
Abstract
Keywords
Schizophrenia is a disorder characterized by disturbances in perception, thought, volition, socialization, psychomotor behaviour and the sense of self. Worldwide prevalence is estimated at between 0.2% and 2% of the general population [1]. The illness is a lifelong debilitating condition for about 40% of patients and is associated with enormous social and economic costs. For these reasons it is critical to understand the aetiologies and pathophysiological mechanisms involved in schizophrenia and to develop effective intervention strategies to prevent the illness or ameliorate its effects.
One of the main difficulties in understanding the neuropathology of this disease is the heterogeneity of the disorder. Despite broad agreement that schizophrenia is associated with a subtle disorder of brain structure [2], the nature, timing and course of the associated neurobiological changes have proven difficult to elucidate [3]. While Kraepelin's notion of dementia praecox suggested a premature and progressive degeneration of the brain [4], the prevailing current view is that schizophrenia is a neurodevelopmental disorder in which structural brain changes, caused by an early prenatal or perinatal insult, confer a predisposition to the development of schizophrenia, but are not progressive beyond the onset of symptoms [5–8].
In this paper we will review the timing of structural changes in schizophrenia and argue that schizophrenia is a neurodevelopmental disorder with limited progressive brain changes occurring during the evolution and early phase of psychosis. Further, we will argue that the functional consequences of structural brain changes in schizophrenia are developmentally moderated such that the later in life a functional component comes ‘online’, the more likely it is to be defective. The implications for early intervention are also briefly considered.
The neurodevelopmental model of schizophrenia: emphasis on early developmental processes
Originally, Weinberger [5] and, separately, Murray and Lewis [9] proposed that schizophrenia was related to a defect in brain development that predisposed patients to a characteristic pattern of brain malfunction in early adult life. In order to explain the delay between an early insult and the onset of symptoms in adolescence, it was hypothesized that the behavioural abnormalities appear later in life, at a time when the maturing brain circuits are placed under functional demand [5]. The motor, cognitive, social and emotional changes that have been described in children who go on to develop schizophrenia may be subtle manifestations of an early neurodevelopmental lesion [10, 11]. Evidence for the neurodevelopmental model of schizophrenia includes findings of tissue loss, reduced cortical folding, and structural changes (e.g. ventricular enlargement) in the absence of any agerelated effects and/or increase in gliosis [3, 12], which is regarded as a necessary neuropathological hallmark of neuronal degeneration [13]. These data have suggested a pathogenesis that is different from other adult-onset and chronic neurodegenerative diseases.
One example of prenatal disturbances to brain development in patients with schizophrenia is provided by our own work examining the surface morphology of the anterior cingulate cortex (ACC). Using magnetic resonance imaging (MRI) we found that, compared with controls, patients with schizophrenia lacked the ‘normal’ leftward ACC sulcal asymmetry, which was explained by reduced folding in the left ACC. These differences were over and above differences in cortical folding across the entire left hemisphere [14]. Given that sulcal/gyral folding is almost complete by the third trimester of gestation [15, 16] and remains relatively stable from soon after birth [17, 18], these anomalies of ACC folding are likely to reflect early (prenatal) neurodevelopmental contributions to the aetiology of schizophrenia. Such findings are in accord with other work that has identified morphological anomalies in schizophrenia [19–21], thereby implicating early neurodevelopmental factors.
Influence of late developmental processes
While such evidence is consistent with an early neurodevelopmental insult, this does not provide a convincing explanation for the long delay between the proposed insult and the onset of symptoms. Despite the fact that proponents of the neurodevelopmental hypothesis have tended to emphasize the influence of pre- or perinatal factors [8, 22], there is likely to be an interaction between such early neurodevelopmental influences and dynamic changes occurring as the brain develops into adolescence [5]. Given that neurodevelopment does not end with birth, a model that incorporates possible anomalies of later developmental processes such as myelination or synaptic pruning is required. Studies that have examined such processes have provided evidence that they are disrupted in schizophrenia [23, 24].
Like Weinberger [5], Benes [25] and McGlashan and Hoffman [24] proposed a model of schizophrenia that involves an early (prenatal) neurodevelopmental insult. However, while Weinberger suggested that behavioural abnormalities were seen when compromised regions were placed under functional demand, the latter authors placed a central role on postnatally developed patterns of altered brain interconnectivity. In McGlashan and Hoffman's [24] model, schizophrenia is a disorder of developmentally reduced synaptic connectivity arising from developmental disturbances of synaptogenesis during the prenatal period and/or synaptic pruning during adolescence. Similarly, Benes [25] proposed that schizophrenia is related to a congenital abnormality involving reduced number and altered interconnectivity of neurones. In particular, Benes proposed that discrete alteration of neurones within, and interconnectivity between, the ACC and prefrontal cortices are at the core of schizophrenia. Benes speculates that such abnormalities may give rise to schizophrenia-like symptoms during late adolescence and early adulthood because this is the period of increased myelination of the perforant pathway [23]. This pathway carries fibres from the entorhinal cortex to the hippocampus and when ‘activated’ may trigger the expression of abnormalities in the cortical regions as they impinge on cortico-limbic circuitry.
To this end, a number of functional imaging studies have demonstrated altered activation patterns in patients with schizophrenia that suggest dysconnectivity between cingulate-hippocampal and prefrontal-medial temporal networks [26, 27]. Our own functional (PET) imaging work has also related ACC dysfunction to morphological differences in schizophrenia [28]. It is possible that these findings reflect anomalous patterns of brain development (i.e. synaptic pruning, myelination) during the postnatal period. Such ‘late neurodevelopmental’ abnormalities may provide some explanation for the onset of schizophrenia in late adolescence/early adulthood and explain the nature and extent of functional impairments observed.
Influence of late neurodevelopmental processes: the transitional period into psychosis and beyond
For a number of years researchers have investigated possible progression of brain abnormalities after the onset of frank psychosis, and more recently work has begun to examine patients during the prodromal phase of the illness and through the transition to first-episode. This work has provided new insights into the timing of structural brain changes in schizophrenia.
1. Structural imaging studies in prodromal/high-risk patients
Prodromal studies in Melbourne and Edinburgh have recently investigated brain structure in large numbers of young people at risk for the development of psychosis using MRI [29–32]. The neurodevelopmental models described above predict brain changes in these subjects that are comparable to those seen in patients with first-episode psychoses. Initial results examining the hippocampi (Melbourne) or the amygdalo-hippocampal complex (Edinburgh) support these predictions, since in both cases the high-risk group had smaller volumes than a comparison population [29, 30, 32]. However, this does not necessarily imply that these abnormalities represent lesions associated with psychosis, as not all of these individuals will develop psychosis. To date, only the Melbourne study has been able to report such data, since they used a ‘close-in’ strategy that maximizes the number of participants who make the transition (around 40% in 12 months [33–35]).
Using this strategy, the Melbourne group have identified that, contrary to the models presented above, the high-risk subjects who did not develop acute levels of psychotic symptoms within 12 months of recruitment had smaller hippocampal volumes at intake while the group developing psychosis did not differ from a comparable normal sample [32]. Similarly, a survival analysis indicated that normal (not smaller) left hippocampal volume was predictive of transition to psychosis in the high-risk group.
It is not immediately obvious why non-reduction of left hippocampal volume should be predictive of the transition to psychosis. One possibility is that onset of acute psychosis is preceded by normal hippocampal size with a subsequent decrease in size as acute illness develops. It may be (a) that the process of transition from an at-risk mental state to acute illness is associated with some loss of hippocampal structure, or (b) as predicted by the neurodevelopmental model, the hippocampi of the ultra-high risk (UHR) sample are small initially (as seen in those UHR subjects who did not develop an acute psychosis) but immediately prior to the onset of psychosis there was a physiological change that was manifested as an increase in hippocampal size to within normal limits, or (c) there are developmental abnormalities in the hippocampi of people who eventually develop acute psychosis which make the hippocampal size larger than might otherwise be expected prior to illness onset. This explanation is also consistent with the neurodevelopmental model as this abnormal structure would have been determined early in life.
The above findings suggest that in order to produce the consistent findings of reduced hippocampal volume in first-episode psychosis there must be a reduction in the hippocampal volume of the UHR patients who develop psychosis, assuming that the populations are similar. In order to investigate this possibility, a prospective, longitudinal study is required. To date, only one such study has reported changes in brain volume over the transition phase to illness [36]. In this study, 21 of the 75 high-risk individuals who had a baseline MRI scan were followed up with a second MRI scan, either immediately postpsychosis (psychotic group) or after 12 months (non-psychotic group). The comparison between baseline and follow-up scans for the two groups indicated that both showed a reduction of grey matter volume in the left cerebellum. However, in the UHR-psychotic group, an additional three regions of the left hemisphere were reduced (a left inferior frontal region, a left medial temporal region that included the hippocampus, surrounding parahippocampal gyrus and the fusiform gyrus, and the cingulate bilaterally). This finding, if confirmed by further investigations, implies that there are active brain changes occurring in patients developing schizophrenia, something that could perhaps be prevented, ameliorated or at least delayed by early intervention during or before the first-episode of psychosis [37]. These initial results are suggestive of progressive brain changes that would be consistent with clinical changes manifest in these patients.
2. Structural imaging studies in first-episode psychosis and established schizophrenia
The most important studies necessary to address the issue of progression following the onset of illness are longitudinal within-subject studies that examine patients early in their course and follow them throughout their illness. These are the most difficult studies to undertake as following up patients with schizophrenia often presents a major challenge. Thus, there are relatively few such studies available.
The earliest longitudinal computed tomography studies generally considered ventricular size to be stable over time but they suffered greatly from methodological problems such as small numbers, no comparison group or variable follow-up interval [38]. In addition, there was a great degree of variability in measurement at the two time points, which could mask real change. Despite improvements in technology, some MRI studies have suffered from similar methodological constraints, and the confusion over the issue of progression remains. One study of a unique population of adolescents with childhood-onset schizophrenia has shown widespread changes in cortical and subcortical regions over a period of 3–5 years [39–42]. Using more sophisticated analysis techniques, this cohort showed an age-accelerated greymatter loss that moved in a dynamic pattern across the brain, from parietal regions anteriorly to temporal and then frontal lobes [43]. This pattern of loss was associated with expected patterns of psychotic symptoms and cognitive deficits. Thus, active brain changes may be occurring throughout the early years of psychosis. Taken together with our findings in adult-onset patients, these data suggest that the nature and location of brain changes may depend on the interaction between the disease process and normal brain development.
Two studies of adult-onset patients are worthy of note. The first was conducted by DeLisi and colleagues [44], in which 50 patients with first-episode schizophrenia/ schizoaffective disorder were followed up annually over 4 years. Reductions in both left and right hemispheric volumes were identified over the follow-up interval, but smaller brain regions such as the amygdalo-hippocampal complex, the caudate nuclei and the temporal lobes showed no change. These findings have recently been replicated by data from our own laboratory [45], in which 12 patients with chronic schizophrenia and 30 with first-episode psychosis were followed over a variable period between 1 and 4 years. Again, whole brain volume was found to decline in both patient groups (at a rate of 1–2% per year), but there was no change in temporal or hippocampal volumes.
It is possible that this failure to identify hippocampal or temporal lobe volume change over the years subsequent to illness onset is because such change does not occur. Alternatively, the time frame for these longitudinal studies may have been too short to detect subtle change, or it may be that the region of change was not easily defined by region of interest tracing techniques. One indirect method of investigating these possibilities is to correlate brain volumes with illness duration in patients with long-standing illness. If there were progressive brain changes throughout the disorder, it would be expected that smaller volumes would be identified in patients with the longest illness duration. DeLisi and colleagues have identified such a correlation in the left temporal lobe of chronically ill patients [46], and a similar correlation has been found in the right hippocampus by Velakoulis and colleagues [47]. Both studies utilized region of interest tracing techniques, but more recently the latter group has used an automated methodology to examine correlations across the whole brain without setting a priori regions of interest. This technique has identified a specific inverse correlation between the volume of right medial temporal lobe regions and illness duration [48].
In summary, these data suggest that brain changes may be occurring actively as psychosis is developing, with further changes occurring after illness onset. Taken together with the findings outlined earlier, the implications of such data are that, while an early neurodevelopmental lesion may be necessary, it may not provide an accurate perspective of the nature and extent of brain structural changes in schizophrenia. Further longitudinal data are necessary to elucidate the exact nature, severity and timing of such changes, and their functional sequelae.
Neurodevelopmental disturbances affecting function in schizophrenia
1. Between infancy and illness onset
In a retrospective study, Walker and colleagues [10] used family home movies to study the neurodevelopment of children who later developed schizophrenia. They were able to identify these children because of subtle abnormalities in motor function, suggestive of developmental disturbances. In contrast, at the time of illness onset, prominent disturbances of behaviour and cognitive function were apparent, while motor disturbances are less prominent. Walker et al. [10] attempted to explain both the childhood motor manifestations of schizophrenia and the postmorbid behavioural dysfunctions. Specifically, these authors proposed that normal maturational processes in the brain moderate the behavioural expression of a congenital neuropathology in schizophrenia that primarily affects limbic structures. This presents as a modular developmental trajectory for schizophrenia, in which neuromotor dysfunction is most pronounced in early childhood and late in life, whereas psychotic symptomatology is most pronounced in late adolescence and early adulthood. This modular developmental trajectory is based on the literature on normal brain development suggesting that different circuits within the brain are differentially activated at various points in the life course. During infant life, the fibre tracts between motor cortex and subcortical structures are highly myelinated and the regions they connect are more metabolically active than either frontal or limbic cortices. In contrast, during late adolescence and early adulthood the motor cortex becomes less metabolically active relative to other cortical regions, particularly in limbic and frontal regions. During this period, the frontal and limbic regions are the most active, and myelination of neural pathways linking limbic regions is completed. It is hypothesized that during this period of late adolescence/ early adulthood the abnormalities of limbic interconnectivity are primarily behaviourally expressed in psychotic symptoms. This would also be consistent with the abnormalities in neuropsychological function, particularly those affecting executive functions.
However, what is also apparent and not fully explained by this model is that the early abnormalities are subtler than the later manifestations and are also less prominent features of the disorder. We propose that this and other current neurodevelopmental models are important but not sufficient in understanding the deficits observed in schizophrenia. It is likely that the interactions between brain plasticity and the normal maturational trajectory of various functions define the nature and extent of their impairment over the course of the disorder. In particular, we propose that functions that normally come online early in life (i.e. during infancy) such as sensory, motor and basic memory functions, when the brain is more adaptable, show fewer deficits at illness onset than functions normally coming on later in life (i.e. during puberty and early adulthood) such as executive and working memory functions [49]. Such a hypothesis would be consistent with the observations of Walker and colleagues [10] and also finds support in the high-risk follow-up study by Marcus et al. [50] and in the birth cohort study by Jones et al. [51] in which motor abnormalities were more marked in infancy rather than later, perhaps indicating that early neurodevelopmental delays were ameliorated.
2. First-episode psychosis and established schizophrenia
Support for this model also comes from our own neuropsychological studies in schizophrenia and normal development, using the Cambridge Neuropsychological Test Automated Battery (CANTAB). This computerized battery includes tests of executive function (such as working memory and set-shifting) and learning and memory. From our work on the normal development of executive function, we have found that while set-shifting ability is fairly well established early in development, working memory skills are not fully developed until at least the age of 20 [52]. Our model would suggest that those cognitive functions that emerge earlier (setshifting in this example) should be less compromised in schizophrenia than those that emerge later (such as working memory). Indeed, this is consistent with the available evidence. Using the set-shifting task from the CANTAB, Hutton et al. [53] found that first-episode patients were relatively unimpaired in set-shifting ability, while Elliott and colleagues [54] found that patients with moderately severe schizophrenia, like frontal lesion patients, showed a specific deficit in set-shifting ability due to a tendency to perseverate. In their study, Pantelis and colleagues [55] directly compared an even more chronic group of patients with schizophrenia with a cohort of patients with frontal lobe lesions, and found more severe deficits in the schizophrenia group; these chronic schizophrenia patients manifested perseverative responding, and also had difficulty in generalizing a rule that had been learned (representing failure at a simpler level of set-shifting ability). We have now examined patients very early in the course of the illness (mean illness duration of 95 days), and despite obvious working memory difficulties, set-shifting ability is less impaired [unpublished data]. Although there are no published longitudinal studies available using this task, these findings suggest that there is minimal impairment of setshifting at first presentation, with slow decline over continuing illness.
Spatial working memory, as assessed by the CANTAB, is clearly impaired in patients with established illness [56], and also at the first presentation of the disorder [53, 57], with no progression over time [57]. In addition, there are deficits in young people at ultra high-risk for psychosis prior to the onset of illness [58], indicating that this ability has never been fully functional. Taken together with the relatively late development of spatial working memory in normal individuals [52], these findings suggest that functions that normally develop during the at-risk period for the development of psychosis manifest the most severe deficits.
Conclusion
In summary, we suggest that the behavioural expression of an early lesion is moderated by the plasticity of the developing brain, accounting for the restoration of motor function during early childhood. However, this early lesion also affects brain regions (i.e. the prefrontal cortex), that do not mature and are not placed under functional demand until adolescence (see Fig. 1). Thus, while patients with schizophrenia tend to perform worse than healthy controls on most cognitive tests, they tend to show a greater magnitude of impairments in certain functions (e.g. working memory, attention and executive functions). The functions that tend to be more impaired are also those functions that tend to come online later in life and require the concerted effort of multiple brain regions (typically associated with the prefrontal cortex). We interpret these findings to support a model of schizophrenia in which these more pronounced functional deficits are a consequence of the individuals' reduced capacity to provide the appropriate central nervous system environment for the maturation of complex and multifaceted brain functions that require multiple circuits. These functional deficits are made worse by the individuals' reduced capacity for plasticity in order to reorganize these functions (termed the Kennard Principle [59]), at which time cognitive impairments (and psychotic symptoms) become apparent. In addition, we also propose that the structural brain changes are not limited to an early insult. Instead, an early neurodevelopmental insult interacts with either normal or abnormal postpubertal brain maturation to produce further brain changes at or around the onset of the disorder (a ‘late neurodevelopmental’ insult; see also [60]). Given the brain's reduced capacity for compensatory response at this stage of neurodevelopment, the functional consequences of these ‘late’ structural brain changes (e.g. in the hippocampus) are also severe, resulting in more pronounced functional deficits (e.g. in areas such as declarative memory) at illness onset.
Illustration of our proposed model. Shaded areas represent periods of structural brain change associated with the disorder, with functions maturing at these times shown below. We suggest that the functional consequences of structural brain changes in schizophrenia are developmentally moderated such that the later in life a functional component comes ‘online’, the more likely it is to be defective.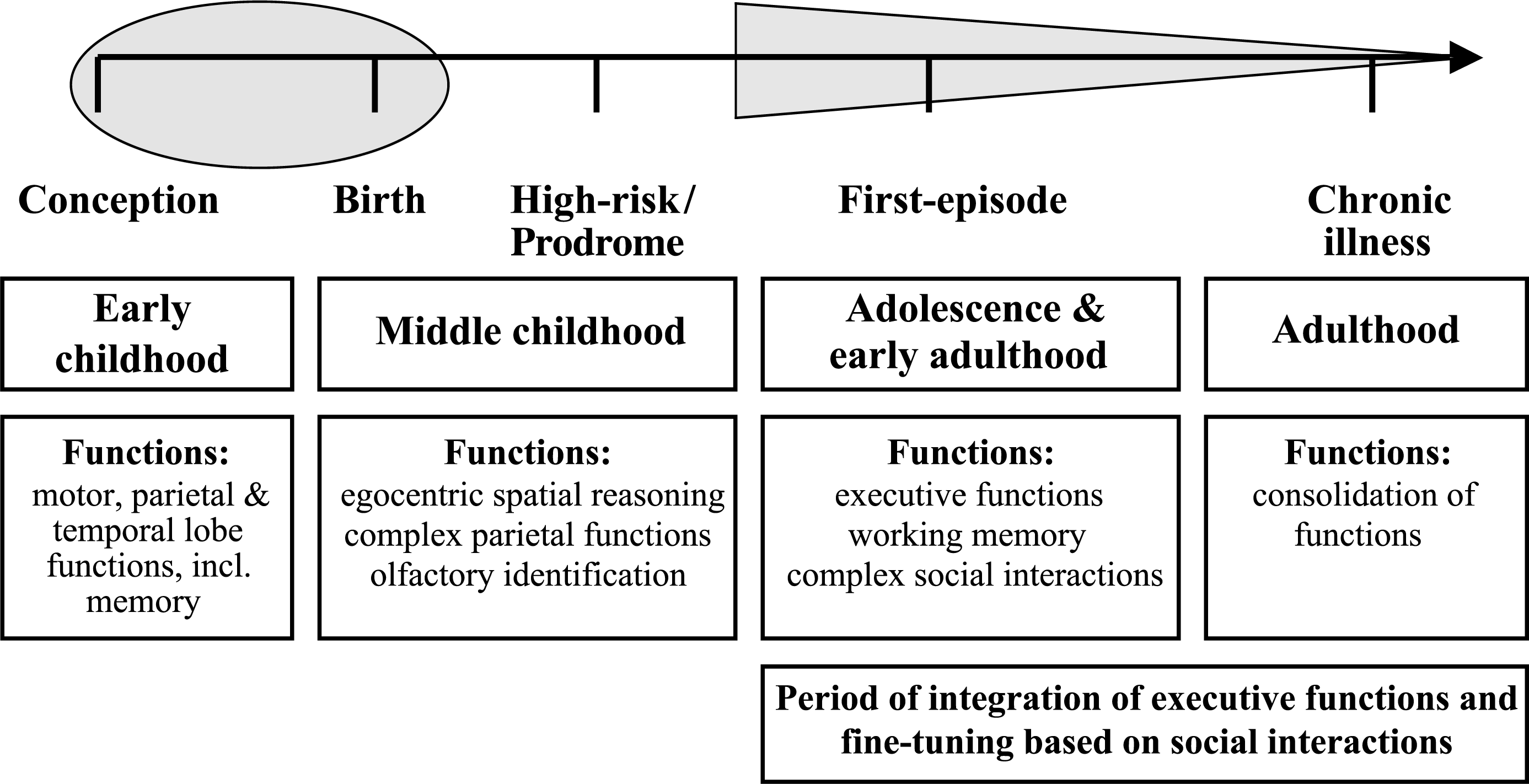
Overall, the model suggests that in order to understand the time course of schizophrenia one must consider relationships between the development of schizophrenia, neurodevelopmental processes and the impact of brain insults occurring in both early and late neurodevelopment. While an early neurodevelopmental lesion prenatally may increase an individual's vulnerability to the development of schizophrenia later in life, the manifestations of the illness may only be understood by considering the interaction of such an early lesion with developmental factors up to and including the period when the illness commences. This may be followed by a further interaction between the neurobiological consequences of the illness itself with the processes relevant to the appropriate neurodevelopmental stage of the individual at illness onset. The heterogeneity of schizophrenia may be a function of this interaction. Finally, the model predicts that the nature and extent of functional deficits observed in schizophrenia depend on the neurodevelopmental stage at which they mature. By further integrating rapidly increasing knowledge from normal human brain development and the developmental course of schizophrenia, we are likely to develop better models for understanding the trajectory of neurological changes in schizophrenia. Such information will help guide early intervention strategies to the most vulnerable brain regions and/or functions, as well as the most vulnerable stages of the illness. Intervention strategies may prevent, ameliorate or delay the structural and functional changes during or before the first-episode of psychosis.
Footnotes
Acknowledgements
This work has been supported by the National Health and Medical Research Council (grant IDs: 145737, 145627, 981112, 970598, 970391, 236175), the Stanley Foundation (USA) and the Ian Potter Foundation. A version of this manuscript was presented as the Novartis Oration awarded to Christos Pantelis at the Australasian Society for Psychiatric Research.