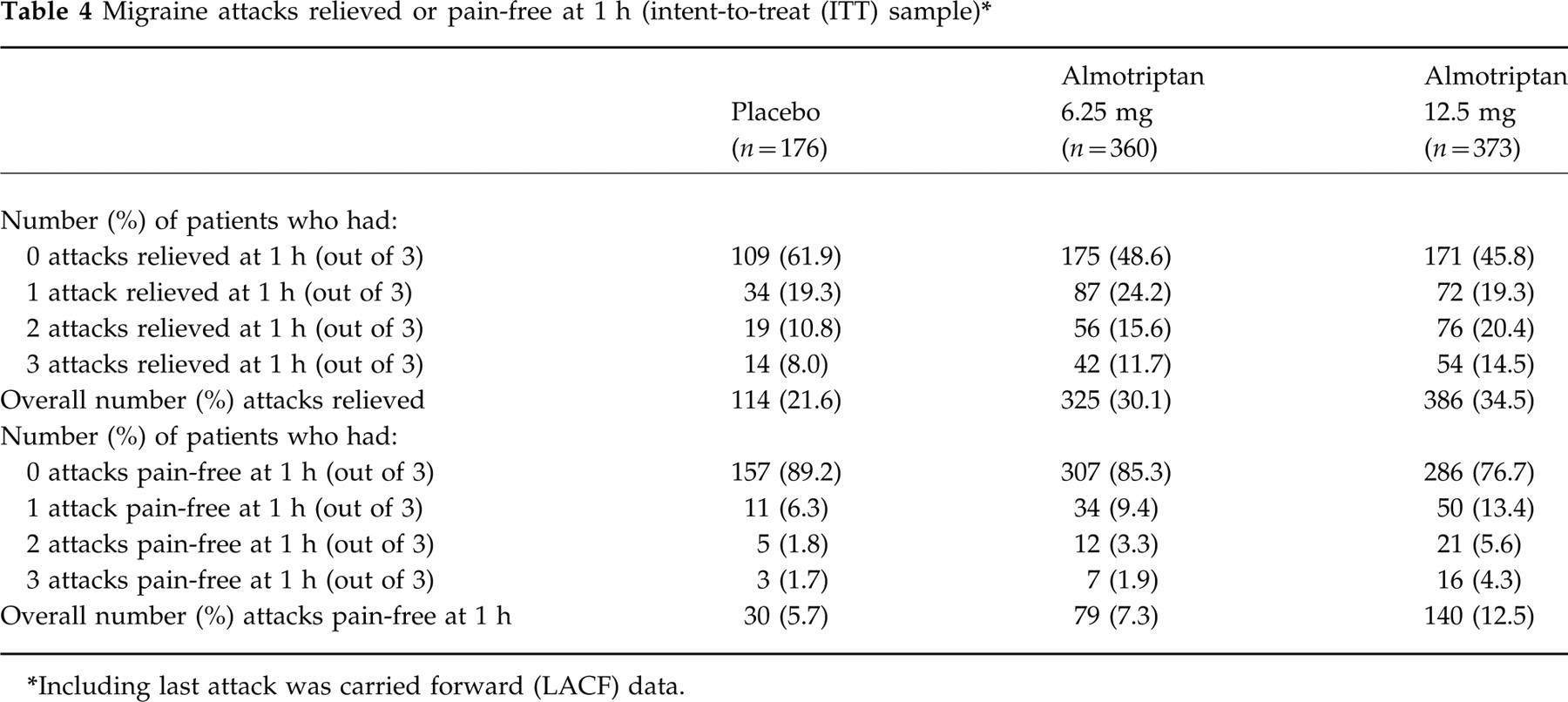
Including last attack was carried forward (LACF) data.

Time between intake of test drug and relief of the migraine attack. P values shown vs. placebo. ∗P = 0.001 vs. almotriptan 6.25 mg.
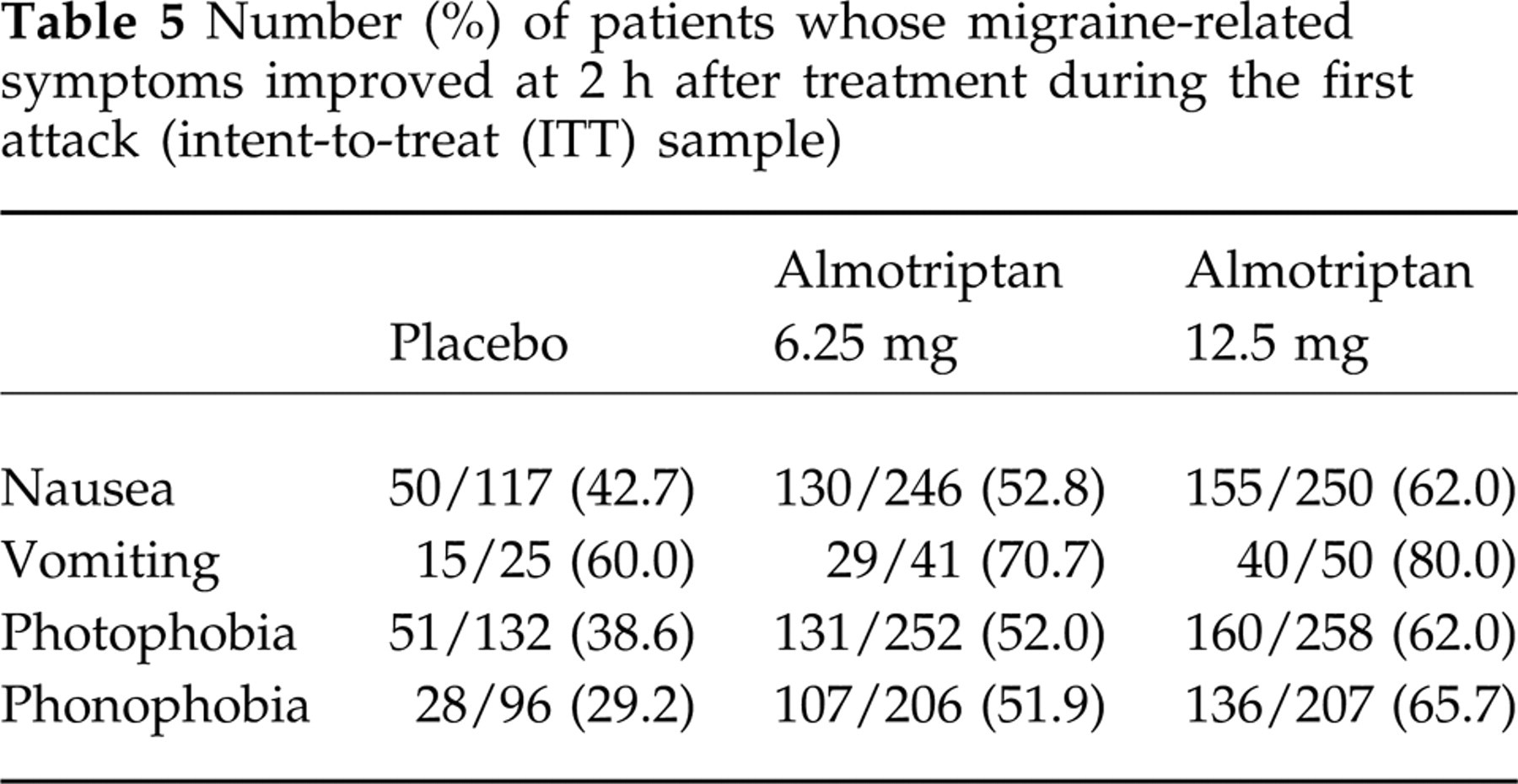
Migraine-related symptoms
Migraine-related symptoms including nausea, vomiting, photophobia and phonophobia were ameliorated in a dose-dependent manner by treatment with almotriptan compared with placebo (Table 5). For nausea, photophobia and phonophobia, there were statistically significant overall differences (all P ≤ 0.001) and pair-wise comparisons were significantly different between almotriptan 12.5 mg and placebo at 2 h for all three attacks (all P < 0.01). For the first attack, the differences between almotriptan 12.5 mg and placebo were statistically significant for all four of the assessed migraine-related symptoms.
Number (%) of patients whose migraine-related symptoms improved at 2 h after treatment during the first attack (intent-to-treat (ITT) sample)
Relapses
Of those patients who had pain relief for at least one attack, the proportion who relapsed (for all three attacks combined) was 37/159 (23.3%) in the placebo group, 160/557 (28.7%) in the almotriptan 6.25-mg group and 200/664 (30.1%) in the almotriptan 12.5-mg group, with no significant differences between the groups. However, a high proportion of these patients responded to the relapse medication. For example, 38/50 (76.0%) and 57/70 (81.4%) of recurrent first attacks were relieved 2 h after the intake of relapse medication in the almotriptan 6.25- and 12.5-mg groups, respectively. The number and percentage of migraine attacks which showed sustained relief (i.e. pain relief at 2 h without subsequent relapse) is shown in Table 6.
Number (%) of migraine attacks with sustained relief∗

Relieved at 2 h without subsequent relapse.
Safety
In total, 210/910 (23.1%) of patients experienced 403 adverse events (Table 7). A higher proportion of patients reported adverse events following the first attack than after the second attack, and even fewer reported adverse events following the third attack. There were no significant differences between treatment groups in the percentages of patients reporting adverse events. The most frequently observed treatment-emergent adverse events were related to the central and peripheral nervous system, the gastrointestinal system and the body as a whole (Table 8). The majority of all adverse events were mild: 37.1% in the placebo group, 50.0% in the almotriptan 6.25-mg group and 60.1% in the almotriptan 12.5-mg group. Severe adverse events were reported by only 3.4%, 3.3% and 2.1% of patients in the placebo, almotriptan 6.25-mg and almotriptan 12.5-mg groups, respectively.
Number (%) of patients with treatment-emergent adverse events∗

According to ‘action taken’ on the adverse event form.
Treatment-emergent adverse events occurring with an incidence of at least 1% of all patients

Adverse events in the almotriptan groups were more often considered to be related to the study medication than those in the placebo group: 10.0%, 16.7% and 23.5% of adverse events in the placebo, almotriptan 6.25- and 12.5-mg groups, respectively, were considered to be probably related to the study medication, whereas 35.7%, 37.7% and 34.4%, respectively, were considered to be possibly related to the treatment. The most frequently reported unexpected treatment-emergent adverse events were allergic reaction (one patient in the 6.25-mg group and two patients in the 12.5-mg group), hyperglycaemia (three patients in the 12.5-mg group) and syncope (one patient in each treatment group). There were no deaths, and three patients experienced serious treatment-emergent adverse events: one patient in the placebo group experienced increased sweating, tremor and vomiting considered to be probably related to treatment; one patient in the 12.5-mg group underwent surgical intervention for gall stones, considered to be improbably related to treatment; and one patient in the 6.25-mg group experienced an episodic coronary artery disorder after the third dose of almotriptan. The latter patient, a 47-year-old woman taking oral contraceptives (ethinylestradiol 30 μg and levonorgestrel 50 μg) for the previous 3 years without a doctor's supervision, experienced nausea, vomiting and discomfort of the epigastrium 27 h after taking 6.25 mg almotriptan. Nevertheless, she slept well and related this discomfort to an alimentary intolerance. An ECG on the following day, performed as part of her routine final treatment visit, was abnormal for repolarization changes posterio-laterally (negative t waves) and the patient was hospitalized for the following 10 days. However, she remained asymptomatic, no abnormalities of cardiospecific enzymes or blood pressure were detected and a control ECG, performed 2 days after her admission, was found to be normal. Both echocardiogram and coronariography were unremarkable. The patient was diagnosed by the investigator with transient posterio-lateral coronary ischaemia, considered possibly related to treatment. Her father had died due to repeated myocardial infarction and her mother suffers from ischaemic heart disease.
Only five patients in the safety sample withdrew due to adverse events: one in the placebo group (due to a single episode of moderate diarrhoea and severe nausea, vomiting and headache); three in the almotriptan 6.25-mg group (one due to nystagmus and abnormal reflexes, one due to a single episode of severe syncope, and one due to a single episode of moderate dizziness and somnolence); and one in the almotriptan 12.5-mg group (due to intermittent moderate hypoaesthesia and leg pain, mild nausea and a single episode of mild headache). These events were all considered possibly related to treatment, with the exception of the dizziness and somnolence which were considered probably treatment-related.
Clinically abnormal laboratory values were found in only nine patients, six of whom had pain relief at follow up; two were considered by the investigator to be possibly related to the study medication and seven improbably related. Of those possibly related to treatment, one patient who received almotriptan 12.5 mg had myalgia and a slight increase in creatine kinase levels during the third visit, 4 days after taking the first dose of study medication. Her ECG remained normal. The patient continued in the study and took the two subsequent almotriptan doses with no clinical or laboratory adverse events. The other patient received almotriptan 12.5 mg and had increased levels of cholesterol during the third and fourth visits. There were no differences between the treatment groups in vital signs, physical findings or other observations related to safety.
Discussion
The results of this large, double-blind, placebo-controlled study demonstrate that almotriptan provides consistently effective relief of acute migraine attacks and is well tolerated. Treatment with a single dose of almotriptan 6.25 or 12.5 mg resulted in statistically significant dose-dependent relief of migraine attacks 2 h after treatment compared with placebo. In addition, almotriptan was significantly more effective thanplacebo in achieving complete freedom from pain at 2 h after administration, with statistically significant improvements for the 12.5-mg dose compared with the 6.25-mg dose. The efficacy of almotriptan was confirmed by the amelioration of the associated symptoms of migraine, with statistically significant improvements in the almotriptan 12.5-mg group compared with the placebo group.
Treatment with almotriptan was also consistently effective across and within patients. Of those patients who had three attacks, at least two of the attacks were relieved in 75% of patients treated with almotriptan 12.5 mg. Furthermore, almotriptan provided good 24-h protection from migraine symptoms. Of those patients who had pain relief within 2 h for at least one attack, 70% had a ‘complete response’, i.e. they did not experience another attack of moderate or severe intensity within 24 h.
The onset of action of almotriptan was rapid, with a significantly higher proportion of attacks relieved or with no pain after only 1 h in patients treated with almotriptan compared with those treated with placebo. Significantly more attacks (out of three) were pain-free at 1 h after almotriptan 12.5 mg compared with almotriptan 6.25 mg.
The 2-h response rates reported here for almotriptan (60% and 70% of attacks relieved for almotriptan 6.25 and 12.5 mg, respectively) are similar to those reported for other members of the triptan drug class. However, the comparative efficacy of the members of the triptan drug class can only really be demonstrated in controlled trials including direct comparisons of the drugs. One such recent study found no significant differences in efficacy between almotriptan 12.5 or 25 mg and sumatriptan 100 mg in 669 patients (Almirall Prodesfarma, data on file).
Almotriptan was well tolerated with no significant differences in the incidence of adverse events between the almotriptan- and placebo-treated patients. The incidence of adverse events after each attack decreased after the first attack and the majority of all adverse events were mild with no serious drug-related adverse events associated with the use of almotriptan. The most common adverse events were dizziness, fatigue and paraesthesia, as reported for other members of the triptan drug class. The recent placebo-controlled comparison of almotriptan and sumatriptan treatment also found that almotriptan 12.5 mg had a tolerability profile similar to that of placebo; however, sumatriptan 100 mg was associated with significantly more adverse events than placebo (14). In the current study, one patient was diagnosed with transient posterio-lateral coronary ischaemia, after receiving a third dose of almotriptan 6.25 mg. However, this patient had a strong family history of ischaemic cardiomyopathy and was also at personal cardiovascular risk due to her use of oral contraceptives without a doctor's supervision. It is also important to note that, in the current study, the global incidence of chest sensations in all patients treated with almotriptan was extremely low (0.4%). Sumatriptan is also rarely associated with serious cardiovascular effects; nevertheless, the majority are observed in patients with some history of cardiovascular disease or with cardiovascular risk factors (11). Chest symptoms however, rarely correlate to cardiac ischaemia, so low incidence does not necessarily suggests a safety feature. These results suggest that, although well tolerated in patients with no cardiovascular risk factors, all anti-migraine 5-HT1B/1D agonists should be avoided in patients with known cardiovascular risk factors.
Overall, the 12.5-mg dose of almotriptan was associated with the most favourable efficacy/tolerability ratio, and is therefore the recommended therapeutic dose.
Conclusion
These results indicate that almotriptan is consistently effective and has a favourable tolerability profile. Therefore, it may provide an alternative to sumatriptan, the current treatment of choice for moderate or severe migraine attacks.