
Abstract
It has long been an assumption that serious and chronic childhood psychiatric disorders reflect, at least in part, relatively subtle abnormalities of brain development. Strong indirect support for this has been provided by the association of childhood psychiatric disorders with numerous neurological and neurodevelopmental disorders [1] and from clinical neuropsychological studies over many decades, indicating abnormal brain function in child psychiatric populations [2]. Because of the limitations of clinical investigation to validate subtle brain abnormalities, child psychiatric research has explored new methods for studying the brain. Brain imaging is an innovative technology that to date has best furthered the goal of understanding normal and psychiatrically abnormal brain structure and function. With the advent of non-invasive brain magnetic resonance imaging (MRI) methodology, imaging data can now be acquired for paediatric populations. Moreover, these data are converging with new information on the organization and function of circuits in the developing brain, and on the molecular mediators of these changes. It is hoped that imaging studies in child psychiatric populations, will not only define the brain systems underlying illness, but also suggest candidate molecules for genetic studies. Based on the nature, location and temporal pattern of these abnormalities, and preclinical findings, we will be able to make more specific and testable hypotheses about the aetiology of these disorders. The quantitative study of brain development during childhood and adolescence with MRI began in the late 1980s (e.g. [3]). Subsequent cross-sectional [4–7] and mixed longitudinal/crosssectional studies (e.g. [8]) have confirmed that although total brain volume changes between ages 5 and 18 are negligible, there are robust and complex changes in white and grey matter. White matter volume increases linearly during this age range, reflecting increasing myelination [4, 9], while grey matter volume increases until early to mid-adolescence before decreasing during late adolescence [8], presumably from synaptic pruning and reduction of neuropil. A special feature of these normative data is that they were acquired in parallel with prospective clinical studies, so that brain development for psychiatrically abnormal populations can be compared. Elegant studies of known chromosomal abnormalities, such as Down syndrome and Rett's syndrome, all testify to abnormal development in these known retardation syndromes [10, 11]. These produce gross disturbances of central nervous system (CNS) development for which the cause is known and, in principle, relatively simple screening could be used to detect and prevent such disorders. More subtle non-dementing disorders, however, have proven more difficult. There remains considerable controversy, for example, about even the validity of the diagnosis of attention deficit hyperactivity disorder (ADHD). Brain morphometric studies may help validate this diagnosis, and are summarized in detail here. Since prospective longitudinal rescan data is now available, we can address not only how the two patient groups, ADHD and childhood onset schizophrenia (COS), differed from controls at their initial evaluation but also examine their differing developmental course.
Anatomic brain magnetic resonance imaging studies of attention deficit hyperactivity disorder and childhood onset schizophrenia
Attention deficit hyperactivity disorder
Table 1 summarizes some representative anatomic brain studies that have been carried out to date in ADHD. As seen in Table 1, several independent studies have found a smaller total brain volume. This represents a global reduction of grey and white equally (not shown here) [12–16]. There are also subtle and not entirely consistent abnormalities of various basal ganglia structures (Table 2) [12, 13, 15, 17] and, most striking, a consistent and significant reduction of the volume of the posterior inferior cerebellar vermis (Table 2) [16, 18, 19]. These findings support other biological models of ADHD implicating frontal–basal ganglia and dopaminergic circuits [20].
Anatomic brain magnetic resonance imaging studies in ADHD
Anatomic brain magnetic resonance imaging studies in ADHD: basal ganglia and cerebellum findings
These abnormalities appear to be a fixed, rather than an ongoing, process. Longitudinal changes during childhood and adolescence did not differ between our 73 ADHD subjects, and 75 healthy matched controls studied prospectively with 2 and 4 year follow-up rescan [21]. These anatomic abnormalities are not due to stimulant drug effects since the 17 medication-naïve patients showed the same brain pattern. Thus, in contrast to COS (described below), the smaller total brain and cerebellar vermis in ADHD, seems due to an earlier process (at least before age 4, the earliest age at which these scans were obtained). Moreover, since the trajectories of the total and regional brain development does not differ between ADHD patients and controls, severe inattention or impulsivity per se is not likely to cause the late progressive abnormalities seen for the schizophrenic group. Based on these findings, candidate processes for the abnormalities in ADHD focus on prenatal or early postnatal events.
Childhood onset schizophrenia
Schizophrenia is a heterogeneous illness both with respect to clinical phenomenology and aetiology. Age of onset of illness has provided an avenue to understanding of disease across all of medicine, with earlier onset cases often having more striking or homogeneous risk factors and/or differing pathophysiologies. For this reason, a study of COS, defined as onset of psychotic symptom by the 13th birthday, has been ongoing since 1990 at the National Institute of Mental Heath. These severely ill patients are less contaminated with substance abuse and other factors seen in later-onset populations. Moreover, the childhood onset illness appears continuous with respect to clinical and neurobiological measures [22].
A comparison of 46 COS and 84 healthy volunteers extends our previous studies [23, 24] showing that COS had decreased total brain volume and increased lateral ventricular volume as seen in adult onset schizophrenia. Unlike the subtle global decrease in grey and white matter seen in ADHD, the decreased brain volume here is due exclusively to the robust 10% decrease in cortical grey matter, as the total white matter volume does not differ significantly between the COS and healthy groups. These findings are summarized in Table 3.
Brain MRI volumes (mL) for childhood onset schizophrenics (n = 46) and healthy controls (n = 82)
In addition, Table 4 and Figure 1 show prospective longitudinal brain MRI rescan measures for these COS cases. There is increasing ventricular volume, and decreasing cortical grey and medial temporal lobe structures across 2, 4 and (for a smaller number of cases), 6 years after their initial scan. Here, too, regional grey–white segmentation showed the progressive loss to be for grey matter only [25–28]. Figure 1 shows this progressive loss during adolescence for regional cortical grey matter.
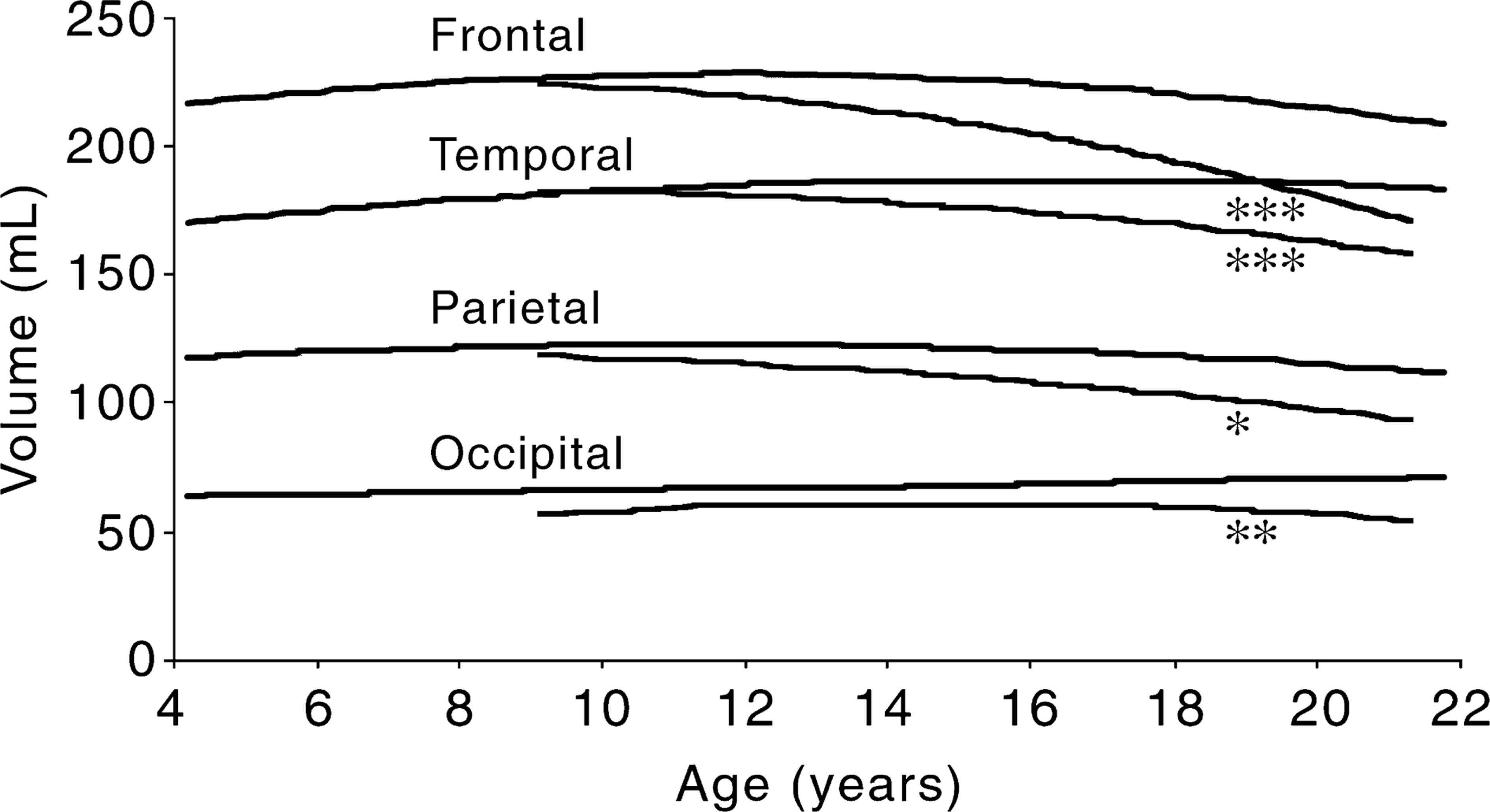
Progressive brain changes during adolescence in COS
These observations have led us to view adolescence as a time-limited window in which progressive brain changes in schizophrenia may be observed.
Clinically these changes parallel a decline in full-scale IQ [29] and lack of normal maturation of neurological status [30] for these patients. These progressive changes are not likely to be due to medication. Data from a contrast group of 17 children with transient psychotic symptoms treated with the same typical and atypical antipsychotic drugs, but without clinical progression to schizophrenia (MDI or ‘multidimensionally impaired’ group) [31] show no progressive ventricular volume increase or grey matter volume loss relative to healthy controls [32].
These findings are consistent with the neuropathology of schizophrenia [33, 34]. The loss of cortical volume is consistent with models of progressive widespread, subtle disruption and decreasing connectivity of multiple cortical regions hypothesized for schizophrenia [35, 36].
Regulation of brain development: clinical implications
Brain plasticity
A combination of genetic and environmental factors controls the development of the CNS. The process is highly plastic, involving a series of sequential and parallel events, and a disruption of one step can greatly influence later processes [37].
Because of high malleability in the human brain, it would seem possible that the stress and abnormal thought and behaviour experienced by psychotic patients might be responsible for the progressive loss of grey matter and cytoarchitectonic deficiencies found in these individuals. For example, could schizophrenic behaviour be a cause rather than an effect of the decreased and decreasing frontal grey brain volume? Could severe inattention and impulsivity produce small grey matter volume? Although the influence of plasticity is most pronounced during the critical periods of postnatal development and declines during adolescence, a significant level of adaptability continues to exist even into adulthood. The fact that developed brains can be induced into schizophrenic states [38] is at least consistent with the idea that changes in synaptic efficacy could play a critical role in the onset of schizophrenia [39, 40].
Important preclinical demonstrations of brain plasticity have suggested two distinct forms. A classic study by Hubel, Wiesel and LeVay exemplifies the first [41]. Specifically, monkeys with one eye completely covered from birth were found to develop a greater number of afferents from the lateral geniculate nucleus to the visual cortex of the open eye than to the sutured eye. In other words, synaptic pathways failing to attain the expected level of stimulation by the environment lost efficiency, while those that were properly activated increased in connectivity and gained efficiency [42]. This process has been called experience-expectant maturation. A second form of plasticity can be demonstrated by studies with rats which have suggested that different levels of general sensory stimulation have distinct influences on brain development. Animals raised in ‘enriched’ environments have been found to have significantly greater cerebral cortical weight and thickness, dendritic length and spine density, total dendritic material per neuron, acetylcholinestrase activity, RNA/DNA ratio, RNA diversity and brain-specific protein concentrations than animals raised in ‘impoverished’ environments [43]. These results suggest that environmental stimuli that are not predetermined and vary between individuals in a species contribute to brain structure: a second form of plasticity described as experience-dependent maturation. Applied to the schizophrenic patient, then, could experiencedependent plasticity cause the cytoarchitechtonic decline and grey matter deficiencies?
Probably not. In general, experiments involving plasticity apply drastic conditions. In the aforementioned examples, animals were exposed to highly unusual if not devastating visual and sensory environments. Other studies involve equally extreme measures. For instance, plasticity is often studied through the induction of lesions. Neuronal input to a target zone is severed, and anatomical, biochemical and electrophysiolocial data are collected to determine whether re-innervation has occurred with other healthy afferent areas [44]. ‘Postlesional plasticity’ has been found in many regions including the cerebral cortex [45] and cerebellum [46].
In considering the relationship between plasticity and the onset of schizophrenia, therefore, it is important to recognize that even the abnormality in environmental input to the brain of a schizophrenic patient is considerably less severe than the conditions involved with most of the experiments exploring plasticity. The possibility that abnormal behaviour per se could induce abnormal connectivity exists, and experimental studies of plasticity using functional imaging may demonstrate such effects. For structural findings reviewed here, however, it seems more probable that schizophrenic behaviours result from these abnormalities rather than the reverse.
Processes that may underlie the brain abnormalities in attention deficit hyperactivity disorder
While total brain volume is slightly but significantly decreased in ADHD, the total and regional growth curves for this group run parallel to normal brain curves [21]. Thus, the events leading to the anatomic differences in ADHD probably occur early in neurodevelopment. The period of critical neurodevelopmental organizational events [47] is influenced by a complex interplay of various genetic and epigenetic factors such as neurotransmitters, neurotrophins and growth factors, and cytokines, along with hormonal influences [48, 49]. Even a subtle injury during this vulnerable process of neurodevelopment and organization in utero (second or third trimester) can affect the brain development and size globally, thus explaining the changes seen in ADHD [50]. Various phenomenological observations in ADHD may help us understand the mechanism of this global size reduction.
Incidence of pregnancy-related complications, and prematurity are slightly higher in ADHD [51]. However, the strongly genetic nature of ADHD, and lack of specificity of findings for what mediates the effects, for example prematurity, leaves us without a specific hypothesis of what might mediate this subtle global reduction in brain volume. The striking volume reduction of the posterior inferior lobule of cerebellar vermis in ADHD are also important in this regard (see Table 1). This region of cerebellar vermis is highly dopaminergic [52] and appears, like most brain volumetric measures to be highly heritable, [Giedd J, Castellanos X: unpublished data]. The posterior inferior vermis, thus, may be an important part of cerebello-striato-frontal circuitry and hence in the aetiopathogenesis of ADHD. Recently, it was observed that brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) mRNAs (important brain growth modulators) are colocalized to specific ventral mesencephalic dopaminergic neurons [53]. Thus any defect in dopaminergic circuitry could alter these growth modulators and ultimately the brain size and development. Allelic variations of several dopaminergic genes (DA transporter, DA receptor 4) have been associated with ADHD [54], however, these alleles have been examined in ADHD populations and show no significant association with any of these brain volumetric measures [55]. Conversely, subtle abnormalities in BDNF, NT-3 and others may cause localized abnormality of dopamine circuitry. This awaits exploration. Finally, differences in the volume of caudate nucleus could also point us toward aetiopathogenic mechanisms.
The caudate nucleus volume is decreased in boys with ADHD [56, 57]. This is interesting as healthy girls have a larger caudate nucleus, probably owing to higher concentration of oestrogen receptors in the region. In the CNS, oestrogen plays a role in regulation of gene transcription, can act as an antioxidant for toxic substances, and can also act as a neuroprotectant [58]. In theory, this might explain the smaller caudate volume in ADHD boys and lower incidence of ADHD in girls. However, this would not account for the fact that brain MRI findings for ADHD boys and girls are very similar (see Table 1).
Candidate processes for childhood onset schizophrenia
The smaller brain sizes in COS and ADHD indicate that some compartment or compartments in the brain are being reduced. Two major theories have been put forth that name different compartments as the source of the loss [59]. The first proposes that the cause is either a developmental failure to produce the proper number of neurons or it is neurodegenerative disease that results in an overall neuronal loss in the brain. The second theory proposes that the differences in brain volume are due to reduction of or reduced formation of the neuropil, which consists of the axonal and dendritic arbours that make up a large fraction of the grey matter in the cortex [34, 60]. This reduction of the neuropil could result from reduction in the numbers of synaptic connections made between neurons. In humans, detailed post-mortem electron microscopy work has shown that there is normally a postnatal increase in synaptic connections followed by a decrease in synaptic connections that extends as far as midadolescence in some parts of the brain [61].
As noted above, the decreased volume in ADHD may be related to the first factor (a reduced number of neurons generated during early neurodevelopment) while COS is due to the second factor (an excessive degree of synapse reduction of an initially fairly normal number of synapses and neurons.
Post-mortem studies to determine if there is a reduced cell number in the brains of people with schizophrenia have not produced consistent results in many parts of the brain [34, 60]. Conversely, a number of studies using stereological methods have found increased densities of neurons in the prefrontal cortex and temporal lobe [62–64]. In addition, these neurons have smaller cell bodies, and it has been shown that the cell bodies are proportional to the level of dendritic and axonal arbourization [60]. Other evidence shows decreased loss of expression of synaptic markers in schizophrenia [65] and most recently, decreased expression of several functional genes important in presynaptic function and development [66]. Thus, while there is not direct evidence for synaptic loss in COS there is a good deal of evidence that supports the reduced neuropil hypothesis. Detailed, quantitative electron microscopy studies are needed to directly address this crucial question as to the cellular locus of the brain abnormality in COS. The two theories are not mutually exclusive, but the evidence is strong in favour of a reduced neuropil playing a prominent role in schizophrenia (see [34, 60] for details).
In normal development it is known that excess numbers of synaptic connections are formed initially and as development progresses the extra connections are eliminated or pruned. Considerable information is available concerning some of the mechanisms regulating the generation of neurons, development of the synapses between the neurons and the process by which some synapses are lost during development. We will briefly review some of this information, focusing primarily on the process of synapse elimination, since the available evidence suggest that this may play a key role in COS.
Synapse elimination is particularly well characterized in two systems, namely in the climbing fibre (CF) system which innervates Purkinje cells (PC) in the cerebellum, and in motor neurons which innervate skeletal muscle fibres, although synapse elimination has been demonstrated in many systems [67]. The possible mechanisms behind pathologic elimination of the synapses are too numerous and complex to thoroughly review here. However, a general idea of the possibilities can be gleaned from the well-studied model of elimination at the neuromuscular junction (NMJ). In this system, at birth in the rodent, all muscle fibres are innervated by at least two axons, and by 3 weeks postnatally all but one axon has been eliminated from all of the muscle cells [68]. This process requires activation of the system because it fails to take place in paralysed preparations. Some of the steps in this coupling between activation and synapse elimination are becoming clarified, and seem to involve a number of protein kinases, enzymes which regulate protein function by altering their state of phosphorylation. In particular, neurotransmitter receptors are known to be targets for kinase action, and their physiology and stability in the membrane of nerve and muscle are greatly affected by the addition of phosphate groups. One major link in the chain leading to synapse elimination, therefore, may be activation of appropriate kinases, phosphorylation of neurotransmitter receptors and subsequent, selective destabilization of the synapses involving those receptors [69]. Loss of synaptic acetylcholine receptors has been shown to be an early step in the loss of synapses at the NMJ [70]. Selective loss of synapses may be due to differential activation of localized kinases, which have different effects on receptor stability.
Evidence for the involvement of a kinase (protein kinase C or PKC) in synapse elimination in the central nervous system has been obtained in experiments on the CF/PC system mentioned above. On average PCs in the cerebellum are innervated by 3.5 CFs at birth but as normal development proceeds, the number of CFs forming a synaptic connection with each PC is reduced to one [67].
Genetic manipulations have given some clues as to the mechanisms involved in synapse elimination. Mutation of one isoform of PKC which inactivates the kinase has been shown to block synapse elimination in the mouse cerebellum [71]. In the mouse cerebellum the expression of the PKC-gamma isoform goes up in PCs during the period during which multiple synapses from CFs are reduced to single innervations. In mutant mice in which PKC-gamma is inactivated there is marked reduction in the elimination of the initial multiple innervations of PCs by CFs. Thus the activity of this particular molecule, PKC, is essential for normal elimination of redundant synapses. It might be that excessive activity of the enzyme would produce the abnormally high degree of synapse elimination postulated to be related to COS.
Trophic factors have been shown to have powerful effects on neuronal survival and synaptic structure and function during development [50,72–74]. Competition for a limited supply of trophic material has been postulated to account for at least a portion of the synapse loss that occurs during development. Blockade of the trophin BDNF has been shown to prevent the normally occurring pruning that is essential for development of the normal architecture of the visual cortex [75] and an inadequate supply could result in inadequate development or survival of cortical synapses.
Cortical neurons are generated during a sharply delimited time window relatively early in development and the total number of neurons can be drastically affected by various manipulations during this period. For instance, it has been shown that a neuropeptide, vasoactive intestinal peptide (VIP) controls the duration of the mitotic cycle in the neuroblasts in the ventricular germinal zone and that this affects the total number of neurons that get born during development. Antagonists of VIP given during the critical neuron-generating period (and only during this period) result in markedly microcephalic animals [74]. Some such interference with the process of neuron generation could be involved in the early deficit in brain size seen in ADHD.
Following on the initial speculation of Feinberg [76], McGlashan and Hoffman [35] and others have used computer modelling of neural networks to test the plausibility of the ‘over-pruning’ hypothesis of schizophrenia. Their experiments characterized the behaviour of over-pruned neural networks and found strong parallels between schizophrenia and the networks’ behaviour.
In summary, as we hope this report illustrates, clinical brain imaging studies are bringing child psychiatry and the developmental neurosciences ever closer. We present here some possible candidates, but expect that much greater specificity and converging information will emerge from genetic linkage and association studies that will be carried out over the next decade. Some of these studies are ongoing with these populations at the NIMH.